
Abstract
A unique feature of female germ cell development in mammals is their remarkably long arrest at the prophase of meiosis I, which lasts up to 50 years in humans. Both dormant and growing oocytes are arrested at prophase I and completely lack the ability to resume meiosis. Here, we show that the prolonged meiotic arrest of female germ cells is largely achieved via the inhibitory phosphorylation of Cdk1 (cyclin-dependent kinase 1). In two mouse models where we have introduced mutant Cdk1T14AY15F which cannot be inhibited by phosphorylation (Cdk1AF) in small meiotically incompetent oocytes, the prophase I arrest is interrupted, leading to a premature loss of female germ cells. We show that in growing oocytes, Cdk1AF leads to premature resumption of meiosis with condensed chromosomes and germinal vesicle breakdown followed by oocyte death, whereas in dormant oocytes, Cdk1AF leads to oocyte death directly, and both situations damage the ovarian reserve that maintains the female reproductive lifespan, which should be around 1 year in mice. Furthermore, interruption of the inhibitory phosphorylation of Cdk1 results in DNA damage, which is accompanied by induction of the Chk2 (checkpoint kinase 2)-p53/p63-dependent cell death pathway, which eventually causes global oocyte death. Together, our data demonstrate that the phosphorylation-mediated suppression of Cdk1 activity is one of the crucial factors that maintain the lengthy prophase arrest in mammalian female germ cells, which is essential for preserving the germ cell pool and reproductive lifespan in female mammals.
Similar content being viewed by others

Introduction
In mammals, female germ cells enter meiosis in the embryonic gonad, but they are subsequently arrested at the dictyate stage of prophase I in the form of dormant oocytes that are enclosed within primordial follicles1. Such meiosis arrest can last for a very long time, which can be, for example, up to 50 years in humans and more than 1 year in mice. In addition, before the oocytes can resume meiotic maturation, they must undergo a prolonged growth period from small primary oocytes to fully grown oocytes. During such growth, which usually takes weeks to months or even longer, the oocytes remain arrested at prophase I2,3,4. In other words, throughout the lengthy dormancy and growth of oocytes, the oocytes are incompetent for meiosis resumption5.
The pool of primordial follicles with dormant oocytes provides the ovarian reserve that maintains the female reproductive lifespan4. Only a limited number of dormant oocytes are recruited into the growth phase, and of these only some will eventually reach the fully grown stage and gain the ability to resume meiosis (illustrated in Figure 1A). Importantly, the maintenance of genomic integrity in the meiotically incompetent oocytes is crucial for generating future healthy progeny. The prophase I arrest of the small oocytes could thus be an essential mechanism for maintaining the genomic integrity of female germ cells.

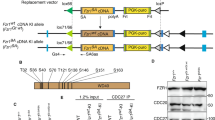
Illustrations of oocyte prophase I arrest and the strategy for expressing the Cdk1AF allele in dormant and growing oocytes. (A) All of the dormant and growing oocytes remain in prophase I arrest with GV and are meiotically incompetent. Only the fully grown oocytes are meiotically competent and have the capability of resuming meiosis. Gdf9-Cre recombinase starts to be expressed in dormant oocytes in primordial follicles, and Zp3-Cre starts to be expressed only in growing oocytes in developing follicles. GV, germinal vesicle. (B) Cdk1 remains in an inactive state due to its phosphorylation at T14 and Y15 by Wee1/Myt1. Cdk1 activation requires dephosphorylation of T14 and Y15 by Cdc25. Cdk1 with mutation of T14 and Y15 to A14 and F15 cannot be phosphorylated and therefore Cdk1 activity cannot be inhibited by Wee1/Myt1. (C) Oocyte-specific expression of the Cdk1AF allele starting from dormant oocytes as mediated by Gdf9-Cre or starting from growing oocytes as mediated by Zp3-Cre.
Although the prophase I arrest in dormant and growing oocytes appears to be similar to that of fully grown germinal vesicle (GV)-stage oocytes, their cell cycle behaviors are fundamentally different. For example, in mice, meiotic resumption can occur in fully grown oocytes either upon stimulation by luteinizing hormone or upon release from the follicular environment in vitro. However, in growing oocytes, such as oocytes smaller than 60 μm or oocytes from the ovaries of postnatal day (PD) 15 or younger mice, release from the follicular environment in vitro does not lead to the resumption of meiosis6,7.
Based on published data, we hypothesize that the molecular regulation of meiotic arrest in dormant and growing oocytes is distinct from mechanisms maintaining the GV-stage of fully grown oocytes. For example, high cyclic AMP (cAMP) levels maintain the meiotic arrest in fully grown oocytes, but they do not maintain the meiotic arrest of smaller oocytes because even if the cAMP levels are decreased in growing oocytes, these oocytes do not resume meiosis8,9. In addition, prophase I arrest in fully grown mouse oocytes is maintained by the constant degradation of cyclin B1 by the anaphase-promoting complex/cyclosome (APC/C)-Cdh1, which keeps the activity of cyclin-dependent kinase 1 (Cdk1) low10. However, the prevention of cyclin B1 degradation in growing Cdh1-null oocytes, where APC/C activity is suppressed, is not sufficient to release the oocytes from prophase I arrest, and these growing Cdh1-null oocytes still do not resume meiosis in vivo (i.e., inside the follicles)11. Moreover, increasing the amount of Cdk1 protein by microinjection of Cdk1 mRNA into growing mouse oocytes does not lead to the resumption of meiosis12. Similarly, more rapid GV breakdown (GVBD) occurs in fully grown mouse oocytes only when the activity of Cdk1 is increased by knocking out the lysine-specific demethylase 113 or by downregulating Wee1B that can phosphorylate and inhibit Cdk114, but GVBD is not seen in growing oocytes in these mutant models. These results raise the important question of whether Cdk1 activity is regulated in dormant and growing oocytes, and if so, what the physiological significance of Cdk1 regulation is in these immature oocytes.
By generating two knock-in mouse models to express a Cdk1 variant that is non-inhibitable by phosphorylation (Cdk1AF) in dormant and growing oocytes, we were able to demonstrate that the simple phosphorylation-mediated suppression of Cdk1 activity plays an important role in maintaining the seemingly complicated lengthy dictyate arrest of mammalian female germ cells. This mechanism also prevents the accumulation of DNA damage and subsequent death of female germ cells, which is essential for maintaining the normal female reproductive lifespan.
Results
Expression of Cdk1AF in dormant mouse oocytes causes DNA damage and rapid oocyte death
To first determine the effects of premature Cdk1 activation on dormant oocytes, we generated a mouse model in which two point mutations lead to substitutions of T14 and Y15 in Cdk1 with alanine 14 and phenylalanine 15 (Figure 1B and Supplementary information, Figure S1). Because these two amino acids are non-phosphorylatable, Cdk1 with these two mutations becomes non-inhibitable by phosphorylation (Cdk1AF)15,16 (Figure 1B). We introduced the T14A and Y15F mutations into one Cdk1 allele to generate the Cdk1T14AY15F (in short, Cdk1AF) allele, but with a loxP-flanked STOP (S) sequence in front of the Cdk1AF allele to prevent its expression in the absence of Cre recombinase. Wild-type Cdk1 is expressed by the other allele (Supplementary information, Figure S1). The mice carrying one floxed STOP-Cdk1AF (SAF) allele and one wild-type Cdk1 allele are referred to as Cdk1+/SAF mice. The Cdk1+/SAF mice displayed normal ovarian development and fertility (see below) and were used as controls.
By crossing Cdk1+/SAF mice with growth differentiation factor 9 (Gdf-9)-Cre mice in which the Cre recombinase is efficiently expressed in dormant oocytes of primordial follicles17,18 (Figure 1C), we specifically deleted the STOP sequence and induced the expression of the Cdk1AF in one allele of the Cdk1 gene (referred to as OoCdk1+/AF; Gdf9-Cre) in dormant and growing oocytes (illustrated in Figure 1C), while the other allele carried the wild-type Cdk1 gene (OoCdk1+).
To maintain the normal length of female reproductive life, the majority of primordial follicles must be maintained in a dormant state throughout the reproductive lifespan. In comparison with the healthy ovaries from control Cdk1+/SAF mice at PD8 (Figure 2A and 2C) and PD16 (Figure 2E), the mutant OoCdk1+/AF; Gdf9-Cre ovaries contained only a few follicles at PD8 (Figure 2B and 2D, arrows, and Supplementary information, Figure S2G), and they were depleted of almost all oocytes by PD16 (Figure 2F and Supplementary information, Figure S2G). These results indicate that suppression of Cdk1 kinase activity is needed to maintain the survival of dormant oocytes. This early loss of all female germ cells makes the OoCdk1+/AF; Gdf9-Cre mice infertile (Figure 2G).

Expression of Cdk1AF in dormant oocytes causes DNA damage and oocyte depletion in OoCdk1+/AF; Gdf9-Cre mice. (A-F) Hematoxylin-stained sections of mouse ovary at PD8 and PD16. By PD8, only a few follicles were left in OoCdk1+/AF; Gdf9-Cre ovaries (B arrows), and the oocytes of surviving primordial follicles remained arrested at GV stage (D, arrows). The follicular structures had mostly disappeared in the mutant ovaries by PD16 (F). As controls, normal ovaries of PD8 (A and C) and PD16 (E) Cdk1+/SAF mice containing healthy follicles are shown. The experiments were repeated more than three times each, and for each time and each age ovaries from one mouse of each genotype were used. (G) Comparison of the average cumulative number of pups per OoCdk1+/AF; Gdf9-Cre female (dotted blue line) and per Cdk1+/SAF female (solid black line), indicating that the OoCdk1+/AF; Gdf9-Cre females were infertile. The numbers of females used are shown as n. (H and I) Prominent γ-H2AX staining was observed in dormant oocytes enclosed in primordial follicles of PD6 OoCdk1+/AF; Gdf9-Cre ovaries (I, arrows) but not in the control oocytes (H, arrowheads). DDX4 was used to label the oocytes and the same sections were counterstained with hematoxylin to visualize ovarian histology. The experiments were repeated more than three times each.
We checked whether the elevated Cdk1 activity in dormant oocytes causes oocyte death via increased DNA damage, because in human KBM-7 cell line elevated Cdk1 kinase activity can cause DNA damage19. One of the first cellular responses to DNA double-strand breaks is the phosphorylation of histone H2AX20. Immunostaining for phosphorylated histone H2AX at Ser139 (known as γ-H2AX) in PD6 OoCdk1+/AF; Gdf9-Cre ovarian sections revealed substantially increased DNA damage in the dormant oocytes (Figure 2I, arrows). In contrast, almost no γ-H2AX foci were seen in control Cdk1+/SAF oocytes (Figure 2H, arrowheads). These results demonstrate that the direct suppression of Cdk1 kinase activity by its phosphorylation is essential for protecting genomic integrity of dormant oocytes arrested at prophase I, thereby maintaining the female reproductive lifespan.
Premature meiotic resumption in growing oocytes expressing Cdk1AF
The rapid depletion of all of the oocytes in OoCdk1+/AF;Gdf9-Cre ovaries makes it impossible to obtain mutant oocytes to study the underlying mechanisms of oocyte depletion caused by Cdk1AF. Therefore, we crossed the Cdk1+/SAF mice with Zona pellucida 3 (Zp3)-Cre mice21 to introduce the expression of Cdk1AF starting in the growing oocytes but not in the dormant oocytes (Figure 1A and 1C). The resulting mice are referred to as OoCdk1+/AF; Zp3-Cre mice. We have previously validated that the Zp3 promoter-mediated Cre recombinase expression is oocyte-specific and is expressed only in growing oocytes but not in dormant oocytes22. We used small growing oocytes from PD14 ovaries from OoCdk1+/AF; Zp3-Cre mice in the subsequent experiments.
We first isolated growing oocytes from the ovaries of PD14 OoCdk1+/AF; Zp3-Cre and control Cdk1+/SAF mice and measured their Cdk1 kinase activity. As shown in Figure 3A, Cdk1 kinase activity was significantly elevated in the PD14 OoCdk1+/AF; Zp3-Cre oocytes, and as expected it was accompanied by a decrease in phosphorylation at Y15 of Cdk1 due to the expression of a Cdk1AF allele in OoCdk1+/AF; Zp3-Cre oocytes (Figure 3A), and the stability of Cdk1 protein in growing oocytes seems to be positively affected by its dephosphorylation (Figure 3A). We found that the OoCdk1+/AF; Zp3-Cre mice were completely infertile (Figure 3B). To study how the expression of Cdk1AF in oocytes leads to infertility, we compared the follicular development in OoCdk1+/AF; Zp3-Cre mice with that in Cdk1+/SAF mice. No apparent morphological difference was found in PD8 ovaries of Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre mice, where ovaries of both genotypes had mostly primordial and some growing follicles (Supplementary information, Figure S2A and S2B) and contained comparable numbers of total follicles (Supplementary information, Figure S2H). By PD14, however, the OoCdk1+/AF; Zp3-Cre ovaries (Supplementary information, Figure S2D) appeared smaller than the Cdk1+/SAF ovaries (Supplementary information, Figure S2C) and contained less number of follicles (Supplementary information, Figure S2H). By PD24, most of the growing follicles in OoCdk1+/AF; Zp3-Cre ovaries were undergoing atresia and contained degenerating oocytes (Supplementary information, Figure S2F, arrows) as compared with the healthy follicles in the Cdk1+/SAF ovaries (Supplementary information, Figure S2E, arrows). Quantification results showed that PD24 OoCdk1+/AF; Zp3-Cre ovaries contained significantly less number of follicles than in the Cdk1+/SAF ovaries (Supplementary information, Figure S2H). Compared with the healthy control Cdk1+/SAF ovaries at 6 weeks (Figure 3C) and 3 months (Figure 3E) of age, the mutant OoCdk1+/AF; Zp3-Cre ovaries contained only a few follicles at 6 weeks of age (Figure 3D, arrows and Supplementary information, Figure S2H), and they were depleted of almost all oocytes by 3 months of age (Figure 3F and Supplementary information, Figure S2H).

Premature in vivo resumption of meiosis in small growing oocytes in OoCdk1+/AF; Zp3-Cre mice. (A) Elevated Cdk1 kinase activity and total Cdk1 level but decreased inhibitory phosphorylation of Cdk1 (p-Cdk1, Y15) were observed in OoCdk1+/AF;Zp3-Cre oocytes, suggesting that Cdk1AF was successfully introduced into the growing oocytes in the mutant mice. β-Actin was used as the loading control. For the Cdk1 kinase assay, 10 oocytes per reaction were used. For Western blots (WB), lysate from 200 oocytes was loaded in each lane. The experiments were repeated more than three times each. (B) Comparison of the average cumulative number of pups per OoCdk1+/AF; Zp3-Cre female (red dotted line) and per Cdk1+/SAF female (solid blue line). All OoCdk1+/AF; Zp3-Cre females were infertile. The numbers of females are shown as n. (C-F) Hematoxylin-stained sections from mouse ovary at 6 weeks and 3 months. By 6 weeks of age, only a few follicles were observed in OoCdk1+/AF; Zp3-Cre ovaries (D, arrows), and healthy follicular structures had mostly disappeared in 3-month-old OoCdk1+/AF;Zp3-Cre ovaries (F). Control Cdk1+/SAF ovaries (C and E) contained healthy oocytes and follicles. CL, corpus luteum. The experiments were repeated more than three times each, and for each time and each age; ovaries from one mouse of each genotype were used. (G, H) Premature in vivo GVBD and chromosome condensation in small primary oocytes of OoCdk1+/AF; Zp3-Cre mice. Ovaries from PD14 OoCdk1+/AF; Zp3-Cre and Cdk1+/SAF females were embedded in paraffin, and serial sections of 8-μm thickness were prepared and stained with hematoxylin. (G) A wild-type primary oocyte in a secondary follicle showing the GV (G, arrowhead) from a PD14 Cdk1+/SAF ovary. (H) A representative growing primary oocyte from a PD14 OoCdk1+/AF; Zp3-Cre ovary where GVBD and chromosome condensation had already occurred (H, arrow). The experiments were repeated more than three times each, and for each time and each age ovaries from one mouse of each genotype were used. (I) Quantification of the percentages of oocytes that resumed meiosis in vivo in PD14 ovaries within primary, secondary, and preantral follicles. Numbers of each type of follicles included (n) are shown. (J-Q) Condensed chromosomes with cohesin localization at the chromosome axis in PD14 OoCdk1+/AF; Zp3-Cre oocytes. Staining with DAPI for DNA (L, M, blue), SMC3 for cohesin (N, O, green), CREST for kinetochores (P, Q, red), and merge (J, K) in OoCdk1+/AF; Zp3-Cre and Cdk1+/SAF oocytes. The experiments were repeated more than three times each, and the representative images from one experiment are shown. (R, S) Increased GVBD in mutant OoCdk1+/AF; Zp3-Cre oocytes upon release from the follicular environment. In wild-type PD14 Cdk1+/SAF oocytes, GV was maintained even if the oocytes were released from the follicles by enzymatic digestion (R, arrowheads). However, an increased rate of GVBD was seen in PD14 OoCdk1+/AF; Zp3-Cre oocytes upon release from the follicles (S, arrows) by enzymatic digestion of ovaries from PD14 mice. Representative Hoechst-stained oocyte nuclei are shown to the right. Quantification of GV and GVBD rates of oocytes immediately after their isolation from follicles in PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre mice (T) and after further in vitro culture (U). The numbers of oocytes analyzed (n) are shown.
In the control mouse ovary, all analyzed oocytes within small or large follicles remained arrested at the GV stage at any given age, as seen in the ovarian section of PD14 control Cdk1+/SAF mice (Figure 3G, arrowhead). However, we found that the young PD14 OoCdk1+/AF; Zp3-Cre oocytes, where Cdk1AF was expressed, had already undergone GVBD within the follicles in vivo and their chromosomes were condensed (Figure 3H, arrow).
Quantification results showed that within the primary, secondary, and preantral follicles in PD14 OoCdk1+/AF; Zp3-Cre ovaries, 1.5% (n = 220), 46.5% (n = 97), and 64.4% (n = 78) of the oocytes had already resumed meiosis, respectively (Figure 3I). This was in sharp contrast to the control PD14 Cdk1+/SAF ovaries in which all oocytes were at the meiotically arrested GV stage (Figure 3I).
Using chromosome spreads, we observed condensed chromosomes in PD14 OoCdk1+/AF; Zp3-Cre oocytes that had already undergone GVBD (Figure 3K and 3M, arrows). In sharp contrast, the chromosomes remained decondensed in the control oocytes (Figure 3J and 3L, arrowheads). This indicates that meiosis had already resumed in the small oocytes of PD14 OoCdk1+/AF; Zp3-Cre ovaries. The homologous chromosome arms are held together by cohesin during meiosis I until late metaphase I when cohesin is cleaved by separase23. Simultaneous staining of SMC3 (structural maintenance of chromosome 3) (Figure 3O, arrows), and CREST (Figure 3Q, arrows) showed that SMC3 was localized along the chromosome axis, indicating that the chromosomes were at prometaphase I in the OoCdk1+/AF; Zp3-Cre oocytes. These results demonstrate that these growing oocytes expressing Cdk1AF are not arrested at the diplotene stage of prophase I.
Earlier studies have shown that oocytes isolated from prepubertal mouse ovaries or oocytes that are smaller than 60 μm in diameter are meiotically incompetent because they fail to resume meiosis spontaneously when denuded and cultured in vitro6,7. This was observed in oocytes isolated from PD14 control Cdk1+/SAF ovaries where all oocytes were arrested at the GV stage and contained decondensed DNA (Figure 3R, arrowheads, and Figure 3T control oocytes, n = 320). We also isolated oocytes from PD14 mutant OoCdk1+/AF; Zp3-Cre ovaries and quantified the proportion of oocytes that had resumed meiosis as evidenced by GVBD. We observed premature in vivo resumption of meiosis in PD14 mutant OoCdk1+/AF; Zp3-Cre oocytes as small as 25 μm in diameter. The results showed that 48% of the oocytes isolated from PD14 mutant OoCdk1+/AF; Zp3-Cre ovaries, which were all smaller than 60 μm in diameter, had undergone premature GVBD with condensed DNA (Figure 3S, arrows, and quantification in Figure 3T, n = 450).
Notably, when the isolated growing OoCdk1+/AF; Zp3-Cre oocytes were cultured in vitro for 16 h, the proportion of mutant oocytes undergoing GVBD increased to 68% (Figure 3U, n = 450), whereas none of the control Cdk1+/SAF oocytes underwent GVBD in the in vitro culture (Figure 3U, n = 340). Therefore, the Cdk1AF increases the rate of meiosis resumption in otherwise meiotically incompetent mouse oocytes. These results indicate that the inhibition of Cdk1 kinase activity by its inhibitory phosphorylation in growing mouse oocytes is important for sustaining arrest in the middle of prophase I.
Accumulation of DNA damage in growing OoCdk1+/AF; Zp3-Cre oocytes
Increased Cdk1 activity led to premature resumption of meiosis in the OoCdk1+/AF; Zp3-Cre oocytes (Figure 3G-3U), which was followed by oocyte depletion (Figure 3F), and immunostaining for γ-H2AX in PD14 ovarian sections revealed increased DNA damage in the OoCdk1+/AF; Zp3-Cre oocytes (Figure 4B, arrows). In contrast, γ-H2AX foci were mostly absent in control Cdk1+/SAF oocytes (Figure 4A, arrowheads).

Expression of Cdk1AF leads to DNA damage and ATM activation in growing OoCdk1+/AF; Zp3-Cre oocytes. (A) Absence of γ-H2AX staining in the oocytes of control PD14 Cdk1+/SAF ovarian sections (arrowheads). (B) Prominent γ-H2AX staining in the oocytes of PD14 OoCdk1+/AF; Zp3-Cre ovaries (arrows). DDX4 was used to label the oocytes and the same sections were counterstained with hematoxylin to visualize ovarian histology. The experiments were repeated more than three times each, and representative images are shown. (C) Absence of γ-H2AX staining in isolated oocytes of control PD14 Cdk1+/SAF mice (arrowhead). (D) Strong γ-H2AX staining in isolated oocytes from PD14 OoCdk1+/AF; Zp3-Cre ovaries (arrow). (E) Quantification of γ-H2AX-positive oocytes that were isolated from PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre ovaries. Numbers of each genotype of oocytes checked (n) are shown. (F) Western blot for γ-H2AX and 53BP1 levels in oocytes isolated from PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre ovaries. β-actin was used as the loading control. Lysate from 200 oocytes was loaded in each lane. The experiments were repeated more than three times. (G-L) High level of p-ATM (S1981) accompanied by increased γ-H2AX staining in PD14 OoCdk1+/AF; Zp3-Cre oocytes (J-L, arrows), in comparison with PD14 control Cdk1+/SAF oocytes where there was almost no signal for p-ATM (S1981) or γ-H2AX (G-I, arrowheads). The experiments were repeated more than three times each, and representative images are shown.
We next isolated oocytes from PD14 OoCdk1+/AF; Zp3-Cre and control Cdk1+/SAF ovaries and immunostained for γ-H2AX. The DNA in OoCdk1+/AF; Zp3-Cre oocytes showed intense γ-H2AX staining (Figure 4D, arrow), whereas the γ-H2AX signal was mostly absent in the DNA of control Cdk1+/SAF oocytes (Figure 4C, arrowhead). Quantification of the γ-H2AX foci-positive oocytes indicated that 94% (n = 85) of the OoCdk1+/AF; Zp3-Cre oocytes were positive for nuclear γ-H2AX foci compared with only 4% (n = 80) of the control Cdk1+/SAF oocytes (Figure 4E). Western blot analysis also confirmed the increased level of γ-H2AX in OoCdk1+/AF; Zp3-Cre oocytes (Figure 4F). The level of 53BP1, another marker of DNA double-strand break24 was also increased in OoCdk1+/AF; Zp3-Cre oocytes (Figure 4F). These results indicate that the elevated Cdk1 kinase activity leads to DNA damage in the growing OoCdk1+/AF; Zp3-Cre oocytes and that this is apparently the cause of the accelerated oocyte death.
The Cdk1-ATM cascade activates Chk2 in OoCdk1+/AF; Zp3-Cre oocytes
In somatic cells, DNA double-strand breaks cause the activation of ATM, the master regulator of the DNA damage checkpoint. ATM is the major kinase involved in the phosphorylation of γ-H2AX during the cellular response to double-strand breaks25,26. However, the induction of DNA damage in fully grown mouse oocytes does not lead to their death or prevent the oocytes from maturing because in fully grown oocytes the DNA damage-response pathway is not activated effectively27,28.
To test whether the DNA damage-response pathway in growing OoCdk1+/AF; Zp3-Cre oocytes is triggered, we analyzed the levels of p-ATM (S1981) in relation to γ-H2AX in these oocytes. We found that both p-ATM (Figure 4J, arrow) and γ-H2AX (Figure 4K, arrow) foci were present in the nuclei of GVBD oocytes in PD14 OoCdk1+/AF; Zp3-Cre ovaries, whereas staining for p-ATM (Figure 4G, arrowhead) and γ-H2AX foci (Figure 4H, arrowhead) were absent in control Cdk1+/SAF oocytes. In addition, we also observed DNA damage and increased p-ATM signal in analyzed GV stage OoCdk1+/AF; Zp3-Cre oocytes (Supplementary information, Figure S3B, arrows). Thus, these results suggest that elevated Cdk1 activation in growing oocytes triggers DNA damage, and unlike in fully grown oocytes, the DNA damage in growing oocytes lead to oocyte death.
Ablation of Chk2 prevents the death of OoCdk1+/AF; Zp3-Cre oocytes
Chk2 is a downstream effector of ATM kinase that responds to DNA damage and it is a component of the DNA damage-sensing checkpoint29,30,31. ATM activates Chk2 by phosphorylating it at T6832. We found that although p-Chk2 (T68) and total Chk2 levels were almost undetectable in control PD14 Cdk1+/SAF oocytes, the p-Chk2 (T68) and total Chk2 levels were elevated in OoCdk1+/AF; Zp3-Cre oocytes (Figure 5A), showing that the DNA damage in OoCdk1+/AF; Zp3-Cre oocytes leads to the activation of the Chk2 pathway. We also found that the level of active caspase 3 was elevated in OoCdk1+/AF; Zp3-Cre oocytes as compared with the control PD14 Cdk1+/SAF oocytes (Figure 5B).

Cdk1AF triggers the Chk2-p63/p53 and cell death pathways in OoCdk1+/AF; Zp3-Cre oocytes. (A) Western blot for p-Chk2 (T68) and Chk2 in oocytes isolated from PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre ovaries. (B) Western blot for active caspase 3 in oocytes isolated from PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre ovaries. β-actin was used as the loading control. Lysate from 200 oocytes was loaded in each lane. The experiments were repeated more than three times each. (C, D) Hematoxylin-stained sections of mouse ovary at 2 month of age. Rescue of follicle depletion by the concomitant deletion of Chk2 in OoCdk1+/AF; Zp3-Cre ovaries. By 2 months of age, although healthy follicular structures and oocytes had mostly disappeared in mutant OoCdk1+/AF; Zp3-Cre ovaries (C), concomitant deletion of Chk2 largely prevented the death of follicles in double-mutant OoCdk1+/AF; Zp3-Cre ; Chk2−/− ovaries, and many surviving oocytes could be observed (D, arrows). The experiments were repeated more than three times each, and for each time and each age ovaries from one mouse of each genotype were used. (E) Surviving oocytes in PD60 double-mutant OoCdk1+/AF; Zp3-Cre ; Chk2−/− ovaries stained positive for γ-H2AX (arrows) indicating that DNA damage is still present. (F) Control Cdk1+/SAF ovaries with γ-H2AX-negative oocytes. The experiments were repeated more than three times, and representative images are shown. (G) Western blot for p63 and p53 levels in oocytes isolated from PD14 Cdk1+/SAF, OoCdk1+/AF; Zp3-Cre, and OoCdk1+/AF; Zp3-Cre ; Chk2−/− ovaries. β-actin was used as the loading control, and lysate from 200 oocytes was loaded in each lane. The experiments were repeated more than three times.
To provide in vivo evidence that Cdk1AF activates the Chk2 pathway and leads to the death of OoCdk1+/AF; Zp3-Cre oocytes, we generated OoCdk1+/AF; Zp3-Cre oocytes in a Chk2-null background (referred to as OoCdk1+/AF; Zp3-Cre; Chk2−/− mice). We found that although the ovaries in OoCdk1+/AF; Zp3-Cre mice were depleted of almost all follicles by 2 months of age (Figure 5C and Supplementary information, Figure S3C), OoCdk1+/AF; Zp3-Cre; Chk2−/− ovaries of the same age had many healthy-looking follicles (Figure 5D, arrows and Supplementary information, Figure S3C). These results suggest that the loss of Chk2 largely prevented the death of oocytes caused by the Cdk1AF in OoCdk1+/AF; Zp3-Cre oocytes.
We further stained for γ-H2AX in ovarian sections from 2-month-old OoCdk1+/AF; Zp3-Cre; Chk2−/− mice and found that the surviving oocytes still exhibited DNA damage (Figure 5E, arrows). As a control, γ-H2AX foci were mostly absent in the oocytes in 2-month-old control Cdk1+/SAF ovarian sections (Figure 5F, arrowheads). Thus, the prevention of oocyte death was mostly due to the lack of Chk2 or its downstream effectors but not due to the repair of DNA because the DNA damage was already present in OoCdk1+/AF; Zp3-Cre ; Chk2−/− oocytes.
We checked whether the activated Chk2 in mutant OoCdk1+/AF; Zp3-Cre oocytes (Figure 5A) triggered the accumulation of p63 and p53 which may mediate the depletion of oocytes. Indeed, the levels of p63 and p53 in OoCdk1+/AF; Zp3-Cre oocytes were much higher than in the control Cdk1+/SAF oocytes (Figure 5G). Moreover, the levels of p63 and p53 in OoCdk1+/AF; Zp3-Cre ; Chk2−/− oocytes returned to the level in Cdk1+/SAF oocytes (Figure 5G). Thus, DNA damage in OoCdk1+/AF; Zp3-Cre oocyte leads to the Chk2-dependent activation of p53 and p63, which in turn leads to oocyte elimination. These results show that the phosphorylation-mediated suppression of Cdk1 in small oocytes is essential in preventing loss of oocytes prematurely.
Discussion
The mechanisms sustaining the prolonged meiotic arrest in female germ cells are of great importance and far from clear. In the present study, we have demonstrated that the maintenance of meiotic arrest in mammalian female germ cells relies on the constant suppression of Cdk1 activity, which is largely achieved by inhibitory phosphorylation of Cdk1. This constant suppression of Cdk1 activity is not only needed for maintaining the arrested meiotic cell cycle, but it is also essential for safeguarding the integrity of the DNA in the dormant and growing oocytes. These molecular mechanisms are essential for preserving the germ cell pool and maintaining the female reproductive lifespan.
There are at least three processes known to be required for Cdk1 activity: the availability of cyclin, dephosphorylation of T14 and Y15, and phosphorylation of the T-loop. In fully grown mouse oocytes, the activity of Cdk1 remains suppressed by the maintenance of low levels of cyclin B1 and cyclin B210,33 or through Wee1B/Myt1-mediated phosphorylation of Cdk114. A previous study has shown that growing oocytes lacking Cdh1 accumulate higher levels of cyclin B1 due to low APC/C activation, but higher levels of cyclin B1 are not sufficient to drive smaller oocytes into premature resumption of meiosis in vivo within the follicles11. Even so, it is still likely that Cdk1-activating cyclins in growing oocytes have not reached high enough levels to activate Cdk1. This is because that although the growing, meiotically incompetent oocytes already contain higher concentration of cyclin B1 protein than Cdk1, the level of cyclin B1 further increases several folds by the time the oocytes are fully grown34.
Similarly, downregulation of Wee1B in mouse oocytes causes more rapid GVBD in fully grown oocytes, but GVBD is not seen in growing oocytes, which implies that Wee1B is not important for regulating Cdk1 phosphorylation in growing oocytes14. However, the lack of GVBD in growing oocytes by knockdown of Wee1B in this transgenic mouse model might be due to the fact that Wee1B was only partially depleted14. In addition, another kinase in oocytes, Myt1, might still be able to maintain meiotic arrest in the absence of Wee1B14. It is also possible that the growing oocytes might not sufficiently activate Cdc25 for the removal of Cdk1 inhibitory phosphorylation. Therefore, whether Cdk1 is regulated in smaller oocytes is far from understood. However, our direct genetic approach revealed that the phosphorylation-mediated suppression of Cdk1 activity is one of the crucial factors for the prolonged prophase I arrest in dormant and growing oocytes.
When fully grown oocytes resume meiosis after Cdk1 activation, it does not lead to DNA damage. Even the induction of DNA damage in fully grown mouse oocytes does not lead to their death; instead, these oocytes still progress through meiosis. This is because DNA damage in fully grown oocytes does not effectively activate the DNA damage-response pathway27,28. In contrast, as we have shown in this study, premature activation of Cdk1 in meiotically incompetent dormant and growing oocytes cannot be tolerated and it triggers massive DNA damage and oocyte death. This is because as we showed the activated Cdk1 in smaller oocytes leads to active response to DNA damage through the ATM-Chk2-p53/p63-mediated pathway and activation of this pathway quickly causes oocyte loss. However, the mechanism for increased level of Chk2 protein observed in growing mutant oocytes remains unclear. It is possible that the degradation of Chk2 might be affected in the mutant oocytes as ubiquitin-mediated Chk2 degradation has been recently reported to regulate Chk2 protein level in other cell lines35.
Interestingly, we found that genetic ablation of Chk2 largely prevented the death of oocytes, which is in line with a previous study showing that the death of mouse oocytes due to ionizing radiation-induced DNA double-strand breaks is prevented by concomitant ablation of Chk229. Nevertheless, a possibility for a contribution of ATR (ataxia telangiectasia and Rad3-related)-Chk1 pathway36 for death of the mutant oocytes cannot be ruled out. Our results thus show that a direct phosphorylation-mediated suppression of Cdk1 activity is essential for safeguarding the DNA integrity of meiotically arrested female germ cells.
Studies in both yeast and mammals have revealed that Cdk1 is involved in the regulation of DNA double-strand break processing, DNA repair by homologous recombination, and DNA damage checkpoint activation, through which the DNA-repair capacity is coordinated with progression through the cell cycle37. In human MCF-7 cell lines, inhibition of Wee1 increases Cdk1 activity and results in the inactivation of DNA repair38. In yeast, the detection of DNA damage in G2 activates a checkpoint that prevents entry into mitosis through the maintenance of the phosphorylation of Y15-Cdc2p (the single Cdk in yeast); however, inactivation of Wee1 and Cdc25 abolishes the G2 checkpoint control39. Similarly, Cdk1 hyperactivation in the Drosophila imaginal wing disc also causes genome instability and chromosomal abnormalities40. These findings suggest that overactivated Cdk1 activity leads to genome instability, indicating that Cdk1 activity needs to be properly suppressed in order to maintain genome stability.
Due to the multiple downstream substrates of Cdk1, elevated Cdk1 activity might cause DNA damage through various routes. For example, Cdk1 has been shown to be involved in the regulation of DNA damage-related molecules such as polo-like kinase 1 (PLK1) in U2OS and HeLa cells41, making it attractive to hypothesize that Cdk1 activation in quiescent and growing oocytes might enhance PLK1 activity and affect the DNA damage-repair response. Previous studies showed that Wee1 inhibition (and thus elevated Cdk1 activity) in mitotic cells leads to dysregulated endonuclease activity and might cause DNA damage42. More in-depth studies are needed to elucidate the mechanisms behind the DNA damage in dormant and growing oocytes caused by active Cdk1. Understanding the response to DNA damage by mouse oocytes in a developmental stage specific manner may have important implications in helping identify additional targets for fertility preservation therapies in cancer patients who are at the verge of losing ovarian reserve.
The rapid loss of growing follicles in OoCdk1+/AF; Zp3-Cre ovaries seems to indirectly increase the activation and subsequent depletion of primordial follicles in the adult ovaries. It is known that the larger follicles secrete inhibitory signals such as Anti-Müllerian hormone to prevent primordial follicle recruitment43, and it is likely that in the absence of growing follicles in OoCdk1+/AF; Zp3-Cre ovaries, the activation of the primordial follicle pool is accelerated (Supplementary information, Figure S4). In summary, our data demonstrate that the Cdk1 inhibitory phosphorylation sustains the prolonged period of meiotic arrest in female germ cells and protects the small dormant and growing oocytes from accumulating DNA damage and their death. Therefore, the phosphorylation-mediated direct suppression of Cdk1 kinase activity is essential for preserving the oocyte pool for maintaining the reproductive lifespan of female mammals
Materials and methods
Generation of Cdk1AF conditional expression knock-in mice
Mouse genomic DNA harboring the Cdk1 locus was retrieved from the clone pBeloBACII 305J21 (ResGen) and inserted into the pBlight-TK vector as described previously44. Using recombineering methods45, a floxed neomycin-selection cassette adjacent to a strong splice acceptor site (derived from exon 2 of the mouse Engrailed gene) with multiple stop codons was introduced to the 5′ end of exon 3 of Cdk1, which had been modified by site-directed mutagenesis to introduce the point mutations into the Cdk1 coding sequence to generate the T14A and Y15F substitutions. The resultant targeting vector (PKB945) was linearized by NotI digestion and electroporated into ES cells. After positive and negative selection with Geneticin and ganciclovir, respectively, genomic DNA from resistant ES cell colonies was used to screen for homologous recombination by Southern hybridization. Correctly targeted ES cell clone #2681 was used for generation of the Cdk1AF conditional expression knock-in mouse strain. The presence of the point mutations in the knock-in mice was verified by sequencing of the DNA from tail snips (see Supplementary information, Figure S1). Cdk1+/SAF mice carry one allele of wild-type Cdk1 and one allele of Cdk1SAF.
Cdk1+/SAF mice of mixed background (129S1/SvlmJ and C57BL/6) were backcrossed to C57BL/6J mice for four generations and then crossed with transgenic mice carrying Gdf-9 promoter-mediated Cre recombinase in a C57BL/6J background18 to induce the expression of the Cdk1AF allele in the oocytes of primordial follicles and further developed follicles (OoCdk1+/AF;Gdf-Cre mice). The Cdk1+/SAF mice were also crossed with transgenic mice carrying Zp3 promoter-mediated Cre recombinase in a C57BL/6J background21 to obtain mutant female mice expressing the Cdk1AF allele only in their growing oocytes but not in the primordial oocytes (OoCdk1+/AF; Zp3-Cre mice). Littermates that expressed a wild-type Cdk1 allele and a Cdk1SAF allele, but not the Cre transgene, were used as controls (Cdk1+/SAF mice). OoCdk1+/AF; Zp3-Cre ; Chk2−/− mice were obtained by multiple rounds of crossings among Cdk1+/SAF; Zp3-Cre, and Chk2−/− mice46.
All mice were housed under controlled environmental conditions with free access to water and food. Illumination was on between 0600 hours and 1800 hours. Experimental protocols were approved by the regional ethical committee of the University of Gothenburg, Sweden, and by the Institutional Animal Care and Use Committee at the Biological Research Centre mouse facility at Biopolis, Singapore.
Reagents, antibodies, and immunological detection methods
Rabbit polyclonal antibodies to γ-H2AX (phospho S139), DDX4, and Chk2 were purchased from Abcam (Cambridge, UK). Rabbit polyclonal antibodies to phospho-Chk2 (T68) and 53BP1 were purchased from Novus Biologicals (Littleton, CO, USA). Mouse monoclonal antibodies against Cdk1, p63, and p-ATM (S1981) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit polyclonal antibodies against phospho-Cdk1 (Y15) and mouse monoclonal antibodies against p53 were obtained from Cell Signaling Technologies (Beverly, MA, USA). Mouse monoclonal antibodies against β-actin, M2, and M16 media, dibutyryl cyclic AMP (dbcAMP), 4′,6-diamidino-2-phenylindole (DAPI), and mineral oil were purchased from Sigma (St. Louis, MO, USA). Human anti-CREST serum was from Antibodies (Davis, CA, USA). Western blots were performed according to the instructions of the suppliers of the different antibodies and visualized using the ECL Prime Western Blotting Detection System (GE Healthcare, UK).
Isolation of oocytes from postnatal mouse ovaries
Mice were killed and their ovaries were dissected free of fat and connective tissue using a stereo microscope (Carl Zeiss, Germany). The ovaries were then minced with a pair of dissection scissors before being incubated in 0.05% collagenase dissolved in Dulbecco's modified Eagle's medium-F12 (DMEM/F12; Invitrogen) supplemented with 4 mg/ml bovine serum albumin (BSA), 100 U/ml penicillin, and 100 μg/ml streptomycin with frequent agitation and pipetting. After the tissues had mostly been digested by collagenase, usually within 45-60 min, EDTA was added to this mixture to a final concentration of 40 mM, and the mixture was incubated at 37 °C with frequent pipetting for another 15-20 min until clusters of granulosa cells or other cells were completely dispersed. The mixture of cells and oocytes was then washed once with PBS by centrifugation, and pellets were resuspended in 5 ml of the above-mentioned serum-free DMEM/F12 medium and poured into 6-cm culture dishes. Oocytes were picked by mouth pipetting. For Western blots, oocytes were washed in HBSS, and lysed in a buffer containing 50 mM Tris HCl (pH 8.0), 120 mM NaCl, 20 mM NaF, 20 mM β-glycerophosphate, 1 mM EDTA, 6 mM EGTA (pH 8.0), 1% NP-40, 1 mM DTT, 5 mM benzamidine, 1 mM PMSF, 250 μM sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 μg/ml pepstatin and lysate from 200 growing oocytes was loaded in each lane.
For the isolation of fully grown oocytes, 3- to 4-week-old female mice were injected with 7.5 IU pregnant mare serum gonadotropin intraperitoneally, and 42–44 h later the mice were killed and their ovaries were collected in 100 μg/ml dbcAMP-containing M2 medium. Fully grown GV-stage oocytes surrounded by cumulus cells were released by puncturing the ovaries in M2 medium supplemented with dbcAMP to maintain meiotic arrest during the in vitro operation. Oocytes were freed from the attached cumulus cells by repetitive pipetting through a narrow-bore glass pipette.
Oocyte immunofluorescence and confocal microscopy
Oocytes obtained by collagenase-EDTA digestion were fixed in 4% paraformaldehyde (PFA). After the fixed oocytes were permeabilized with 0.5% Triton X-100 at room temperature (RT) for 20 min, they were blocked with 1% BSA-supplemented PBS for 1 h and incubated at 4 °C with anti-γ-H2AX (phospho S139) antibody (1:500 dilution) and anti-p-ATM (S1981) antibody (1:500 dilution) overnight followed by an incubation with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG (Invitrogen Molecular Probes, Carlsbad, CA, USA) for 1 h at RT. DNA was stained with DAPI (1 μg/ml in PBS). Finally, the oocytes were mounted on glass slides with DABCO-containing mounting medium and examined by laser-scanning confocal microscopy (Zeiss LSM700 Inverted).
Chromosome spreads
For chromosome spreads, the oocytes isolated from PD14 Cdk1+/SAF and OoCdk1+/AF; Zp3-Cre ovaries were first freed of the zona pellucida with acidic Tyrode's solution (Sigma-Aldrich). After a brief recovery in M2 medium, the oocytes were transferred onto glass slides and fixed in a solution of 1% PFA in distilled H2O (pH 9.2) containing 0.15% Triton X-100 and 3 mM dithiothreitol. The slides were left to dry and then blocked with 1% BSA in PBS for 1 h at RT. The spreads were incubated with primary antibodies against CREST and SMC3 (Abcam) overnight at 4 °C using a dilution of 1:100. After three washes, the slides were incubated with the corresponding secondary antibodies (Invitrogen) at 1:500 dilution for 1 h at RT. DNA on the slides was stained with DAPI, and the slides were mounted for observation with laser-scanning confocal microscopy (Zeiss LSM700 Inverted).
Histone H1 kinase assay
Cdk1 activity was measured using histone H1 kinase assays. This assay was performed using 10 oocytes in a 10 μl reaction volume based on a published protocol23. In brief, the reaction was carried out in a buffer containing 25 mM HEPES (pH 7.4), 15 mM MgCl2, 20 mM EGTA, 50 mM NaCl, 0.05% NP-40, 0.1 mM ATP, 100 mM PMSF, 1 mM DTT, 1 μg histone H1 (Roche, Mannheim, Germany), and 3 μCi of (γ-32P) ATP (Perkin-Elmer, Boston, MA, USA) and incubated at 37 °C for 30 min. Samples were boiled in SDS sample buffer and then separated on a 15% SDS-polyacrylamide gel. The radioactive signal was detected by exposing the gels on an FLA 3000 phosphorimager (Fuji, Tokyo, Japan).
Histological analysis of ovarian tissues and immunofluorescence staining
Histological analysis of ovarian tissue was performed as previously described17. Briefly, ovaries were fixed in 4% PFA, dehydrated, and embedded in paraffin. Paraffin-embedded ovaries were serially sectioned at 8 μm thickness and stained with hematoxylin for morphological observation. To quantify total number of follicles per ovary, every fifth section was counted throughout the entire ovary, beginning with the first section, and the final count was multiplied by 5 as a correction factor. To calculate the ovarian follicles at different stages of development, all the follicles were counted in all sections of an ovary based on the well-accepted standards established by Pedersen and Peters47. Oocytes that had resumed meiosis were identified by observation of the condensed chromosomes.
To detect the DNA damage in ovarian sections by immunofluorescence staining, the sections were treated with 0.01% sodium citrate buffer (pH 6.0) for antigen retrieval. After blocking with 1.5% goat serum in PBS, the sections were incubated with anti-γ-H2AX (phospho S139) and DDX4 primary antibodies overnight at 4 °C followed by an incubation with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG (Invitrogen Molecular Probes) for 1 h at RT. The sections were then stained with DAPI for 5 min. The fluorescent images were taken with a Zeiss Axio Scope A1 upright microscope. After imaging for γ-H2AX, DDX4, and DAPI, the same sections were stained with hematoxylin to clearly visualize the histology.
Statistical analysis
All experiments were repeated at least three times. For comparisons between groups, the differences were calculated with Student's t-test, and a difference was considered to be significant if p< 0.05.
Author Contributions
DA, PK, and KL designed the study. DA, KB, JZ, MH, SR, MBB, MS, and MKD performed the experiments. DA, PK, and KL wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
Nagaoka SI, Hassold TJ, Hunt PA . Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 2012; 13:493–504.
Zheng W, Zhang H, Gorre N, Risal S, Shen Y, Liu K . Two classes of ovarian primordial follicles exhibit distinct developmental dynamics and physiological functions. Hum Mol Genet 2014; 23:920–928.
McGee EA, Hsueh AJ . Initial and cyclic recruitment of ovarian follicles. Endocr Rev 2000; 21:200–214.
Adhikari D, Liu K . Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr Rev 2009; 30:438–464.
Eppig JJ, Viveiros MM, Bivens CM, De La Fuente R . Regulation of mammalian oocyte maturation. In: Leung PC, Adashi EY, eds. The Ovary. Academic Press: Amsterdam 2004:113–129.
Sorensen RA, Wassarman PM . Relationship between growth and meiotic maturation of the mouse oocyte. Dev Biol 1976; 50:531–536.
Hirao Y, Miyano T, Kato S . Acquisition of maturational competence in in vitro grown mouse oocytes. J Exp Zool 1993; 267:543–547.
Nishimura T, Fujii W, Kano K, Sugiura K, Naito K . Analyses of the involvement of PKA regulation mechanism in meiotic incompetence of porcine growing oocytes. Biol Reprod 2012; 87:53.
Goren S, Piontkewitz Y, Dekel N . Meiotic arrest in incompetent rat oocytes is not regulated by cAMP. Dev Biol 1994; 166:11–17.
Reis A, Chang HY, Levasseur M, Jones KT . APCcdh1 activity in mouse oocytes prevents entry into the first meiotic division. Nat Cell Biol 2006; 8:539–540.
Holt JE, Tran SM, Stewart JL, et al. The APC/C activator FZR1 coordinates the timing of meiotic resumption during prophase I arrest in mammalian oocytes. Development 2011; 138:905–913.
de Vantery C, Stutz A, Vassalli JD, Schorderet-Slatkine S . Acquisition of meiotic competence in growing mouse oocytes is controlled at both translational and posttranslational levels. Dev Biol 1997; 187:43–54.
Kim J, Singh AK, Takata Y, et al. LSD1 is essential for oocyte meiotic progression by regulating CDC25B expression in mice. Nat Commun 2015; 6: 10116.
Han SJ, Chen R, Paronetto MP, Conti M . Wee1B is an oocyte-specific kinase involved in the control of meiotic arrest in the mouse. Curr Biol 2005; 15:1670–1676.
Norbury C, Blow J, Nurse P . Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J 1991; 10:3321–3329.
Krek W, Nigg EA . Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J 1991; 10:3331–3341.
Reddy P, Liu L, Adhikari D, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 2008; 319:611–613.
Lan ZJ, Xu X, Cooney AJ . Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod 2004; 71:1469–1474.
Heijink A, Blomen V, Bisteau X, et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc Natl Acad Sci USA 2015; 112.
Rogakou EP, Boon C, Redon C, Bonner WM . Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905–916.
de Vries WN, Binns LT, Fancher KS, et al. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 2000; 26:110–112.
Adhikari D, Diril MK, Busayavalasa K, et al. Mastl is required for timely activation of APC/C in meiosis I and Cdk1 reactivation in meiosis II. J Cell Biol 2014; 206:843–853.
Kudo NR, Wassmann K, Anger M, et al. Resolution of chiasmata in oocytes requires separase-mediated proteolysis. Cell 2006; 126:135–146.
Panier S, Boulton SJ . Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15:7–18.
Bakkenist CJ, Kastan MB . DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499–506.
Shiloh Y, Ziv Y . The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197–210.
Ma JY, Ou Yang YC, Wang ZW, et al. The effects of DNA double-strand breaks on mouse oocyte meiotic maturation. Cell Cycle 2013; 12:1233–1241.
Marangos P, Carroll J . Oocytes progress beyond prophase in the presence of DNA damage. Curr Biol 2012; 22:989–994.
Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC . Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 2014; 343:533–536.
Hirao A, Cheung A, Duncan G, et al. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol 2002; 22:6521–6532.
Matsuoka S, Huang M, Elledge SJ . Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282:1893–1897.
Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE . Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 2000; 60:5934–5936.
Gui L, Homer H . Hec1-dependent cyclin B2 stabilization regulates the G2-M transition and early prometaphase in mouse oocytes. Dev Cell 2013; 25:43–54.
Kanatsu-Shinohara M, Schultz RM, Kopf GS . Acquisition of meiotic competence in mouse oocytes: absolute amounts of p34(cdc2), cyclin B1, cdc25C, and wee1 in meiotically incompetent and competent oocytes. Biol Reprod 2000; 63:1610–1616.
Garcia-Limones C, Lara-Chica M, Jimenez-Jimenez C, et al. CHK2 stability is regulated by the E3 ubiquitin ligase SIAH2. Oncogene 2016.
Chen Y, Poon RY . The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci 2008; 13:5016–5029.
Trovesi C, Manfrini N, Falcettoni M, Longhese MP . Regulation of the DNA damage response by cyclin-dependent kinases. J Mol Biol 2013; 425:4756–4766.
Krajewska M, Heijink AM, Bisselink YJ, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene 2013; 32:3001–3008.
Raleigh JM, O'Connell MJ . The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J Cell Sci 2000; 113:1727–1736.
Ayeni JO, Varadarajan R, Mukherjee O, et al. Dual phosphorylation of cdk1 coordinates cell proliferation with key developmental processes in Drosophila. Genetics 2014; 196:197–210.
Benada J, Burdova K, Lidak T, von Morgen P, Macurek L . Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015; 14:219–231.
Dominguez-Kelly R, Martin Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol 2011; 194:567–579.
Durlinger AL, Kramer P, Karels B, et al. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 1999; 140:5789–5796.
Diril MK, Ratnacaram CK, Padmakumar VC, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA 2012; 109:3826–3831.
Lee EC, Yu D, Martinez de Velasco J, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 2001; 73:56–65.
Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000; 287:1824–1827.
Pedersen T, Peters H . Proposal for a classification of oocytes and follicles in the mouse ovary. J Reprod Fertil 1968; 17:555–557.
Acknowledgements
This study was supported by grants to KL from the Jane and Dan Olssons Foundation, the LUA/ALF-medel Västra Götalandsregionen, AFA Insurance, the Swedish Research Council, and the Swedish Cancer Foundation, Sweden. PK and MKD were supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore.
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Generation of Cdk1+/SAF knockin mice. (PDF 200 kb)
Supplementary information, Figure S2
Comparison of ovarian morphology and quantification of ovarian follicles. (PDF 358 kb)
Supplementary information, Figure S3
Increased ATM activation in GV stage OoCdk1+/AF; Zp3-Cre oocytes and quantification of ovarian follicles in OoCdk1+/AF; Zp3-Cre; Chk2−/− mice. (PDF 202 kb)
Supplementary information, Figure S4
Schematic diagram of oocyte development in Cdk1+/SAF, OoCdk1+/AF; Gdf9-Cre and OoCdk1+/AF; Zp3-Cre mouse ovaries. (PDF 168 kb)
Rights and permissions
This license allows readers to copy, distribute and transmit the Contributionas long as it attributed back to the author. Readers are permitted to alter, transform or build upon the Contribution as long as the resulting work is then distributed under this is a similar license. Readers are not permitted to use the Contribution for commercial purposes. Please read the full license for further details at - http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Adhikari, D., Busayavalasa, K., Zhang, J. et al. Inhibitory phosphorylation of Cdk1 mediates prolonged prophase I arrest in female germ cells and is essential for female reproductive lifespan. Cell Res 26, 1212–1225 (2016). https://doi.org/10.1038/cr.2016.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.119
Keywords
This article is cited by
-
The transcriptome-wide N6-methyladenosine (m6A) map profiling reveals the regulatory role of m6A in the yak ovary
BMC Genomics (2022)
-
MDC1 is essential for G2/M transition and spindle assembly in mouse oocytes
Cellular and Molecular Life Sciences (2022)
-
Hormesis of low-dose inhibition of pAkt1 (Ser473) followed by a great increase of proline-rich inositol polyphosphate 5-phosphatase (PIPP) level in oocytes
In Vitro Cellular & Developmental Biology - Animal (2021)
-
Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality
Oncogene (2019)
-
CFP1 coordinates histone H3 lysine-4 trimethylation and meiotic cell cycle progression in mouse oocytes
Nature Communications (2018)
