
Abstract
Toxoplasma gondii is an intracellular protozoan parasite that infects rodents as part of its natural transmission cycle and induces disease in humans, an end-stage host. As one of the natural hosts of T. gondii, the mouse has been used extensively for elucidating the cellular and molecular basis of immunity to this pathogen while relatively few studies have focused on the response of humans. In our recent work, we identified CD16+ monocytes and DC1 dendritic cells as the major myeloid cell populations that respond to T. gondii in human peripheral blood. Interestingly, these myeloid subsets represent the opposite counterparts from those triggered by the parasite in mice. Moreover, whereas the innate cytokine response to T. gondii in the mouse involves stimulation of Toll-like receptors by a soluble parasite ligand, the response of human cells instead requires phagocytosis of the live pathogen. We speculate that these marked distinctions in the pathways utilized for innate recognition of toxoplasma in mouse and man reflect the differing roles of the two hosts in the biology of this parasite.
Similar content being viewed by others
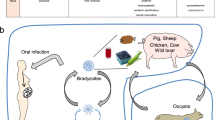


Introduction
Experimental animal models have played a major role in the elucidation of innate recognition pathways for human pathogens. In the case of parasitic protozoa, murine infection with toxoplasma has been one of the most powerful in vivo models for revealing fundamental aspects of the innate immune system–pathogen interaction. Toxoplasma gondii is an obligate intracellular protozoan that can infect a wide variety of mammals and is thought to be maintained as a zoonosis in which the parasite cycles between its feline definitive hosts and their major prey, small rodents.1 When humans acquire a T. gondii infection, they can develop a variety of pathologies but are thought to be an end-stage host not required for maintaining the life cycle.2 Thus, although inbred laboratory mice differ genetically from their wild outbred counterparts, the study of T. gondii infection in this experimental model should largely mirror a physiologic host–parasite interaction occurring naturally. However, the use of the mouse to model human toxoplasma infection and disease has never been formally justified and, as is the case with other clinically relevant pathogens, is based solely on the common mammalian phylogeny of the two species.
Intermediate hosts such as mice and humans acquire T. gondii through ingestion of parasite-containing cysts that convert into the fast-dividing tachyzoites that initially invade the gut epithelium and lamina propria, replicate asexually through a process called endodyogeny, then egress to infect new cells and disseminate systemically to multiple tissue sites in the body.3 The immune response then kicks in to control the infection and protect the host from acute death. However, a few tachyzoites escape by infecting long-lived cells such as neurons and myocytes where they convert into the slow-dividing bradyzoite stage and form tissue cysts, which normally remain dormant throughout the lifetime of the host. The induction of this chronic state is thought to be important in promoting transmission, which in the natural life cycle typically occurs through feline predation. The stimulation of a rapid and persistent immune response is thus critical for both host survival and parasite propagation.4 Early sensing of the protozoan by the innate immune system has a key role in this process.
The cell biology of the tachyzoite–host cell interaction is important in understanding the mechanisms by which innate immunity to the parasite is triggered. Unlike many other intracellular pathogens, T. gondii does not infect cells by being phagocytized or by escaping into the cytosol. Rather, toxoplasma infects host cells through an active process dependent on the actin cytoskeleton5, 6 of the parasite as well as secretory organelles called rhoptries and micronemes at the apical end of the tachyzoite, which are discharged during the invasion process.7 The parasite then forms an invagination within the host cell membrane and later encases itself in a parasitophorous vacuole (PV)8 from which most membrane proteins of host origin are excluded, thereby impairing lysosomal fusion.9 Toxoplasma is able to survive and replicate in the PV environment whereas the parasite is killed when entering the host cell through phagocytosis.10
Innate immunity against toxoplasma: the murine perspective
Early studies established the mouse model as a powerful tool for deciphering the mechanisms of host resistance to T. gondii and revealed a critical role for CD4+ dependent cell-mediated immunity in this process.11 The relevance of this pathway to humans was driven home by the observation of re-activated toxoplasmosis in AIDS.12 It subsequently became clear that interferon-γ (IFN-γ) is critical for host control of T. gondii in the mouse13 and that its production by both T helper (Th1) cells and NK cells is dependent on the innate cytokine interleukin-12 (IL-12).14, 15 As a result, the attention became focused on both the early cellular source of IL-12 and the parasite-sensing mechanism that triggers its production. A major role for dendritic cells (DCs) was suggested by experiments in which these sentinels were shown to produce high levels of the cytokine in response to a soluble tachyzoite extract (STAg) or live tachyzoites themselves both in vitro and in vivo.16 When examined in naive mouse spleen, the entire IL-12 p40 response was shown to be mediated selectively by DCs belonging to the CD8α+ subset.16 The importance of this DC population was later confirmed in studies in BATF3−/− mice that lack functional CD8α+ DCs and were shown to fail to develop IL-12-dependent immunity against the parasite following intraperitoneal (i.p.) tachyzoite inoculation.17 Nevertheless, an additional role for monocyte-derived CD11b+ CD8α− DCs as an early source of IL-12 was revealed in in vivo experiments during i.p. infection with parasite cysts (the life cycle stage that mediates natural infection of intermediate hosts). However, in contrast to IL-12 production by CD8α+ DCs, the response of this subset was dependent on prior priming by IFN-γ from NK cells.18
A second important innate cell type whose role in host resistance to T. gondii was revealed through studies in the murine model is the inflammatory monocyte. These cells, which possess a Gr-1+ Ly6C+ phenotype produce nitric oxide (NO) and tumor necrosis factor-α (TNF-α) and are recruited from the bone marrow in a CCR2-dependent manner in response to both oral and parenteral T. gondii infections.19, 20, 21, 22 Indeed, in CCR2−/− (as well as MCP-1−/−) toxoplasma-exposed mice, inflammatory monocytes fail to exit the bone marrow and migrate to the site of infection leading to increased parasite burden and mortality.21 As a consequence, Ly6C+ monocytes are necessary for the early control of T. gondii replication. Although these cells produce IL-12 and can give rise to CD11b+CD8α− (Tip) DCs, which as noted above can also produce the cytokine, their IL-12 synthesis does not appear necessary for their protective function.21 Instead, inflammatory monocytes appear to be directly involved in limiting parasite growth.
The ability of a STAg extract, to stimulate IL-12 production from murine DCs in vitro allowed the purification of the major parasite agonist for this important innate response. The molecule in question is the T. gondii homolog of profilin, a protein that is required for tachyzoite actin remodeling in the parasite during host cell invasion and egress.23, 24 Purified or recombinant profilin in low doses stimulates high levels of IL-12 production in vitro from DCs and IFN-γ-activated macrophages, in vivo as assayed in serum or in situ in lymphoid tissue. The fact that profilin is a protein lacking significant carbohydrate or lipid modification led to the rapid identification of the host pattern recognition receptors (PRRs) through which it signals. The first PRR was the Toll-like receptor TLR11.23 Mice deficient in TLR11 failed to produce IL-12 following T. gondii infection or injection with STAg or profilin, and this response defect was also observed in isolated DCs. Later, TLR12 was identified as a second receptor for T. gondii profilin, and TLR12 KO mice were shown to have defects in IL-12 production to the parasite that were similar to those of TLR11-deficient animals.25 The discovery that TLR11 and TLR12 can form functional heterodimers25, 26, 27 is the probable explanation of their similar role in host resistance. Nevertheless, TLR12-deficient mice appear to be more susceptible to T. gondii infection than their TLR11 counterparts, an observation that we proposed may result from the expression of TLR12 but not TLR11 homodimers on plasmacytoid DCs, which in turn promotes the induction of IFN-γ from NK cells.25
TLR11 and TLR12 together with the nucleic acid-sensing TLR3, TLR7 and TLR9 share an endosomal location in the host cell. UNC93B1 is a chaperone required to bring all five of these TLRs from the endoplasmic reticulum (ER) to the endocytic system.28 UNC93B1-deficient mice were found to be highly susceptible to toxoplasma infection29 and considerably more so than TLR11-deficient mice. Interestingly, quadruple KO mice deficient in TLR3, TLR7, TLR9 and TLR11 closely resemble UNC93B1 mutant mice in their complete loss of resistance to T. gondii.26 This observation suggested that the extreme phenotype of the UNC93B1 mutant mice is due to a combined defect in the nucleic acid-sensing and profilin-sensing TLRs, thereby implicating TLR3, TLR7 and/or TLR9 in the innate immunity to T. gondii.26
Because of their importance in the innate response of toxoplasma, the genomes of other mammalian host species were examined for the presence of TLR11 and 12 homologs. Unlike most other TLRs, TLR11 and TLR12 were found to be restricted to mammals and present only in a subset of species within this phylogenetic class.30 Although clearly expressed by rodents and other feline prey, the two receptors are notably absent in cats and birds, which can also serve as intermediate hosts for toxoplasma. Importantly, as introduced above, functional TLR11 and 12 genes appear to be totally lacking in humans and all other primates. Thus, although the segment of mouse chromosome 4 that contains the TLR11 gene is found on human chromosome 1, the human TLR11 gene contains three stop codons and does not encode a functional protein. Moreover, the TLR12 gene, located on chromosome 14 of the mouse, is entirely absent in the human genome.30, 31 This observation predicted that the recognition of profilin, which is a strong activator of the innate immune system in the mouse, may play no role in the innate response of humans to T. gondii infection.
The human innate response to T. gondii: a gaping hole in our knowledge
Studying the immunology of toxoplasma infection in humans poses major challenges not encountered in the murine model. First, as with most other human immunological studies, the measurement of immune responses is largely confined to the peripheral blood and, in the case of innate responses, there is no way to examine cellular immunity at the site of initial infection. A second shortcoming is that although latently infected seropositive individuals are many, it is very hard to obtain patients during the brief period of acute exposure to the parasite. Nevertheless, numerous studies have been performed on patients with ocular toxoplasmosis and other parasite-induced pathologies.32, 33, 34 In general, these studies support the general Th1/Th17 character of the adaptive immune response to T. gondii and an effector role for T-cell-derived IFN-γ in the intracellular control of parasite growth in vitro.35 However, until recently, there have been only a few publications addressing the response and function of human innate cells to toxoplasma and their mode of recognition of the parasite.36, 37
We have attempted to address this deficit by systematically studying the production of innate cytokines by primary myeloid cell populations in human peripheral blood following infection with live tachyzoites or exposure to parasite products. Monocytes and DCs are the major human leukocytes that secrete innate cytokines, such as IL-1, IL-6, IL-12 and TNF, in response to microbial stimulation. Human monocytes are heterogeneous and consist of three major subsets. The largest subset consists of the classical (CL) monocytes, which make up to 85% of the total population and are CD14+ CD16neg. This subset is the human equivalent of murine inflammatory monocytes (Ly6C+)38 and enters into inflamed tissues. The two smaller subsets are the intermediate (IM) monocytes and non-classical (NC) monocytes, which both express CD16 and are CD14+ and CD14dim, respectively. The NC subset is the human equivalent of the murine patrolling monocyte subset (Ly6Clo) that responds to viral pathogen-associated molecular patterns (PAMPs), recruits neutrophils to sites of inflammation and clears cellular debris from the luminal side of blood vessels.39 The myeloid DCs (mDCs) in human peripheral blood can also be divided into two major subsets: the CD1c+ mDC1 and the CD141 mDC2 that have unique functions and are the human equivalent of murine CD8α− CD11b+ DCs40 and CD8α+ DCs,41 respectively.
When exposed to live tachyzoites, unfractionated elutriated monocytes from normal donors produced the important innate cytokines TNF and IL-12 p40 (as well as p70) along with a select group of other cytokines, chemokines and growth factors.42 Because of its major role in Th1 immunity, we decided to use IL-12 as the primary read-out in our subsequent experiments. The bioactivity of the IL-12 produced was confirmed in co-culture experiments in which stimulated monocytes as well as their supernatant were shown to trigger IFN-γ production from purified NK cells in an IL-12-dependent manner. The above experiments established a model for studying innate recognition of T. gondii by human myeloid cells.42
T. gondii induced IL-12 production in mice and humans originates from distinct myeloid subsets
Because of the well-defined function of DCs in innate immunity to T. gondii in the mouse, we next determined whether this myeloid cell type responds to tachyzoite stimulation in humans. Both monocyte-derived DCs (differentiated in vitro) and primary mDCs FACS-purified from total elutriated monocytes produced high levels of IL-12 p40 following parasite exposure in vitro. Interestingly, when mDCs were subfractionated into their major CD1c+ mDC1 and the CD141+ mDC2 subsets, only the DC1 cells responded.
As mentioned above, in the murine system, CD8α+ DCs produce high levels of IL-12 when exposed to T. gondii. By contrast, their human counterpart, the mDC2 subset, failed to produce significant levels of the cytokine following comparable parasite stimulation.
In parallel studies, elutriated monocytes were sorted by FACS into their major CL, NC, and IM subsets defined above. Surprisingly, we found that the minor IM and NC monocyte subsets produce the majority of the IL-12 (and TNF), whereas the CL monocytes are largely non-responsive (Figure 1). Again, this result is in sharp contrast to the mouse in which inflammatory monocytes (the CL equivalent) have been documented as major responders at T. gondii tissue infection sites19, 21 and in the circulation of naive mice (unpublished observations by the authors). The failure of the mDC2 and CL myeloid cells to produce IL-12 following T. gondii stimulation could not be accounted for by a general defect in the synthesis of the cytokine by these subsets as they were fully responsive when stimulated with LPS and R848.42

Discordance in human and murine equivalent myeloid subsets producing IL-12 in response to T. gondii. Peripheral blood murine monocytes are defined phenotypically as CD115+ CD11b+ and further subdivided into inflammatory (Ly6Chi) or patrolling (Ly6Clow) subsets. Similarly, human monocytes are defined as HLA-DR+ and subdivided into classical (CD16neg CD14+), intermediate (CD16+ CD14+) and non-classical (CD16+ CD14dim) subsets. In the mouse, lymphoid resident conventional DCs are CD11c+ MHC II+ and are subdivided into CD8α+ CD11bneg or CD8α+CD11b+ subsets. Human peripheral blood myeloid DCs are CD11c+ HLA-DR+ and defined as either mDC1 (CD141neg CD1c+) or mDC2 (CD141+ CD1cneg). In this figure, the murine subsets and their human counterparts are indicated by similar coloring. The myeloid subsets that produce IL-12 in response to T. gondii tachyzoites are highlighted with a gray shadow.
The response of human myeloid cells to T. gondii requires phagocytosis of live tachyzoites
As noted above, the innate cytokine response of murine myeloid cells to T. gondii is mediated primarily by the TLR11- and TLR12-dependent recognition of profilin, a molecule that appears to be released by tachyzoites in a soluble form. By contrast, earlier studies had suggested that in the case of human cells, live tachyzoites are needed to stimulate cytokine production in vitro.36, 37 We confirmed and extended these observations showing that while live parasites trigger IL-12 and TNF production from human monocytes and DCs, neither recombinant profilin, STAg nor heat-killed tachyzoites do so and that tachyzoites genetically deficient in profilin induce normal cytokine secretion. Moreover, we observed that direct parasite-host cell contact is critical for TNF and IL-12 induction.42 These findings are at odds with a recent study by Salazar Gonzalez et al.,43 who showed that T. gondii profilin stimulates human monocytes to secrete IL-6 and IL-12 in a TLR5-dependent manner. The basis of this discrepancy is unclear but may relate to the purity of the profilin or cell populations employed by the two laboratories.
As introduced above, tachyzoites can enter host cells either by invasion where they can establish a replicative infection or by phagocytosis where they are rapidly killed. Surprisingly, when we treated tachyzoites with either of two irreversible inhibitors that block their invasion capacity without affecting parasite viability, an enhancement of cytokine production was indeed observed. By contrast, when either parasite phagocytosis or endosomal acidification was inhibited by drug treatment of the host cells, the tachyzoite stimulated cytokine response was completely suppressed.42 Thus, in the absence of the TLR11 and 12-profilin recognition pathway present in the mouse, human monocytes and DCs sense T. gondii by a distinct mechanism that requires the phagocytic uptake and endosomal processing of the live pathogen (Figure 2). Although it is possible that a related phagocytosis-dependent cytokine triggering mechanism may also exist in the mouse and other TLR11 and 12-expressing host species, its function is likely redundant because of the presence of the more dominant profilin recognition driven pathway.

Proposed T. gondii sensing mechanism(s) utilized by human monocytes. Although T. gondii tachyzoites can infect host cells either actively by invasion or passively by being phagocytized, only the latter process results in proinflammatory cytokine secretion by human myeloid cells. Once the live parasite has been engulfed (1), endosomal acidification and recruitment of lysosomes to the phagosomes leads to the degradation of the parasite (2). Released parasite molecules may be recognized either by PRR-contacting endosomes trafficking to the phagolysosome (3a) or by PRR in the cytosol detecting parasite components leaked from the phagosome (3b). In the scenario/model indicated in 3a, T. gondii DNA and RNA interaction with endosomal TLR will activate the canonical NF-κB signaling pathway and trigger TNFα and IL-12 production. In the scenario/model shown in 3b, as a consequence of phagolysosome leakage, parasite RNA is recognized by cytosolic RLR, such as RIG-I or MDA-5, which signals through the MAVS-dependent pathway to promote secretion of these cytokines.
In murine myeloid cells, direct infection with live tachyzoites is associated with the suppression of NF-κB-dependent cytokine production induced by exogenous agonists.44, 45 Interestingly, we observe a similar phenomenon in human monocytes that become actively infected with the parasite (unpublished observations). Thus, the responsiveness of human cells to tachyzoites is unlikely due to the absence of this downregulatory pathway but rather to the dominance of phagocytosis-dependent cytokine induction in this species.
How do human myeloid cells sense T. gondii?
If not through TLR11 and 12, what is the innate recognition mechanism by which human cells detect and respond to T. gondii? Because phagocytosis of heat-killed tachyzoites fails to trigger cytokine production, the phagocytic event in itself is not the critical signal. Similarly, it is unlikely that PRR on the host cell plasma membrane are involved as they should be triggered normally when phagocytosis is blocked. Instead, PRR present in the endosomal membrane or cytosol would seem more likely candidates. Indeed, as noted above, T. gondii DNA and mRNA stimulate IL-12 production from total human peripherial blood mononuclear cells when the cells were primed with IFN-γ, suggesting the involvement of nucleic acid-sensing PRR known to be located at these two intracellular sites.26 As a labile molecule likely to be sensitive to digestion in heat-killed parasites or tachyzoite extracts, parasite RNA is attractive as a candidate PAMP for cytokine induction. Preliminary experiments in which inhibitors of the endosomal TLR (that is, TLR3, TLR7 and TLR9) failed to suppress the cytokine response of human monocytes to tachyzoites raise the interesting possibility that cytosolic sensors of parasite nucleic acids or other components may have a role in this pathway. In this scenario, the relevant parasite PAMPS would reach the cytoplasm through leakage of the phagolysosomal membrane. The possible downstream involvement of autophagy-associated signals also deserves investigation.
Evolutionary implications
Although the findings summarized here cast a strong cautionary shadow on the use of mice to model mechanisms of human innate immunity to T. gondii, they pose a series of fascinating questions related to the development of pathogen recognition mechanisms during mammalian evolution. As discussed above and in a previous publication,30 rodents are more than just a convenient experimental model for studying infection with this protozoan pathogen. They are a major intermediate host involved in maintaining the life cycle of the parasite. Humans, by contrast, are thought to be an end-stage host for toxoplasma and are not essential for its natural transmission. Nevertheless, because they both display a high prevalence of infection, rodents and humans need to be able to control T. gondii and clearly develop strong adaptive immunity as evidenced by the extreme susceptibility of T-cell-compromised mice and patients to the parasite. A critical difference is that in the case of rodents the development of strong host resistance is also of enormous benefit to the parasite. This is because felines acquire the infection largely through predation rather than scavenging, and it is thus in toxoplasma’s self-interest to be controlled by the rodent immune system rather than induce rodent death. In so doing, it also induces neurologic changes in its hosts that make them easier prey for cats.46 Because of the importance of this adaptation, T. gondii may have evolved a specialized profilin protein that is a strong agonist of TLR11 and TLR12, thereby ensuring the induction of a highly effective innate and adaptive response that prevents it from overwhelming its critical intermediate hosts. Indeed, T. gondii profilin appears to be considerably more potent than phylogenetically related profilins in stimulating TLR11-/TLR12-dependent cytokine production in mouse myeloid cells.23, 47
TLR11/TLR12 is expressed functionally in many lower and higher mammalian species that are not normal hosts for T. gondii; thus, it is unlikely that these receptors evolved solely in response to selection by the parasite. The explanation for the loss of the two receptors in primates is unclear. One hypothesis30 is that they were selected against because of their potential to elicit host immunopathology either in response to foreign or possibly self-profilin (in the event of mutation). Given the preservation of TLR4 and other PRR with similar pathologic signaling potential in humans, this explanation seems unlikely. More plausible is that the loss in TLR11 and TLR12 occurred spontaneously after the evolution of a redundant and similarly protective innate-sensing mechanism for triggering cell-mediated immunity to T. gondii. As proposed here, this parasite recognition pathway appears to be linked to tachyzoite phagocytosis, thus avoiding the potential deleterious effects of sensing of the parasite 'at a distance' associated with profilin recognition by TLR11/TLR12. Indeed, it is interesting that while IL-12 production by myeloid cells in humans requires live tachyzoite uptake, the cells responding in the mouse are those that have never made direct contact with the parasite.48
More clues to this fascinating evolutionary puzzle should emerge as the nature of the mechanism responsible for innate recognition of T. gondii in humans is elucidated. It will be equally exciting to learn whether the phagocytosis-dependent pathway summarized here has a role in the innate sensing of other human pathogens and whether it functions in more host species in which TLR11/TLR12 receptors have been lost. Finally, having characterized a mechanism for innate recognition of T. gondii in humans, it will be important to elucidate its role in both the induction of adaptive immunity and in clinical disease.
References
Hill D, Dubey JP . Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect 2002; 8: 634–640.
Tenter AM, Heckeroth AR, Weiss LM . Toxoplasma gondii: from animals to humans. Int J Parasitol 2000; 30: 1217–1258.
Harker KS, Ueno N, Lodoen MB . Toxoplasma gondii dissemination: a parasite’s journey through the infected host. Parasite Immunol 2015; 37: 141–149.
Buzoni-Gatel D, Werts C . Toxoplasma gondii and subversion of the immune system. Trends Parasitol 2006; 22: 448–452.
Dobrowolski JM, Sibley LD . Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell 1996; 84: 933–939.
Meissner M, Schluter D, Soldati D . Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 2002; 298: 837–840.
Carruthers VB, Sibley LD . Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur J Cell Biol 1997; 73: 114–123.
Suss-Toby E, Zimmerberg J, Ward GE . Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc Natl Acad Sci USA 1996; 93: 8413–8418.
Mordue DG, Desai N, Dustin M et al. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med 1999; 190: 1783–1792.
Sibley LD, Weidner E, Krahenbuhl JL . Phagosome acidification blocked by intracellular Toxoplasma gondii. Nature 1985; 315: 416–419.
Gazzinelli R, Xu Y, Hieny S et al. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J Immunol 1992; 149: 175–180.
Kodym P, Maly M, Beran O et al. Incidence, immunological and clinical characteristics of reactivation of latent Toxoplasma gondii infection in HIV-infected patients. Epidemiol Infect 2015; 143: 600–607.
Suzuki Y, Orellana MA, Schreiber RD et al. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 1988; 240: 516–518.
Gazzinelli RT, Hieny S, Wynn TA et al. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci USA 1993; 90: 6115–6119.
Gazzinelli RT, Wysocka M, Hayashi S et al. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J Immunol 1994; 153: 2533–2543.
Reis e Sousa C, Hieny S, Scharton-Kersten T et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med 1997; 186: 1819–1829.
Mashayekhi M, Sandau MM, Dunay IR et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011; 35: 249–259.
Goldszmid RS, Caspar P, Rivollier A et al. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 2012; 36: 1047–1059.
Dunay IR, Damatta RA, Fux B et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 2008; 29: 306–317.
Dunay IR, Fuchs A, Sibley LD . Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect Immun 2010; 78: 1564–1570.
Robben PM, LaRegina M, Kuziel WA et al. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med 2005; 201: 1761–1769.
Mordue DG, Sibley LD . A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol 2003; 74: 1015–1025.
Yarovinsky F, Zhang D, Andersen JF et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 2005; 308: 1626–1629.
Plattner F, Yarovinsky F, Romero S et al. Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe 2008; 3: 77–87.
Koblansky AA, Jankovic D, Oh H et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 2013; 38: 119–130.
Andrade WA, Souza Mdo C, Ramos-Martinez E et al. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 2013; 13: 42–53.
Raetz M, Kibardin A, Sturge CR et al. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J Immunol 2013; 191: 4818–4827.
Kim YM, Brinkmann MM, Paquet ME et al. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008; 452: 234–238.
Melo MB, Kasperkovitz P, Cerny A et al. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog 2010; 6: e1001071.
Gazzinelli RT, Mendonca-Neto R, Lilue J et al. Innate resistance against Toxoplasma gondii: an evolutionary tale of mice, cats, and men. Cell Host Microbe 2014; 15: 132–138.
Roach JC, Glusman G, Rowen L et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA 2005; 102: 9577–9582.
Yamamoto JH, Vallochi AL, Silveira C et al. Discrimination between patients with acquired toxoplasmosis and congenital toxoplasmosis on the basis of the immune response to parasite antigens. J Infect Dis 2000; 181: 2018–2022.
de-la-Torre A, Sauer A, Pfaff AW et al. Severe South American ocular toxoplasmosis is associated with decreased Ifn-gamma/Il-17a and increased Il-6/Il-13 intraocular levels. PLoS Negl Trop Dis 2013; 7: e2541.
de-la-Torre A, Pfaff AW, Grigg ME et al. Ocular cytokinome is linked to clinical characteristics in ocular toxoplasmosis. Cytokine 2014; 68: 23–31.
Meira CS, Pereira-Chioccola VL, Vidal JE et al. Cerebral and ocular toxoplasmosis related with IFN-gamma, TNF-alpha, and IL-10 levels. Front Microbiol 2014; 5: 492.
Denney CF, Eckmann L, Reed SL . Chemokine secretion of human cells in response to Toxoplasma gondii infection. Infect Immun 1999; 67: 1547–1552.
Subauste CS, Wessendarp M . Human dendritic cells discriminate between viable and killed Toxoplasma gondii tachyzoites: dendritic cell activation after infection with viable parasites results in CD28 and CD40 ligand signaling that controls IL-12-dependent and -independent T cell production of IFN-gamma. J Immunol 2000; 165: 1498–1505.
Ingersoll MA, Spanbroek R, Lottaz C et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2010; 115: e10–e19.
Auffray C, Fogg D, Garfa M et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007; 317: 666–670.
Bachem A, Guttler S, Hartung E et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207: 1273–1281.
Watchmaker PB, Lahl K, Lee M et al. Comparative transcriptional and functional profiling defines conserved programs of intestinal DC differentiation in humans and mice. Nat Immunol 2014; 15: 98–108.
Tosh KW, Mittereder L, Bonne-Annee S et al. The IL-12 response of primary human dendritic cells and monocytes to Toxoplasma gondii is stimulated by phagocytosis of live parasites rather than host cell invasion. J Immunol 2016; 196: 345–356.
Salazar Gonzalez RM, Shehata H, O’Connell MJ et al. Toxoplasma gondii-derived profilin triggers human Toll-like receptor 5-dependent cytokine production. J Innate Immun 2014; 6: 685–694.
Butcher BA, Kim L, Johnson PF et al. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-κB. J Immunol 2001; 167: 2193–2201.
Butcher BA, Denkers EY . 2002. Mechanism of entry determines the ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect Immun 2002; 70: 5216–5224.
Berdoy M, Webster JP, Macdonald DW . Fatal attraction in rats infected with Toxoplasma gondii. Proc Biol Sci 2000; 267: 1591–1594.
Kucera K, Koblansky AA, Saunders LP et al. Structure-based analysis of Toxoplasma gondii profilin: a parasite-specific motif is required for recognition by Toll-like receptor 11. J Mol Biol 2010; 403: 616–629.
Christian DA, Koshy AA, Reuter MA et al. Use of transgenic parasites and host reporters to dissect events that promote interleukin-12 production during toxoplasmosis. Infect Immun 2014; 82: 4056–4067.
Acknowledgements
We thank Drs Ricardo Gazzinelli, Lis Antonelli, Carl Feng, Jonathan Howard, Steve Singer and Tom Nutman for helpful discussions. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sher, A., Tosh, K. & Jankovic, D. Innate recognition of Toxoplasma gondii in humans involves a mechanism distinct from that utilized by rodents. Cell Mol Immunol 14, 36–42 (2017). https://doi.org/10.1038/cmi.2016.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.12
