
Abstract
Recent success in the derivation of haploid embryonic stem cells (haESCs) from mouse via parthenogenesis and androgenesis has enabled genetic screening in mammalian cells and generation of gene-modified animals. However, whether haESCs can be derived from primates remains unknown. Here, we report the derivation of haESCs from parthenogenetic blastocysts of Macaca fascicularis monkeys. These cells, termed as PG-haESCs, are pluripotent and can differentiate to cells of three embryonic germ layers in vitro or in vivo. Interestingly, the haploidy of one monkey PG-haESC line (MPH1) is more stable compared with that of the other one (MPH2), as shown by the existence of haploid cells for more than 140 days without fluorescence-activated cell sorting (FACS) enrichment of haploid cells. Importantly, transgenic monkey PG-haESC lines can be generated by lentivirus- and piggyBac transposon-mediated gene transfer. Moreover, genetic screening is feasible in monkey PG-haESCs. Our results demonstrate that PG-haESCs can be generated from monkeys, providing an ideal tool for genetic analyses in primates.
Similar content being viewed by others

Introduction
Haploid cells are amenable for genetic analyses because they contain only one set of chromosomes. For example, the budding yeast Saccharomyces cerevisiae has been largely utilized as a model organism to understand important biological processes1. Recently, mouse haploid embryonic stem cells (haESCs) have been successfully derived from parthenogenetic (PG)2,3 and androgenetic (AG)4,5 blastocysts, and applied to forward or reverse genetic screening2,3 and production of genetically modified mice4,5, showing a great potential in genetic studies in mammalian organisms6. The success in mouse raises a particular challenging question of whether haESCs can be established in non-human primates. Owing to the dramatic genetic and physiologic similarities to human7, non-human primates8 represent the most ideal experimental models for basic and applied biomedical research. Normal diploid embryonic stem cell (ESC) lines have been generated from different types of non-human primate embryos, including rhesus9, marmoset10 and cynomolgus (Macaca fascicularis)11 monkeys. However, different from murine, but similar to human ESCs, non-human primate ESCs proliferate at a low rate and need to be manually dissociated into small clumps for propagation12,13. This property of non-human primate ESCs may hinder the derivation of haESCs, as the establishment of mouse haESCs is based on the haploid cell enrichment using single-cell-sorting technology3,5.
In this study, we have established protocols for the derivation of haESCs from Macaca fascicularis monkey PG blastocysts. These cells display pluripotency in vitro and in vivo. Interestingly, the haploidy can be well maintained in MPH1 for more than 140 days without FACS enrichment of haploid cells. More importantly, we provide the evidence that monkey PG-haESCs can be genetically manipulated readily.
Results
Derivation of haESC lines from monkey PG blastocysts
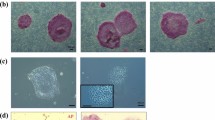
To generate PG haploid embryos from monkey, we adopted an artificial activation protocol, which was originally developed by Mitalipov et al.14. Briefly, cumulus-free mature metaphase II (MII) oocytes from Macaca fascicularis monkey were treated sequentially with ionomycin, followed by cycloheximide (CHX). Ten hours after the activation, the second polar body and one female pronucleus could be observed (Figure 1A). Among 181 activated oocytes, 167 (92%) divided and 70 (39%) developed into blastocysts in vitro (Table 1 and Figure 1A), similar to the developmental efficiency of intracytoplasmic sperm injection (ICSI) embryos (Table 1). All blastocysts derived from activated oocytes and ICSI controls were used for ESC derivation. After removal of zona pellucida, inner cell mass (ICM) was isolated via immunosurgery15, plated on mitotically inactivated human fibroblast feeder (HFF)16 feeder layers and cultured in a standard monkey ESC culture system15,17, with the addition of ROCK inhibitors, 1 μM Thiazovivin and 10 μM Y-2763218,19. ICM outgrowths were individually manually dissociated into small clumps and replated on new HFF feeder layers. The resulting colonies were further expanded by mechanical dissociation for several passages and then enzymatically dispersed, as described for human ESC derivation and passaging18,20,21. Four diploid ESC lines were established from ICSI-derived blastocysts (referred to as MES1-MES4) (Table 1). Among 10 ESC lines that we established from PG blastocysts, two lines (referred to as MPH1 and MPH2), originated from two individual monkeys (183 and 118) (Table 1), contained haploid cells in the initial cell sorting. Strikingly, compared with the low ratio of haploid cells (< 5%) at the first sorting during mouse AG-haESC derivation5, the percentage of haploid cells in both MPH1 and MPH2 (Figure 1B and Supplementary information, Figure S1A) was substantially higher (about 30%). The haploidy of monkey PG-haESCs could be well maintained with regular FACS (Figure 1B and Supplementary information, Figure S1A) for over 30 passages. Karyotyping of these PG-haESCs revealed that both of the cell lines had a haploid set of 21 chromosomes (Figure 1C and Supplementary information, Figure S1B). Comparative genomic hybridization (CGH) of monkey PG-haESC lines confirmed that the haploid cells sustained genome integrity (Figure 1D, Supplementary information, Figure S1C and Table S1).

Derivation of monkey PG-haESCs. (A) Representative images of different stages of monkey ESC line derivation. PB, polar body; PN, pronucleus; ICM, inner cell mass. Scale bar, 100 μm (top, middle panel and bottom left) and 500 μm (bottom right). (B) Establishment of PG-haESC line (represented by MPH1) after three times of FACS enrichment for haploid cells. Right is FACS data of diploid control ESCs for comparison. D62, 62 days after the ICM was plated for ESC formation. (C) Karyotype of MPH1 (passage 7) and MPH2 (passage 5) showing the normal haploid complement of 21 chromosomes (20 + X). (D) CGH analysis of PG-haESCs (MPH1, passage 11) and female monkey adipose cells. Upper panel, MPH1 (p11) vs adipose cells. Lower panel, adipose cells vs adipose cells. No major genomic alterations (amplifications or losses) were detected in monkey PG-haESCs.
Pluripotency of monkey PG-haESCs
Monkey PG-haESCs showed the colony morphology similar to that of normal ICSI-derived diploid ESCs. Immunostaining analyses detected the expression of typical monkey ESC markers, including NANOG, OCT4, SSEA4, TRA-1-60 and TRA-1-81 in PG-haESC colonies (Figure 2A) and diploid ESCs (Supplementary information, Figure S2A). Next, we compared the gene expression profiles of PG-haESCs with those of normal ESCs and monkey fibroblasts (MFs) from female individuals. Clustering of these cells based on microarray expression results showed a high correlation between PG-haESCs and control diploid ESCs, but not MFs (Figure 2B and Supplementary information, Figure S3A). To characterize the differentiation potential of monkey PG-haESCs and ICSI-derived ESCs, we suspended ESCs for the formation of embryoid body (EB) in vitro. We observed that PG-haESCs, similar to ICSI control ESCs (Supplementary information, Figure S2B), could differentiate into various cell types originating from all three embryonic germ layers, as demonstrated by the expression of TUJ1, MAP2 (ectoderm markers), ACTININ, VIMENTIN (mesoderm markers), SOX17, GATA6 and FOXA2 (endoderm markers) (Figure 2C and Supplementary information, Figure S3B). To test the differentiation potential of PG-haESCs in vivo, we injected undifferentiated PG-haESCs and ICSI control ESCs into NOD SCID mice for teratoma formation. The formation of teratoma is considered as the most stringent standard of pluripotency for monkey ESCs13 to date, as the recent report showed that monkey ESCs failed to support the generation of chimeras22. Two or three months after injection, well-shaped teratomas were observed from both haploid and diploid ESCs. Histochemical analyses indicated the existence of a variety of tissues from all three embryonic germ layers, such as neural epithelium, pigment cells (ectoderm), cartilage, muscle (mesoderm), intestinal and respiratory epithelium (endoderm) (Figure 2D, Supplementary information, Figures S2C and S3C) in the teratomas, validating the pluripotency of monkey PG-haESCs.

Pluripotency of monkey PG-haESCs. (A) Expression of primate ESC markers in monkey PG-haESCs, including Nanog, Oct4, SSEA4, TRA-1-60 and TRA-1-81. Mouse-specific ESC marker, such as SSEA1, was not detected. Shown are the representative immunostaining images of monkey PG-haESC colonies. Scale bar, 100 μm. (B) Gene expression profiles of PG-haESCs. Scatter plots showing high correlation in gene expression between PG-haESCs and ICSI-derived diploid ESCs. Diploid ESCs, three replicates (MES1, MES2 and MES3). Haploid, five replicates (MPH1, passages 7, 8, 9 and 11; MPH2, passage 4). Fibroblasts, monkey somatic fibroblasts (MF1 and MF2 from female individuals, two replicates). (C) Expression of markers of various cells from different germ layers in EBs formed from PG-haESCs, including TUJ1 (ectoderm), ACTININ (mesoderm) and SOX17 (endoderm). Scale bar, 50 μm. (D) Histochemical analysis of teratomas from PG-haESCs. Representative images indicated the existence of tissues from all three embryonic germ layers, such as neural epithelial (ectoderm), cartilage (mesoderm) and intestinal epithelium (endoderm). Scale bar, 100 μm.
Cellular origin of monkey PG-haESCs
As the haploid genome of MPH1 and MPH2 PG-haESC lines was from activated oocytes containing only one copy of genome, all chromosomes might be expected to display 100% of homozygosity in diploidized daughter cells. To test this hypothesis, we conducted short tandem repeat (STR) genotype and single-nucleotide polymorphism (SNP) analyses23 using genomic DNA extracted from PG-haESCs and oocyte donor monkeys. The analysis of 24 STR loci confirmed that MPH1 originated from the egg donor 183, and MPH2 was from egg donor 118 (Figure 3A and Supplementary information, Table S2). As expected, both MPH1 and MPH2 cells exhibited homozygosity at all tested loci (Supplementary information, Table S2). Consistently, all 23 detected SNPs in PG-haESC lines were precisely inherited from the corresponding egg donors. Moreover, both cell lines displayed homozygosity for all tested SNPs (Supplementary information, Figure S3B and Table S3).

Cellular origin of MPH1 and MPH2. (A) Representative images of STR locus (D6S501) of MPH1 and MPH2 and their egg donors (no. 183 and no. 118). (B) Representative images of SNP locus (rs5436199) of MPH1 and MPH2 and their egg donor monkeys (no. 183 and no. 118). (C) Expression of the imprinted genes measured by qPCR. The expression levels in PG-haESCs were relative to those in diploid ESCs, which were set to 1. *0.01 < P < 0.05; **0.001 < P < 0.01. (D) Methylation analysis of SNRPN and H19 in monkey skin, sperm, diploid ESCs (MES2, passage 4 for SNRPN, and passage 10 for H19) and PG-haESCs (MPH1, passage 5 for H19 and passage 7 for SNRPN; MPH2, passage 9 for H19 and passage 8 for SNRPN). Open and filled circles represent unmethylated and methylated CpG sites, respectively.
As genomic materials of PG-haESC lines originated from mature oocytes, in which maternal imprints were established during oogenesis, we anticipated that PG-haESCs might maintain maternal imprints. We first analyzed the expression of maternal and paternal imprinted genes in PG-haESCs and diploid ESCs from microarray results. As expected, mRNA levels of all detected paternally imprinted genes (expressed from the paternal allele) were lower in PG-haESCs than in diploid ESCs (Supplementary information, Table S4). In contrast, transcripts from most paternally imprinted genes (expressed from the maternal allele) in PG-haESCs had similar levels to those in diploid ICSI-derived ESCs (Supplementary information, Table S4). Quantitative RT-PCR results verified the different expression patterns of maternally and paternally imprinted genes in PG-haESCs (Figure 3C). Interestingly, a similar expression pattern was observed in monkey PG diploid ESCs23. To further assess epigenetic inheritance, we performed bisulfite sequencing to analyze the methylation profiles of one maternally imprinted gene, SNRPN, and one paternally imprinted gene, H19. Consistent with the extremely low methylation level of SNRPN in sperm, the differentially methylated region (DMR) of SNRPN largely retained methylation in MPH1 and MPH2 (Figure 3D). In contrast, H19, which is normally unmethylated on the maternal allele, was partially methylated in both PG-haESC lines (Figure 3D). These results are consistent with the observations of epigenetic instability in monkey9,24, human25,26,27 and mouse28,29 ESCs, particularly at the H19 locus5. Taken together, these data support the idea that monkey PG-haESCs are derived from parthenogenetically activated oocytes.
Haploidy in monkey PG-haESCs
MPH1 and MPH2 were derived from two independent PG blastocysts developed from two oocytes harvested from two individual donor monkeys (no. 183 and no. 118). The haploidy could be well maintained in both cell lines by regular FACS, and PG-haESCs were propagated without any signs of proliferative crisis. Interestingly, MPH1 and MPH2 showed distinct features regarding the maintenance of haploidy. We observed the high ratio of haploid cells (around 30%) in first cell sorting during the process of MPH1 and MPH2 formation (Figure 1B and Supplementary information, Figure S1A). We hypothesized that the haploidy could be stably maintained in MPH1 and MPH2 lines. To test this hypothesis, some cells were used for haploid cell enrichment in the first FACS, whereas others were directly replated without cell sorting and the haploidy was traced to determine when haploid cells would completely convert into the diploid state. Strikingly, haploid cells persisted for almost 140 days without sorting enrichment in MPH1 from the day when the ICM was plated on HFF feeder layers for ESC formation (Figure 4A and data not shown). After the second FACS enrichment for haploid cells, the haploid cells from MPH1 (MPH1a) were replated on HFF feeder layers and the haploidy could also be maintained for more than 100 days in vitro (Figure 4A). In contrast, the MPH2 line showed less stability in the haploidy maintenance compared with the MPH1 line. Without any cell sorting, haploid cells disappeared in MPH2 on day 58 after ICM was plated for ESC derivation. Consistently, enriched haploid cells from MPH2 (MPH2a) completely converted into the diploid state after 50 days in culture.

Haploidy in monkey PG-haESCs. (A) Maintenance of haploidy in MPH1 and MPH2. MPH1a and MPH2a represented subclones formed after replating haploid cells enriched via FACS from MPH1 and MPH2, respectively. (B) Growth curves of monkey PG-haESCs (MPH1 and MPH2), monkey diploid ESCs (MES1 and MES2) and mouse haESCs. MTT assay for monitoring cell growth was performed for three times. **P < 0.01. (C) Flow cytometry analysis of DNA content of cells isolated from two teratomas. D70, 70 days after injection of PG-haESCs into NOD SCID mouse. (D) Expression of the differentiated genes measured by qPCR. The expression levels in diploid cells isolated from teratomas were relative to those in PG-haESCs, which were set to 1.
We then addressed whether the cell growth rate accounted for the different haploidy stability between MPH1 and MPH2 lines. To this end, we performed MTT assay, which is widely used to determine the cell growth rate30,31. As shown in Figure 4B, the cells from MPH1 and MPH2 lines displayed a comparable growth rate. Furthermore, the growth rate of both MPH1 and MPH2 was similar to that of monkey diploid ESCs (MES1 and MES2, Figure 4B). Therefore, the distinct haploidy feature of MPH1 and MPH2 is not dependent on the cell growth rate, implying the possible existence of some essential differences between two individual donor monkeys that contributed to the difference in the haploidy maintenance between these two cell lines.
Mouse haploid ESCs quickly convert to diploid cells during development and differentiation5. We next investigated whether monkey haploid cells could persist in teratomas derived from PG-haESCs. In two tested teratomas, haploid cells could not be detected (Figure 4C). Quantitative RT-PCR analyses showed that marker genes of various cell types from three embryonic germ layers, such as BRACHURY (mesoderm), SOX17, FOXA2, FOXA1 (endoderm), MAP2 and PAX6 (ectoderm) were highly expressed in these diploid cells (Figure 4D). Thus, monkey haploid ESCs spontaneously converted into diploid cells during differentiation. Taken together, the haploidy maintenance in monkey PG-haESCs, similar to that in mouse haESCs3,4,5,22, relies heavily on repeated cell sorting.
Genetic manipulation in monkey PG-haESCs
We next examined the feasibility of genetic manipulation in monkey PG-haESCs. We initially applied three different strategies to establish transgenic PG-haESCs. First, 5 × 105 monkey PG-haESCs were infected with FUGW-lentivirus vectors carrying green-fluorescent protein (GFP)33. Seven days after infection, FACS analysis showed that about 29% of cells were GFP-positive, in which 15% were haploid (Supplementary information, Figure S4A). Infected cells were replated on fresh feeder cells and GFP positive ESC colonies were observed (Supplementary information, Figure S4A). Second, we employed the transposon system, which has been applied for a variety of genetic manipulation in mouse34,35 and human36 cells, for producing transgenic monkey PG-haESCs. Donor PB (Act-RFP) element and the helper plasmid ACT-PBase34 were co-transfected into PG-haESCs by lipofectamine. FACS analysis showed that about 29% of cells carried RFP transgene, in which 20% were haploid (Supplementary information, Figure S4B). We replated RFP-positive haploid cells and observed RFP-positive colonies after culturing for one week (Supplementary information, Figure S4B). Third, we tested the feasibility of drug selection in the derivation of monkey transgenic haploid cells. To this end, donor PB (Act-RFP) carrying a neomycin-resistant gene (neor) and the helper plasmid ACT-PBase were co-transfected into PG-haESCs by lipofectamine. As expected, after 10 days of antibiotic (G418) selection, 84% of neomycin-resistant cells were RFP positive. FACS analysis indicated that almost all survived haploid cells were RFP positive (Figure 5A, left). G418-resistant cells were replated and RFP-positive colonies were randomly picked for expansion, leading to 12 transgenic haploid ESC lines (Figure 5A, right).

Genetic manipulation in monkey PG-haESCs. (A) Generation of transgenic PG-haESCs via drug selection. After co-transfection of donor PB (Act-RFP) carrying a neomycin-resistant gene (neor) and the helper plasmid ACT-PBase into PG-haESCs and antibiotic (G418) selection, almost all neomycin-resistant cells were RFP positive (left). G418-resistant cells were replated and RFP-positive clones were picked for expansion, leading to 12 haploid ESC lines (right). Scale bars, 200 μm. (B) Diagram of PB insertional mutagenesis screening in monkey PG-haESCs. After introducing both PB (PGK-neo) and ACT-PBase into PG-haESCs and G418 selection, survived clones were picked for expansion. Clones with haploid population were selected for further analyses. (C) Four mutated cell lines were established from G418-resistant clones through multiple passages and FACS enrichment for haploid cells. (D) Confirmation of three genes (PRKD1, NIF3L1 and LIPC) mutated in four clones, respectively. Normal PG-haESCs were used as control (lane one). (E) Expression of the NIF3L1 gene measured by qPCR. The expression levels in mutated PG-haESCs were relative to those in ha-ESCs, which were set to 1. **P < 0.01.
Efficient generation of transgenic monkey PG-haESC lines promoted us to explore the possibility of using monkey PG-haESCs for genome-wide genetic screenings (Figure 5B). To this end, we applied insertional mutagenesis by introducing both PB (PGK-neo) and ACT-PBase into PG-haESCs. After G418 selection, survived clones were individually picked and replated for expansion. FACS analysis confirmed that 18 out of 40 clones, derived from two independent experiments, contained haploid cells (Figure 5C), indicating that these clones originated from single neomycin-resistant haploid cell. Inverse PCR analyses revealed that 16 clones contained PB transposon integration sites (Supplementary information, Figure S5A) at an efficiency similar to PB insertion in human ESCs37. DNA sequencing of PCR products showed that three genes (PRKD1, NIF3L1 and LIPC) were mutated in four clones (Supplementary information, Figure S5B), respectively. Further genotyping analyses demonstrated that these clones contained only the mutated allele, whereas the control cells harbor only the wild-type allele (Figure 5D), supporting early observations that the mutagenesis occurred in haploid cells. As LIPC and PRKD1 were not expressed in monkey ESCs (Supplementary information, Figure S5C), we then examined the expression of NIF3L138 in established mutated PG-haESC lines. As expected, the disruption of NIF3L1 by PB resulted in extremely low levels of NIF3L1 mRNA in both clones 206 and 224 (Figure 5E) compared with that in the wild-type haploid ESCs. These data provide the evidence that monkey PG-haESCs can be used for mutagenesis screening and for creating homozygous mutations.
Discussion
Near-haploid cell lines have been established from human tumors39,40 and applied to genetic screenings for host genes required for the action of toxins or viruses41,42,43. However, the aneuploidy and cancerous characters of these cells have limited applications6 in both basic and applied research. Here, we established two haploid ESC lines from monkey PG blastocysts, indicating the feasibility of generation of normal haploid cell lines from primates. Our PG-haESCs carry 21 chromosomes and largely sustain genomic integrity. These cells express typical diploid markers of monkey diploid ESCs and can differentiate into various cell types of all three embryonic germ layers in vitro and in vivo. Importantly, genetic manipulation is feasible in these PG-haESCs.
Monkey ESC colonies are usually propagated by manual dissociation to small cell clusters13,44 rather than single cells, as enzymatic treatment results in dramatic decrease in plating efficiency (< 1%)13. However, to enrich haploid cells, monkey PG-haESCs should be disaggregated into single cells for FACS. To solve this problem, we applied a modified single-cell enzymatic dissociation method employed in human ESC expansion20,21,45,46, in which Accutase, instead of collagenase IV, was chosen for cell dissociation. Accutase is a formulated mixture of digestive enzymes, which can gently dissociate sensitive cells, such as human ESCs19,47. Meanwhile, ROCK inhibitors, Thiazovivin and Y-27632, which have been recently used in human ESC derivation18 and single-cell expansion19, were also employed in our PG-haESC-derivation procedure. Presumably due to the use of these new methods, PG-haESCs derived from monkey oocytes can now be expanded for over 30 passages.
Interestingly, two monkey haploid ESC lines established in this study showed different capability in maintaining the haploidy. Consistently, their subclones (MPH1a and MPHH2a, Figure 4A), which were from the first FACS-enriched haploid cells, exhibited a similar diploidization tendency to their originators. We have ruled out the possibility that cell growth rate accounts for the difference, as MPH1 and MPH2 displayed a similar cell growth rate (Figure 4B). Meanwhile, the cell growth rates of monkey haploid ESCs were not significantly different from that of monkey diploid ESCs, implying that this distinction is not related with ploidy of the cells. As MPH1 and MPH2 were generated from two individual donor monkeys, it is likely that the genetic background of these two monkeys may account for the difference. At present, we cannot exclude the possibility that the stability of haploidy in MPH1 is a chance event. More monkey haploid ESC lines need to be established in the future to address this issue. Nevertheless, further investigation into the underlying differences between MPH1 and MPH2, and differences between their donor monkeys may not only aid in our understanding of the haploidy maintenance, but also yield clues for improving the maintenance of haploid cells in vitro, allowing more efficient genome-wide screenings.
Most importantly, our PG-haESCs could be randomly mutated, and all mutations analyzed so far were homozygous, indicating that monkey haploid ESCs can be utilized for whole-genome screenings for elucidating the mechanisms involved in different biological processes. One intriguing application is to use our PG-haESCs for screening the key factors that may be employed for generation of naive human or non-human primate ESCs.
In summary, we have demonstrated that haploid ESCs can be generated from non-human primates and used as a tool for genetic screening. The next challenge is to establish haploid ESCs from non-human primate AG blastocysts, which can be used as a genetically tractable fertilization agent, like mouse AG-haESCs4,5, for generating genetically modified monkey via injection into oocyte.
Materials and Methods
Animal use and care
All animal procedures were performed under the ethical guidelines of the Institute of Biochemistry and Cell Biology, and the Institute of Neurosciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Parthenogenetic activation
Cumulus-free MII oocytes, 35-36 h post-hCG injection, were activated by exposure to 5 mM ionomycin for 5 min in TALP/HEPES medium supplemented with 1 mg/ml fatty acid-free BSA and then cultured in HECM-9 medium containing 7.5 μg/ml CHX at 37 °C in 6% CO2 for 5 h14. Activated oocytes were transferred to four-well dishes containing protein-free HECM-9 medium covered with paraffin oil and cultured at 37 °C in 6% CO2, 5% O2 and 89% N2. Embryos that reached the eight-cell stage were transferred to four-well dishes with fresh HECM-9 medium supplemented with 5% fetal bovine serum (FBS) (Invitrion) and cultured for a maximum of 9 days to reach blastocyst stage, with medium change every other day.
ESC derivation
Blastocysts generated by PG activation and ICSI were selected to establish ESC lines as described15. Zona pellucidae of PG blastocysts were removed by brief exposure (45-60 s) to 0.5% pronase in ESC-derivation medium. For immunosurgical isolation of ICMs, zona-free blastocysts were exposed to rabbit anti-rhesus spleen serum (Axell Labs, Westbury, NY, USA) (1:5) for 30 min at 37 °C. After extensive washing in ESC derivation medium, embryos were incubated in guinea pig complement (Sigma) (1:5) for an additional 30 min at 37 °C. Partially lysed trophectodermal cells were mechanically dispersed by gentle pipetting with a small-bore pipette followed by the rinse of ICM three times with ESC-derivation medium. Each isolated ICM was transferred into one well of the four-well dishes (Nunc) seeded with mitotically inactivated HFF feeders16 in ESC-derivation medium, composed of F-12 medium (Invitrogen) supplemented with 1% nonessential amino acids (Invitrogen), 2 mM L-glutamine (Invitrogen), 0.1 mM β-mercaptoethanol, 50 units/ml penicillin, 50 μg/ml streptomycin (Invitrogen), 20% KO-SR replacement, 40 ng/ml bFGF (Invitrogen), 2 mM Thiazovivin (Selleck) and 10 mM Y-27632 (Selleck). After 10-14 days in culture, outgrowths were manually dissociated into small-cell clumps with a microscalpel and transferred to a four-well plate with a fresh feeder layer in fresh medium. After the first several passages, colonies with ESC-like morphology were selected for further propagation, characterization and low-temperature storage. Established ESCs were expanded in ESC culture medium, which is similar to ESC-derivation medium, with modification of supplementation with 10 ng/ml bFGF. The culture medium was changed daily, and ESC colonies were split every 5-7 days manually or disaggregated by Accutase (Millipore).
Karyotype analysis
ESCs were incubated with 400 ng/ml demecolcine (Sigma) for 4 h. After being scraped off the ESCs were resuspended in 0.075 M KCl at 37 °C for 30 min. Hypotonic solution-treated cells were fixed in methanol:acetic acid (3:1 in volume) for 30 min and dropped onto precleaned slides. Chromosome spreads were Giemsa-banded and photographed. Karyotypes were assessed by normal G-banding procedure, and a normal karyotype would show normal chromosome numbers and G-banding patterns in the spreads examined. More than 10 M-phase spreads were analyzed.
Immunostaining
Cells on glass coverslips were fixed in PBS supplemented with 4% paraformaldehyde for 15 min at room temperature (RT). The cells were then permeabilized using 0.2% Triton X-100 in PBS for 15 min at RT. The cells were blocked for 30 min in 1% BSA in PBS. All primary antibodies against Oct4 (1:100) (sc-5279, Santa Cruz), Nanog (1:100) (RCAB002P-F, Reprocell), SSEA-1 (1:100) (mab4301, Millipore), SSEA-4, TRA-1-60, TRA-1-81 and Sox2 (1:100) (ab5603, Millipore) were diluted in the same blocking buffer and incubated with the samples overnight at 4 °C. The cells were treated with a fluorescently coupled secondary antibody and then incubated for 1 h at RT. The nuclei were stained with Hoechst 33342 (Sigma) for 10 min at RT.
FACS
To sort haploid cells, ESCs were disaggregated by Accutase, washed by Dulbecco's Phosphate-Buffered Saline (GIBCO) and then incubated with 15 mg/ml Hoechst 33342 in a 37 °C water bath. Subsequently, the haploid 1n peak was purified using BD FACS AriaII for further culturing. For analysis, after fixation in 4% paraformaldehyde, cells were digested by 20 mg/ml RNase A and stained with 50 mg/ml propidium iodide (PI). Analytic flow profiles were recorded by BD LSRII SORP.
Quantitative reverse transcription PCR
Total RNA was isolated from the cells using Trizol reagent (Invitrogen). One microgram of total RNA was reverse transcribed using a First Strand cDNA Synthesis kit (TOYOBO). Real-time quantitative PCR reactions were set up in triplicate using the SYBR Green Realtime PCR Master Mix (TOYOBO) and run on a Bio-Rad CFX96. All of the gene expression levels were normalized to the internal standard gene, GADPH. Primer information is presented in Supplementary information, Table S5.
Bisulphite sequencing
To obtain monkey sperm and tissue genomic DNA, samples were pretreated with tissue digest buffer, supplemented with dithiothreitol and 1% SDS (for sperm only) for 3 h, followed by proteinase K lysis and phenol-chloroform extraction. For DNA methylation analysis in ESCs and fibroblast, bisulphite conversion was performed in agarose beads as described48. The PCR products were cloned into pMD19-T vectors (Takara), and individual clones were sequenced by Invitrogen, Shanghai, China. Bisulphite primer information is presented in Supplementary information, Table S5.
Microarray analysis
RNAs from three monkey diploid ESC lines (MES1, MES2 and MES3), two haploid ESC lines (MPH1 passages7, 8, 9 and 11, and MPH2 passage 4) and duplicates of MFs (MF1 and MF2) from female individuals were extracted using TRIZOL Reagent (Life Technologies) and checked for a RIN number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent Technologies). Qualified total RNA was further purified by RNeasy micro kit (QIAGEN) and RNase-Free DNase Set (QIAGEN). Affymetrix Rhesus Macaque Gene Chip was used for gene expression analysis. RNA amplification, labeling, array hybridization and data acquisition were performed at the Shanghai Biochip Company following the manufacturer's instructions. The data were analyzed using Genespring GX software (Agilent Technologies). The relatedness of transcription profiles was determined by calculating the Pearson's correlation coefficient (r).
Comparative genomic hybridization
DNA samples (MPH1 passages 11 and 20, MPH2 passage 5 and adipose tissue from a female monkey) for CGH experiments were extracted using DNeasy Blood & Tissue Kit (QIAGEN). The DNA was purified by using 1% agarose gel electrophoresis, and quality of DNA was checked using NanoDrop (ThermoFisher). Agilent Acgh G3 Rhesus Macaque 4×180k was used for CGH analyis. Genomic DNA digestion, sample labeling, array hybridization and data acquisition were performed at the Shanghai Biochip Company following the manufacturer's instructions.
Embryoid body formation
Monkey PG-haESCs were digested into small clumps by using 5 mg/ml collagenase IV (GIBCO), and suspension culture was performed with these small clumps in low-attachment dishes for 9 days. Then, formed EBs were replated onto matrigel-coated glass slides for additional 3 days for immunofluorescence staining. The culture medium was DMEM/F12 (GIBCO) with 20% FBS (Hyclone), 2 mM L-glutamine (Invitrogen), 1% nonessential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen).
Teratoma formation
For teratoma induction, we harvested 2 × 107 cells of each PG-haESC line in 400 μl F12/DMEM (30% matrigel). We injected 200 μl of the cell suspension (1 × 107 cells) intramuscularly into NOD SCID mice with matrigel. After 8-12 weeks, teratomas were harvested. Sections were stained with hematoxylin and eosin.
Growth curve detection
Cells were plated onto 96-well plate in a density of 500 cells per well. Ten microlitre 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) (Sangon Biotech) with a concentration of 5 mg/ml was added into each well on day 1, 3, 5, 7, 9, 11 and 12 ys. MTT assays were performed as reported previously30.
PB transfection and insertion site characterization
PB (PGK-neo) and ACT-PBase were co-transfected into cells by lipofectamine (Invitrogen). Transfectants were cultured and screened by using G418 with a concentration of 250 μg/ml. To construct PB-PGK-neo-f2, the PGK-neo gene, a 1.7-kb SpeI-MunI fragment from pBigT was cloned into PB (ACT-RFP)DS to make PB-PGK-neo-f1, and thena 300-bp fragment from pBigT being cloned into SpeI sites of PB-PGK-neo-f1. For characterization of the insertion sites of PB element, genomic DNA was digested with HaeIII in 37 °C and the products were self-ligated. Then, ligated DNA was used as a template for PCR detection with primers PB-RF and PB-RR to detect the insertion sites as reported previously34.
Statistical analysis
Differences of gene expression levels between groups were analyzed by means of Student's t-test. All statistical analyses were done by SPSS software 13.0.
Accession numbers
Gene expression and CGH data sets can be accessed as the GEO reference series GSE43980 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=rbsffoccawogyxw&acc=GSE43980). This series includes the GSE43978 (mRNA expression datafrom monkey PG-haESC, diploid ESC and fibroblast) and the GSE43979 (CGH analysis of monkey PG-haESCs) data sets.
References
Zhang N, Bilsland E . Contributions of Saccharomyces cerevisiae to understanding mammalian gene function and therapy. Methods Mol Biol 2011; 759:501–523.
Elling U, Taubenschmid J, Wirnsberger G, et al. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell 2011; 9:563–574.
Leeb M, Wutz A . Derivation of haploid embryonic stem cells from mouse embryos. Nature 2011; 479:131–134.
Li W, Shuai L, Wan H, et al. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 2012; 490:407–411.
Yang H, Shi L, Wang BA, et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell 2012; 149:605–617.
Shi L, Yang H, Li J . Haploid embryonic stem cells: an ideal tool for mammalian genetic analyses. Protein Cell 2012; 3:806–810.
Sun Q, Dong J, Yang W, et al. Efficient reproduction of cynomolgus monkey using pronuclear embryo transfer technique. Proc Natl Acad Sci USA 2008; 105:12956–12960.
Ebeling M, Kung E, See A, et al. Genome-based analysis of the nonhuman primate Macaca fascicularis as a model for drug safety assessment. Genome Res 2011; 21:1746–1756.
Thomson JA, Kalishman J, Golos TG, et al. Isolation of a primate embryonic stem cell line. Proc Natl Acad Sci USA 1995; 92:7844–7848.
Thomson JA, Kalishman J, Golos TG, et al. Pluripotent cell lines derived from common marmoset (Callithrix jacchus) blastocysts. Biol Reprod 1996; 55:254–259.
Suemori H, Tada T, Torii R, et al. Establishment of embryonic stem cell lines from cynomolgus monkey blastocysts produced by IVF or ICSI. Dev Dyn 2001; 222:273–279.
Byrne JA, Mitalipov SM, Wolf DP . Current progress with primate embryonic stem cells. Curr Stem Cell Res Ther 2006; 1:127–138.
Wolf DP, Kuo HC, Pau KY, Lester L . Progress with nonhuman primate embryonic stem cells. Biol Reprod 2004; 71:1766–1771.
Mitalipov SM, Nusser KD, Wolf DP . Parthenogenetic activation of rhesus monkey oocytes and reconstructed embryos. Biol Reprod 2001; 65:253–259.
Mitalipov S, Kuo HC, Byrne J, et al. Isolation and characterization of novel rhesus monkey embryonic stem cell lines. Stem Cells 2006; 24:2177–2186.
Li C, Yu H, Ma Y, et al. Germline-competent mouse-induced pluripotent stem cell lines generated on human fibroblasts without exogenous leukemia inhibitory factor. PLoS One 2009; 4:e6724.
Byrne JA, Pedersen DA, Clepper LL, et al. Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 2007; 450:497–502.
Noggle S, Fung HL, Gore A, et al. Human oocytes reprogram somatic ells to a pluripotent state. Nature 2011; 478:70–75.
Li X, Krawetz R, Liu S, et al. ROCK inhibitor improves survival of cryopreserved serum/feeder-free single human embryonic stem cells. Hum Reprod 2009; 24:580–589.
Chen AE, Egli D, Niakan K, et al. Optimal timing of inner cell mass isolation increases the efficiency of human embryonic stem cell derivation and allows generation of sibling cell lines. Cell Stem Cell 2009; 4:103–106.
Cowan CA, Klimanskaya I, McMahon J, et al. Derivation of embryonic stem-cell lines from human blastocysts. N Engl J Med 2004; 350:1353–1356.
Tachibana M, Sparman M, Ramsey C, et al. Generation of chimeric rhesus monkeys. Cell 2012; 148:285–295.
Dighe V, Clepper L, Pedersen D, et al. Heterozygous embryonic stem cell lines derived from nonhuman primate parthenotes. Stem Cells 2008; 26:756–766.
Fujimoto A, Mitalipov SM, Kuo HC, Wolf DP . Aberrant genomic imprinting in rhesus monkey embryonic stem cells. Stem Cells 2006; 24:595–603.
Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA . Epigenetic status of human embryonic stem cells. Nat Genet 2005; 37:585–587.
Frost J, Monk D, Moschidou D, et al. The effects of culture on genomic imprinting profiles in human embryonic and fetal mesenchymal stem cells. Epigenetics 2011; 6:52–62.
Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA . Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum Mol Genet 2007; 16 Spec No. 2:R243–R251.
Dean W, Bowden L, Aitchison A, et al. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development 1998; 125:2273–2282.
Humpherys D, Eggan K, Akutsu H, et al. Epigenetic instability in ES cells and cloned mice. Science 2001; 293:95–97.
Wang BB, Lu R, Wang WC, Jin Y . Inducible and reversible suppression of Npm1 gene expression using stably integrated small interfering RNA vector in mouse embryonic stem cells. Biochem Biophys Res Commun 2006; 347:1129–1137.
Hansen MB, Nielsen SE, Berg K . Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 1989; 119:203–210.
Schimenti J . Haploid embryonic stem cells and the dominance of recessive traits. Cell Stem Cell 2011; 9:488–489.
Lois C, Hong EJ, Pease S, et al. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 2002; 295:868–872.
Ding S, Wu X, Li G, et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005; 122:473–483.
Li X, Zhu L, Yang A, et al. Calcineurin-NFAT signaling critically regulates early lineage specification in mouse embryonic stem cells and embryos. Cell Stem Cell 2011; 8:46–58.
Woltjen K, Michael IP, Mohseni P, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009; 458:766–770.
Chen YT, Furushima K, Hou PS, et al. PiggyBac transposon-mediated, reversible gene transfer in human embryonic stem cells. Stem Cells Dev 2010; 19:763–771.
Tascou S, Uedelhoven J, Dixkens C, et al. Isolation and characterization of a novel human gene, NIF3L1, and its mouse ortholog, Nif3l1, highly conserved from bacteria to mammals. Cytogenet Cell Genet 2000; 90:330–336.
Kotecki M, Reddy PS, Cochran BH . Isolation and characterization of a near-haploid human cell line. Exp Cell Res 1999; 252:273–280.
Sukov WR, Ketterling RP, Wei S, et al. Nearly identical near-haploid karyotype in a peritoneal mesothelioma and a retroperitoneal malignant peripheral nerve sheath tumor. Cancer Genet Cytogenet 2010; 202:123–128.
Carette JE, Guimaraes CP, Wuethrich I, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol 2011; 29:542–546.
Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011; 477:340–343.
Carette JE, Guimaraes CP, Varadarajan M, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009; 326:1231–1235.
Tachibana M, Ma H, Sparman ML, et al. X-chromosome inactivation in monkey embryos and pluripotent stem cells. Dev Biol 2012; 371:146–155.
Brook FA . Single-cell enzymatic dissociation of hESC lines OxF1-OxF4 and culture in feeder-free conditions. Methods Mol Biol 2012; 873:209–215.
Oh SK, Kim HS, Park YB, et al. Methods for expansion of human embryonic stem cells. Stem Cells 2005; 23:605–609.
Bajpai R, Lesperance J, Kim M, Terskikh AV . Efficient propagation of single cells Accutase-dissociated human embryonic stem cells. Mol Reprod Dev 2008; 75:818–827.
Hajkova P, el-Maarri O, Engemann S, et al. DNA-methylation analysis by the bisulfite-assisted genomic sequencing method. Methods Mol Biol 2002; 200:143–154.
Acknowledgements
We thank Dr DS Li, Dr MM Poo and Dr GL Xu for critical reading of the manuscript. This study was supported by grants from the Ministry of Science and Technology of China (2009CB941101 to JL, 2010CB945201 and 2013CB966800 to YJ), the National Natural Science Foundation of China (31225017 to JL, 91019023 to YJ), the 'Strategic Priority Research Program' of the Chinese Academy of Sciences (XDA01010403 to JL) and the Shanghai Municipal Commission for Science and Technology (12JC1409600 and 13XD1404000 to JL).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Derivation of monkey PG-haESCs. (PDF 259 kb)
Supplementary information, Figure S2
Pluripotency of ICSI-derived ESCs. (PDF 336 kb)
Supplementary information, Figure S3
Pluripotency of monkey PG-haESCs. (PDF 175 kb)
Supplementary information, Figure S4
Generation of transgenic PG-haESCs via different strategies. (PDF 160 kb)
Supplementary information, Figure S5
Insertional mutagenesis in monkey PG-haESCs. (PDF 267 kb)
Supplementary information, Table S1
Segmentation table of the CGH analysis of monkey PG-haESCs (PDF 79 kb)
Supplementary information, Table S2
Short tandem repeat genotypes in monkey oocyte donors and MPH cell lines (PDF 22 kb)
Supplementary information, Table S3
SNP analysis of monkey oocyte donors and MPHcell lines (PDF 24 kb)
Supplementary information, Table S4
Imprinted gene expression in MPH ESCs and normal diploid ESCs* (PDF 19 kb)
Supplementary information, Table S5
Primer information (PDF 208 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Yang, H., Liu, Z., Ma, Y. et al. Generation of haploid embryonic stem cells from Macaca fascicularis monkey parthenotes. Cell Res 23, 1187–1200 (2013). https://doi.org/10.1038/cr.2013.93
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.93
Keywords
This article is cited by
-
Epigenetic integrity of paternal imprints enhances the developmental potential of androgenetic haploid embryonic stem cells
Protein & Cell (2022)
-
In vitro expansion of human sperm through nuclear transfer
Cell Research (2020)
-
Current advances in haploid stem cells
Protein & Cell (2020)
-
Derivation and maintenance of mouse haploid embryonic stem cells
Nature Protocols (2019)
-
Mammalian haploid stem cells: establishment, engineering and applications
Cellular and Molecular Life Sciences (2019)
