
Abstract
Breast cancer that is accompanied by a high level of cyclin E expression usually exhibits poor prognosis and clinical outcome. Several factors are known to regulate the level of cyclin E during the cell cycle progression. The transcription factor DEC1 (also known as STRA13 and SHARP2) plays an important role in cell proliferation and apoptosis. Nevertheless, the mechanism of its role in cell proliferation is poorly understood. In this study, using the breast cancer cell lines MCF-7 and T47D, we showed that DEC1 could inhibit the cell cycle progression of breast cancer cells independently of its transcriptional activity. The cell cycle-dependent timing of DEC1 overexpression could affect the progression of the cell cycle through regulating the level of cyclin E protein. DEC1 stabilized cyclin E at the protein level by interacting with cyclin E. Overexpression of DEC1 repressed the interaction between cyclin E and its E3 ligase Fbw7α, consequently reducing the level of polyunbiquitinated cyclin E and increased the accumulation of non-ubiquitinated cyclin E. Furthermore, DEC1 also promoted the nuclear accumulation of Cdk2 and the formation of cyclin E/Cdk2 complex, as well as upregulating the activity of the cyclin E/Cdk2 complex, which inhibited the subsequent association of cyclin A with Cdk2. This had the effect of prolonging the S phase and suppressing the growth of breast cancers in a mouse xenograft model. These events probably constitute the essential steps in DEC1-regulated cell proliferation, thus opening up the possibility of a protein-based molecular strategy for eliminating cancer cells that manifest a high-level expression of cyclin E.
Similar content being viewed by others


Main
DEC1 belongs to a subfamily of bHLH transcription factors that are involved in a number of cell processes, including proliferation, apoptosis and circadian rhythms.1, 2, 3, 4 Several studies, which used protein overexpression and knockdown strategies have revealed the roles of DEC1 in cell cycle arrest, cell senescence and cell survival.5 Previous studies have shown that DEC1 can repress transcription in a HDAC-dependent manner, causing cell growth arrest, as well as demonstrating that DEC1 is a target of the p53 family and mediates cell cycle arrest and DNA damage-induced premature senescence.6, 7, 8 DEC1 also plays a role in cell survival. It mediates TGF-β-induced cell survival in breast cancer cells,9 and DEC1-overexpressing cells can resist oxidative stress-mediated cell death.10 Furthermore, DEC1 also regulates p53-dependent cell survival versus cell death through MIC-1 in response to DNA damage stress.11 Thus, DEC1 has multifaceted roles in cancer progression. However, whether it also affects cancer progression through regulating the cell cycle factors has not yet been clearly established.
Cyclin E, a member of the cyclin family, binds to and activates the Cdk2.12 The level of cyclin E protein oscillates throughout the cell cycle and peaks at around the beginning of the S phase, but subsequent degradation of the cyclin E protein is needed for the orderly cell progression to occur, which is regulated by E2Fs-dependent cyclin E transcription and ubiquitin-mediated cyclin E proteolysis.13 Two types of ubiquitin ligases are known to trigger the ubiquitin-mediated degradation of cyclin E, and these are the Cul1-(SCF) or Cul3-(BCR) dependent ubiquitin ligases.14, 15, 16, 17 Cyclin E that is bound to Cdk2 is targeted for ubiquitination by Cul1-dependent ubiquitin ligase, and this ubiquitination requires the phosphorylation of cyclin E at specific residues (Thr62, Ser372, Thr380 and Ser384).12, 17, 18 During the G1→S phase transition of the cell cycle progression, the formation of cyclin E/Cdk2 complex occurs in the nuclei and it needs to reach certain threshold in order to trigger the initiation of DNA replication.7, 19 However, abnormal stabilization of cyclin E inhibits transcription by increasing the initiation of replication and subsequently induces delay in the S phase.20, 21 Dysregulated activity of cyclin E is known to cause cell lineage-specific abnormalities such as impaired maturation as a result of increased genetic instability, cell proliferation and apoptosis or senescence via several different mechanisms.16, 22
In this study, we showed that DEC1 stabilized cyclin E without affecting its mRNA level. We also demonstrated that DEC1 stabilized cyclin E by blocking the proteasome pathway and hence, repressed the ubiquitination of cyclin E through reducing the interaction between cyclin E and Fbw7α. Furthermore, DEC1 promoted the activity and the formation of cyclin E/Cdk2 complex as well as the localization of cyclin E and Cdk2 in the nucleus, and repressed the subsequent formation of the cyclin A/Cdk2 complex, which led to the cells stalling at the S phase. These findings therefore provided new insight into the mechanism associated with DEC1-regulated cell cycle and proliferation of breast cancer cells.
Results
DEC1 expression is reduced in breast cancer cells and inhibits the proliferation
In order to study the function of DEC1 in breast cancer cells, we examined the expression and subcellular location of DEC1 in breast carcinoma and adjacent normal breast tissue using immunohistochemistry analysis. Overall, 25/30 (83.3%) of the breast carcinoma were negative for DEC1 and 16/18 (88.9%) of the adjacent normal breast tissue were positive for DEC1 (Figure 1a). DEC1 was highly expressed in adjacent noncancerous breast tissue, but only lowly expressed in breast carcinoma (Figures 1a and b; Supplementary Figure S1). The effect of DEC1 on the proliferation of breast cancer cells was investigated by overexpressing DEC1 in MCF-7 and T47D cells assessing their proliferation by MTT and colony formation assays. Overexpression of DEC1 inhibited the proliferation and colony formation of both MCF-7 and T47D cells (Figures 1c and d). Two DEC1 siRNAs (siRNA-1 and siRNA-2) were synthesized and used to knockdown the endogenous DEC1. siRNA-2 appeared to be more effective than siRNA-1 (Figure 1e). Therefore, the DEC1-specific target sequence within siRNA-2 was used to construct the shRNA (shDEC1) for the knockdown of DEC1 expression. The results showed that knockdown of DEC1 in MCF-7 cells increased the number and size of the colonies compared to the control cells (Figure 1f). These data suggested that DEC1 may play a regulatory role in the proliferation of MCF-7 and T47D breast cancer cells.

The expression of DEC1 in human breast tumor tissue samples and the effect of DEC1 on the cell proliferation. (a and b) Immunohistochemistry (IHC) analysis of DEC1 expression in adjacent normal breast tissues (n=18) and breast carcinoma (n=30). Each sample was incubated with antibody against DEC1. Positive staining and negative staining are indicated by brown and blue staining, respectively. (c and d) MCF-7 cells and T47D cells were transfected with Flag-DEC1 or control vectors (Ctrl). Cells were then cultured in selective medium (500 μg/ml G418) and subjected to colony formation and MTT assays (days, time after transfection). Each bar represents the mean±S.D. from five independent experiments. *P<0.05. (e) Western blot analysis of endogenous DEC1 expression. MCF-7 cells were transfected with DEC1 siRNAs (siRNA-1 and siRNA-2) for 24 h and probed with anti-DEC1 and anti-β-actin. Each bar represents the mean±S.D. from three independent experiments. (**P<0.01). (f) Colony formation assay, MCF-7 cells were transfected with the shDEC1 vector, followed by 2-week selection with hygromycin B. Hygromycin-resistant clones are shown in the left panel. The right panel shows the corresponding quantitative analyses. Only representative data from three independent experiments are shown. Scale bar, 500 μm
DEC1 regulates cyclin E at late G1 phase and S phase
To explore whether the levels of DEC1 protein at the various cell cycle stages are differently regulated, we measured the levels of DEC1 protein in MCF-7 cell extracts at every 2 h after 16 h of synchronization by nocodazole (which caused cell arrest at the G2/M stage; Figure 2a, upper panel). The phase of the cell cycle at each time point was determined by the expression profiles of cyclin B, cyclin E and FACS analysis (Figure 1a, bottom). The expression of endogenous DEC1 was increased during the late G1 phase, and peaked at the G1/S boundary before decreasing during the following S phase, which almost precisely overlapped with the expression dynamics of cyclin E (Figure 1a). These data suggested that DEC1 may play an important role in regulating the progression of cell cycle at the G1/S checkpoint.

The expression dynamics of DEC1 in cell cycle and the effects of DEC1 expression on cyclin E stability. (a) Endogenous DEC1 levels during the cell cycle. MCF-7 cells were synchronized at G2/M by treating with 50 ng/ml nocodazole for 16 h. The cells were collected at the indicated time points following the removal of nocodazole. The cells were analyzed by FACS, and DEC1 level in the total cell lysate was determined by western blotting using anti-DEC1 antibody. (b and c) DEC1 affects the level of cyclin E protein but not in its transcription. The transcript and protein levels of cyclin E, p53 and p21 were measured in MCF-7 cells that overexpressed cyclin E only or cyclin E plus DEC1 by using reverse transcription PCR or by western blotting using specific antibodies against cyclin E, p53 and p21. (d) Western blot of cyclin E in DEC1-overexpressing T47D and MCF-7 cells. (e) T47D and MCF-7 cells transfected with DEC1 shRNA and probed with anti-DEC1 and anti-cyclin E antibodies. (f) DEC1 regulates cyclin E in a dose-dependent way. MCF-7 cells were transfected with Myc-tagged cyclin E and either empty vector or 0.5, 1 and 4 μg Flag-DEC1. Lysates were analyzed by western blot with anti-myc, anti-Flag or anti-β-actin. (g) DEC1 regulates the level of cyclin E protein in both the presence and absence of serum. MCF-7 cells were transfected with the indicated plasmids for 12 h and were then starved for 24 h in serum-free medium. The histogram in the right plot shows the quantitative analysis of the bands. (h) Western blot analysis of the effect of DEC1 on the stability of cycin E under serum starvation condition. MCF-7 cells were transfected with or without DEC1 to detect the change in cyclin E protein level. The graph shows the relative intensity of the cycin E bands at different time points. In all experiments (a–h), β-actin expression or GAPDH mRNA level was used as a reference
We next investigated whether the involvement of DEC1 in the cell cycle progression would involve the regulation of these cell cycle factors (such as p53, p21 and cyclin E23) by DEC1. Surprisingly, overexpression of DEC1 led to an obvious increase in the level of endogenous cyclin E, but not of endogenous p53 protein, and a slight decrease in the level of endogenous p21 protein (Figure 2b) without affecting their mRNA levels (Figure 2c). Increased expression of cyclin E was induced by the overexpression of DEC1 and in a dose-dependent manner (Figures 2d and f). In contrast, knockdown of DEC1 decreased the level of endogenous cyclin E, especially in the case of MCF-7 cells (Figure 2e). Subsequent experiments that focused on the mechanism by which DEC1 could stabilize cyclin E were carried out in MCF-7 cells because the effect was more obvious in MCF-7 cells than in T47D cells. These data led us to hypothesize that DEC1 may regulate the expression of cyclin E independently of its transcriptional activity. To detect whether DEC1 could regulate the stability of cyclin E independently of its transcriptional activity, we constructed three truncated versions of DEC1 and tested if they could regulate the stability of cyclin E. Flag-DEC1 (302–412) contained neither the bHLH domain nor the three α-helices, and it could upregulate the stability of cyclin E (Supplementary Figures S2A and B). However, Flag-DEC1 (1–129) and Flag-DEC1 (129–301), which contained only the bHLH domain and the three α-helices, respectively, exhibited little or no effect on the protein level of cyclin E. This suggested that DEC1 may upregulate the stability of cyclin E independently of its transcriptional activity.
Since serum stress condition could block cell proliferation at the G0/G1 stage and decrease cyclin E expression,24, 25 we examined the role of DEC1 on the expression of cyclin E in MCF-7 cells that had been subjected to serum starvation, a condition that would effectively reduce the expression of cyclin E. DEC1 still upregulated the expression of cyclin E regardless of serum starvation (Figure 2g). Furthermore, the level of endogenous cyclin E was more stable in cells that overexpressed DEC1, but decreased sharply in the control cells, especially during the first 8 h of serum starvation (Figure 2h). These findings suggested that DEC1 was necessary for stabilizing cyclin E regardless of whether or not the cells were under serum starvation.
DEC1 inhibits Fbw7-mediated cyclin E ubiquitin-proteasome pathway
The positive effect that DEC1 exerted on the stability of cyclin E suggested that it might increase the half-life of cyclin E. Indeed, DEC1 markedly extended the half-life of cyclin E from 4 to 8 h in MCF-7 (Figure 3a) and T47D (Supplementary Figure S3B) cells. Increase in the half-life of a protein usually involves a reduction of its degradation via the proteasome pathway.26 When the cells were treated with CHX plus the proteasome inhibitor MG132, overexpression of DEC1 did not enhance the half-life of cyclin E (Figure 3b) suggesting that DEC1 may regulate the stability of cyclin E protein via the ubiquitin-proteasome pathway. Subsequent ubiquitination assay showed that in the absence of DEC1 overexpression, cyclin E was heavily ubiquitinated (Figure 3c, lane 2), whereas in the presence of DEC1 overexpression, cyclin E ubiquitination was markedly decreased (Figure 3c, lane 3). Since Fbw7s is the ubiquitination E3 ligase of cyclin E,17 the effects of all three isoforms of Fbw7s on the stability of cyclin E were investigated. Cyclin E level was most significantly affected by Fbw7α, which caused the highest reduction compared to the other two isoforms (Figure 3d). However, this effect of Fbw7α on cyclin E was compromised by DEC1, since cells that overexpressed Fbw7α, cyclin E and DEC1 yielded the same level of cyclin E as cells that overexpressed cyclin E only (Figure 3e). When Fbw7s in the cells was knocked down by shFbw7, overexpression of DEC1 exhibited no increase in the level of cyclin E compared to cells in which Fbw7s was also knocked down, but without overexpression of DEC1 (Figure 3f). Since the effect of DEC1 on the stability of cyclin E depended on Fbw7α, we sought to determine whether DEC1 could affect the interaction between cyclin E and Fbw7α. Co-immunoprecipitation experiment showed that overexpression of DEC1 markedly reduced the interaction between Fbw7α and cyclin E whether or not the cells were under serum starvation (Figures 3g–j). In addition, we also investigated the ubiquitination of endogenous cyclin E in the cells in which either DEC1 or Fbw7 had been knocked down as well as in the cells in which both DEC1 and Fbw7 had been knocked down (Figure 3k). This demonstrated that the inhibition of cyclin E ubiquitination by DEC1 was dependent on the presence of Fbw7. Taken together these results indicated that DEC1 stabilized cyclin E protein through blocking the ubiquitin-mediated proteasomal degradation of cyclin E, which probably occurred through a reduction of interaction between cyclin E and Fbw7α.

DEC1 regulates the stability of cyclin E in MCF-7 via Fbw7-mediated cyclin E ubiquitin-proteasome pathway. (a and b) Western blot analysis of the effect of DEC1 on the half-life of cyclin E. MCF-7 cells were transfected with Myc-cyclin E and Flag-DEC1 or empty vector. The cells were then treated with CHX only (a) or both CHX and MG132 (b), and harvested at the indicated time periods. The cell extract was subjected to western blot with anti-Myc, anti-Flag and anti-β-actin antibody. The histogram in the right panel (a and b) shows the quantitative analysis of cyclin E bands. (c) MCF-7 cells were transfected with HA-tagged cyclin E and Myc-Ub or empty vector, followed by treatment with MG132 for 8 h before harvest. The cell extract was immunoprecipitated with anti-HA antibody and then probed with anti-Myc antibody. (d) MCF-7 cells were transfected with plasmids expressing the indicated Flag-tagged Fbw7 isoforms together with Myc-tagged cyclin E or empty vector or DEC1, followed by western blot analysis with anti-Myc antibody. (e) MCF-7 cells were transfected with Myc-tagged cyclin E, Flag-Fbw7α and Flag-DEC1 or empty vector, followed by treatment with MG132. Clear cell extracts were probed with anti-Myc and anti-Flag antibodies. (f) MCF-7 cells were transfected with Myc-cyclin E only or together with Flag-DEC1 in the presence of sh-Fbw7 or sh-c. The histogram shows the quantitative analysis of cyclin E protein levels after normalization to β-actin (bottom). Data are the means±S.D. (g) MCF-7 cells were transfected with Myc-tagged cyclin E, Flag-Fbw7α and GFP-DEC1 or empty vector, treated with MG132 for 8 h before harvest. The clear cell extracts were immunoprecipitated with anti-Myc antibody, and then probed with anti-Flag, anti-GFP and anti-Myc antibody as indicated. β-Actin was used as a negative control. (h) Effect of DEC1 on the interaction between Fbw7α and cyclin E under serum starvation stress. MCF-7 cells were transfected as in (g), and cultured in serum-free medium, and then treated with 10 μM MG132 for 8 h before harvest. The cells were collected and subjected to immunoprecipitation assay as in (g). (i and j) Effect of DEC1 on the interaction between endogenous Fbw7α and cyclin E. MCF-7 cells were transfected with GFP-DEC1 or empty vector, and cultured with or without serum, and then treated with MG132 for 8 h before harvest. The clear cell extracts were immunoprecipitated with anti-IgG or anti-cyclin E antibody, probed with anti-Fbw7, anti-GFP and anti-cyclin E antibody as indicated. β-Actin was used as a negative control. (k) Ubiquitination status of endogenous cyclin E in DEC1- and Fbw7-silenced cells. MCF-7 cells were transfected with either shDEC1 or shFbw7α, or both. Cell extract was immunoprecipitated with anti-cyclin E antibodies, followed by western blotting using anti-Ub or anti-cyclin E antibody. Total cell lysate was also analyzed by western blotting using anti-β-actin antibody. To test the expression and immunoprecipitation of cyclin E, mouse monoclonal (SB62a) secondary antibody against rabbit IgG light chain (HRP, ab99697) was used at a 1 : 5000 dilution in (g–k)
DEC1 is a cyclin E-interacting protein
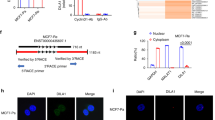
The positive regulation of cyclin E by DEC1 at the protein level suggested that these two proteins might interact with each other. Co-immunoprecipitation experiment showed that DEC1 specifically interacted with cyclin E in MCF-7 cells regardless of whether or not the cells were under serum starvation (Figures 4a and b). The interaction between endogenous DEC1 and cyclin E was also verified in MCF-7 cells, both in the absence and presence of serum (Figures 4c and d). GST pull-down assay and the CheckMate mammalian two-hybrid system (Promega, Madison, WI, USA) also detected the association of cyclin E with DEC1 (Figures 4e–g). Furthermore, DEC1 and cyclin E exhibited a pronounced nuclear co-localization (Figures 4h and i). This indicated the presence of a substantial interaction between DEC1 and cyclin E.

Interaction between DEC1 and cyclin E. (a) MCF-7 cells were transfected with Flag-tagged DEC1 and Myc-tagged cyclin E. After 24 h of transfection, the cell extract was subjected to immunoprecipitation with anti-Flag antibody, followed by western blot analysis with anti-Myc antibody. (b) MCF-7 cells transfected as in (a) were subjected to serum starvation for 24 h. The cell extract was subjected to immunoprecipitation with anti-Flag antibody followed by western blot analysis with anti-Myc antibody. (c and d) CoIP experiment showing the interaction between endogenous DEC1 and cyclin E in serum-plus and serum-free conditions. MCF-7 cells were subjected to immunoprecipitation with anti-cyclin E antibody followed by western blot analysis with anti-DEC1 antibody. Immunoprecipitation carried out with anti-IgG antibody was used as control. Mouse monoclonal (SB62a) secondary antibody against rabbit IgG light chain (HRP) was used at a 1 : 5000 dilution. (e) Western blot analysis of GST pull-down assay showing the interaction between cyclin E and DEC1. The cell extract was incubated with glutathione-agarose beads coated with purified GST or GST-tagged cyclin E as indicated at the top. After extensive washing, bound proteins were eluted, resolved by SDS-PAGE, and probed with anti-DEC1 and anti-GST antibodies. (f and g) Interaction between DEC1 and cyclin E as detected by a mammalian two-hybrid system for cells without and with serum starvation. Cyclin E and DEC1 were expressed from pBIND-cyclin E and pACT-DEC1, respectively. The MCF-7 cells were transfected with pG5-luc and the empty vectors (pACT and pBIND) as indicated. Positive control cells were transfected with pBIND-ID and pACT-MyoD. Each bar represents the mean±S.D. from three independent experiments. **P<0.01 compared with cells transfected with pACT and pBIND. (h and i) Co-localization of DEC1 and cyclin E in MCF-7 cell nuclei. MCF-7 cells were transfected with Myc-cyclin E and Flag-DEC1. Flag-antibody complex and Myc-antibody complex were visualized with FITC and TRITC, respectively. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). (j) CoIP experiments showing the dynamics of the interaction between DEC1 and cyclin E. MCF-7 cells were transfected with Flag-DEC1, and then subjected to immunoprecipitation with anti-Flag antibody followed by western blot analysis with anti-cyclin E antibody. HRP (ab99697) was used as secondary antibody
We then explored the dynamics of the interaction between DEC1 and cyclin E at different stages of the cell cycle. The result showed that the interaction of these two protein displayed cell cycle-dependent dynamics, detected mainly at the G1/S phase 8 h after release from nocodazole treatment (Figure 4j). These data also suggested that DEC1 may play an important role in the regulation of the cell cycle progression by interacting and regulating the activity of cyclin E at the G1 and S phases.
DEC1 promotes the cyclin E/Cdk2 complex formation
Like all cyclin family members, cyclin E forms a complex with cyclin-dependent kinase (Cdk2) at the G1/S phase checkpoint, which needs to be degraded, and this will in turn promote the formation of cyclin A/Cdk2 complex and activity during the transition from S to G2 phase.27, 28, 29 Immunoprecipitation assay showed that DEC1 increased the amount of cyclin E-associated Cdk2 and decreased the interaction between cyclin A and Cdk2 by ~50% (Figures 5a and b). This effect of DEC1 was also confirmed by the CheckMate mammalian two-hybrid system (Promega), which showed that cells that overexpressed DEC1 displayed a significant increase in reporter activity over cells that overexpressed cyclin E and Cdk2 (Figure 5c). Furthermore, the level of cyclin E/Cdk2 kinase activity of cells that overexpressed cyclin E and DEC1 was significantly higher than the level of those that either overexpressed cyclin E or p21 (the Cdk2 inhibitor used as a negative control) alone (Figure 5d). Since, cyclin E functions as a nuclear protein, and is associated with Cdk2 during the G1/S phase transition,30 we tested whether DEC1 would affect the sublocalization of cyclin E and Cdk2. As shown in Figures 5e and f, cyclin E and Cdk2 were localized in both the cytoplasm and the nucleus, but in the presence of DEC1, they were predominantly localized in the nucleus. These data suggested that overexpression of DEC1 may affect the progression of cell cycle S phase.

DEC1 promotes the complex formation and activity of cyclin E/Cdk2 and inhibits cell cycle progression through the S phase. (a) Effect of DEC1 on the formation of cyclin E/Cdk2 complex. MCF-7 cells were transfected with HA-Cdk2 and Myc-cyclin E only or HA-Cdk2, Myc-cyclin E and Flag-DEC1, and the clear cell extract was immunoprecipitated with anti-Myc antibodies and then probed with anti-HA antibody. (b) Effect of DEC1 on the sequent formation of the cyclin A/Cdk2 complex as detected by CoIP. MCF-7 cells were transfected with empty vector only, HA-Cdk2 and Myc-cyclin A only or HA-Cdk2, Myc-cyclin A and Flag-DEC1, and the clear cell extract was immunoprecipitated with anti-Myc antibody and then probed with anti-HA antibody. (c) Mammalian two-hybrid assay analysis of the effect of DEC1 on the interaction between cyclin E and Cdk2. Cyclin E, Cdk2 and DEC1 were expressed from pBIND-cyclin E, pACT-Cdk2 and Flag-DEC1, respectively. Each bar represents the mean±S.D. from three independent experiments. **P<0.01 compared with cells transfected with pACT and pBIND. (d) Effect of DEC1 on the activity of Cdk2/cyclin E kinase. Columns, mean (n=3)±S.D.; *P<0.05 compared with controls. (e) Immunofluorescence assay showing the effect of DEC1 on the sublocation of cyclin E. MCF-7 cells were transfected with Myc-cyclin E only or Myc-cyclin E and Flag-DEC1, and were fixed and stained with rabbit anti-Myc antibody (red) and then counterstained with DAPI (blue) for nucleus detection. (f) Effect of DEC1 on the sublocation of Cdk2. MCF-7 cells were transfected with HA-Cdk2 only or HA-Cdk2 and Flag-DEC1 and stained with mouse anti-HA antibody (green), and then counterstained with DAPI (blue) for nucleus detection. (g and h) CoIP assay showing the interaction between DEC1 and Cdk2. Control cells or cells overexpressing HA-Cdk2 only, Flag-DEC1 only or Flag-DEC1 and HA-Cdk2 were subjected to immunoprecipitation with anti-HA antibody (g) and with anti-Flag antibody (h). (i) Mammalian two-hybrid assay analysis of the interaction between DEC1 and Cdk2. DEC1 and Cdk2 were expressed from pACT-DEC1 and pBIND-Cdk2, respectively. Columns, mean (n=3)±S.D.; **P<0.01 compared with controls
By using co-immunoprecipitation assay and mammalian two-hybrid assay, we showed that DEC1 could bind to cdk2, but without affecting the level of Cdk2 (Figures 5g–i and Supplementary Figure S4E). These data suggested that a high level of cyclin E (induced by DEC1) may stabilize the interaction between cyclin E and Cdk2, consequently interfering with the association between Cdk2 and cyclin A, an event that could lead to the cells stalling at the S phase.
Overexpression of DEC1 causes defect in cyclin E degradation, results in cell cycle S-phase delay
We next synchronized MCF-7 to specifically study the effect of DEC1 on cell cycle from early G2/M phase to late S phase. We noticed that in the cells that expressed DEC1, the level of cyclin E persisted for four more hours while the level of cyclin B was delayed for 4 h compared to the levels of the control cells. This suggested that overexpression of DEC1 could extend the S phase of the cells, raising the possibility that DEC1-induced increased stability of cyclin E could inhibit the progression of the cells through the S phase (Figures 6a and b). To further confirm this possibility, we performed co-immunoprecipitation (CoIP) experiment and found that the interaction between cyclin E and Cdk2 was clearly increased at the G1/S transition checkpoint (from 6 to 10 h after release) and decreased at the late S phase (from 16 h after release; Figure 6c and Supplementary Figure S4A). This observation was further verified by the mammalian two-hybrid system (Figure 6d). However, in DEC1-overexpressing cells, the stabilized cyclin E remained associated with Cdk2 and inhibited the subsequent formation of cyclin A/Cdk2 complex 16 h after release from nocodazole treatment (Figure 6c and Supplementary Figure S4B).

DEC1 inhibits the progression of S phase in the cell cycle. (a and b) Western blot of cyclin E and cyclin B in control and DEC1-overexpressing cells. The plots show the relative intensities of the bands in the blots. MCF-7 cells were transfected with empty vector or Flag-DEC1, and then synchronized at G2/M stage by treating with 50 ng/ml nocodazole for 20 h. The cells were analyzed at different time points after release from nocodazole treatment. The clear cell extract was subjected to western blot analysis with anti-cyclin E, anti-cyclin B or anti-β-actin antibody. (c) MCF-7 cells were treated as in (a and b) and then subjected to immunoprecipitation using anti-cyclin E antibody followed by western blot with anti-Cdk2, anti-DEC1 and anti-β-actin antibody. (d) Effect of DEC1 on the dynamics of cyclin E and Cdk2 as demonstrated by mammalian two-hybrid assay. Cyclin E, Cdk2 and DEC1 were expressed from pACT-cyclin E, pBIND-Cdk2 and Flag-DEC1, respectively. MCF-7 cells were transfected with the indicated plasmids, synchronized and harvested at the indicated time points. Each bar represents the mean±S.D. from three independent experiments. **P<0.01 compared with cells transfected with pACT and pBIND. (e) Effect of DEC1 on the cell cycle progression (on the left panel) as demonstrated by FACS assay. Quantified analysis is shown by histogram in the right panel. MCF-7 cells were transfected with DEC1 or control vector, synchronized and harvested at the indicated time points
Flow cytometric analysis of the synchronized cells indicated that DEC1-overexpressing cells had an extended S phase compared to the control (Figure 6e and Supplementary Figure S4C). Delay in cell cycle may have a substantial negative effect on the genome stability and replication initiation.21 We found that cells overexpressing DEC1 had higher level of γH2AX than control cells, and in a cyclin E-dependent manner (Supplementary Figure S4D). Taken together, these results demonstrated that DEC1 delayed cell cycle progression through the S phase and induced genome instability, eventually resulting in repression of cell proliferation.
DEC1 inhibits the proliferation of cells overexpressing cyclin E and inhibits tumor xenograft growth
The effect exerted by DEC1 on the growth of cyclin E-overexpressing MCF-7 cells was examined. As expected, cells that overexpressed cyclin E had a high growth rate than cells that did not overexpress DEC1 (transfected with empty vector), but the growth rate of MCF-7 cells that overexpressed both DEC1 and cyclin E became partly inhibited over a 5- day period (Figure 7a). Moreover, colony formation assay and soft agar assay showed that cells that overexpressed both DEC1 and cyclin E produced significantly lower number of colonies as well as smaller colonies compared to cells that only overexpressed cyclin E (Figures 7b and c). These results indicated that DEC1 may be used as an inhibitor or suppressor for tumor growth.

DEC1 inhibits the proliferation of cells overexpressing cyclin E and inhibits tumor xenograft growth. (a and b) MCF-7 cells were transfected with Myc-cyclin E only, Myc-cyclin E and Flag-DEC1 or control vector. The cells were then cultured in selective medium (500 μg/ml G418) and subjected to colony formation and MTT assays. Each bar represents the mean±S.D. from five independent experiments. *P-value was determined by ANOVA with Bonferroni test (*P<0.05). (c) Representative colonies of each experimental group are shown. MCF-7 cells transfected with control vector, Myc-cyclin E or Myc-cyclin E and Flag-DEC1 were selected with 800 μg/ml G418 for 15 days. The cells were then collected and suspended in a soft agar. Photographs of the colonies were taken 30 days after seeding. Scale bar, 100 μm. All experiments were repeated at least three times. (d) Tumor growth by subcutaneously implanted MCF-7 cells (6 × 105 cells) transfected with pCMV10-3 × Flag or pCMV10-3 × Flag-DEC1 and screened with G418. P-value was determined by unpaired t-test. (e) Tumor formation 36 days after the mice were injected with the tumor cells. Left: mice injected with control cells. Right: mice injected with DEC1 overexpressed cells. (f and g) Tumor images (f) and tumor weight (g) 48 days after mice were injected subcutaneously with MCF-7 cells overexpressed Flag-DEC1 or empty vector. n=5 mice per group in (d, f and g). (h) Representative immunohistochemical data for H&E and DEC1 on paraffin-embedded section of subcutaneous tumors in (f) and (g), generated by MCF-7 cells overexpressed DEC1 or empty vector. (i) Western blot analysis of the expression of cyclin E and DEC1 in control (Ctrl) and Flag-DEC1-overexpressing tumors
To further investigate the effect of DEC1 on tumorigenicity in vivo, we subcutaneously implanted MCF-7 cells that stably overexpressed DEC1 or those that harbored the empty vector into nude mice and monitored the size of the tumor developed from these cells. Mice that were implanted with DEC1-overexpressing MCF-7 cells showed a much smaller tumor throughout the experimental period than mice implanted with MCF-7 cells that harbored the empty vector (Figures 7d and f). Forty-eight days after tumor cell implantation, a 2.6-fold decrease in the weight of the tumors was achieved for MCF-7 cells that overexpressed DEC1 (Figure 7g). Examination of the expression level of DEC1 in the tumor by IHC and WB showed that DEC1 was successfully expressed in tumors with small sizes and the level of cyclin E in the tumor was also upregulated (Figures 7h and i; Supplementary Figure S6). Overall, these in vitro and in vivo experiments indicated that DEC1 functioned as a tumor suppressor and inhibited cell growth.
Discussion
As an important transcription factor, DEC1 plays an important role in cell differentiation, proliferation and apoptosis.31, 32, 33, 34, 35 In this study, we found in this study that DEC1 could affect the level of cyclin E in a cell, not through its transcriptional activity (Figures 2b and c; Supplementary Figures S2A and D), but through protein-protein interaction, and this effectively allowed DEC1 to regulate the cell cycle progression, consequently resulting in the regulation of cell proliferation. As shown in Figures 2 and 3, DEC1 upregulated the level of cyclin E protein in a dose-dependent pathway and prolonged the half-life of cyclin E, and the underlying mechanism by which it achieved this was through interfering with the interaction between cyclin E and Fbw7, thereby reducing the Fbw7-mediated ubiquitination of cyclin E (Figures 3g–j). In addition to increasing the protein level of cyclin E through inhibiting the Fbw7-mediated ubiquitination and degradation pathway, DEC1 also decreased the level of p21 (Supplementary Figure S5) and enhanced the binding of cyclin E to Cdk2 as well as the kinase activity of the cyclin E/Cdk2 complex (Figures 5a and d), suggesting that multiple mechanisms could be at work.36
More and more evidences have shown that cyclin E may function as a ‘switch’ or a double-edged sword: however, high expression of cyclin E promotes a faster transition from G1 to S phase,37, 38 which is why cyclin E is always expressed at a high level in various types of cancers and its expression correlates with tumorigenesis;39, 40 however, excessive cyclin E would interfere with the assembly of the pre-replication complex and lead to replication stress, DNA damage and genomic instability, which will block the S-phase progression and cause cell cycle arrest.21, 41, 42 In this study, we have verified the following: (i) DEC1 could promote the nuclear accumulation of cyclin E/Cdk2 and inhibit the formation of cyclin A/Cdk2, which would cause the cells to stay in the S phase resulting in cell cycle arrest (Figures 5 and 6). (ii) DEC1 could interact with Cdk2 directly without influencing the level of Cdk2, which may work as a platform to enhance the formation of cyclin E and Cdk2. (iii) Overexpression of DEC1 would increase the level of γH2AX in cells subjected to serum-deprived stress (S4D), and this may cause genetic instability. However, we could not yet determine whether the high level of γH2AX in the cells that overexpressed DEC1 reflected increased DNA damage, impaired DNA repair, or both. (iv) A high level of DEC1 expression in adjacent normal breast tissue compared to the carcinoma (Figure 1a). We speculated that DEC1 may not only regulate cell proliferation but may also be involved in metastasis, which is also a subject for further study.
In addition, although DEC1 could interact with cyclin E both in the presence and absence of serum, noticeable difference in the localization of cyclin E-DEC1 between the two conditions was detected (Figures 4h and i). The co-localization of DEC1 and cyclin E in the nucleus was more obvious under serum starvation condition than under normal culturing conditions. Thus, DEC1 could potentially be involved in the binding of MCM proteins to the replication origin by interacting with cyclin E under serum starvation condition.
In summary, our data demonstrated that DEC1 promoted the formation of cyclin E/Cdk2 complex and inhibited the subsequent formation of cyclin A/Cdk2 complex leading to the stalling of S phase of the cell cycle and inhibition of cell proliferation. This valuable insight would help us to conceive a way to combat cancers; especially those mediated by cyclin E dysregulation, and may even provide a unique therapeutic strategy to control the growth of these cancer cells.
Materials and Methods
Cell culture and transfection
MCF-7 and T47D cells have been used in our previous study and were maintained in our laboratory as standard human breast cancer cell lines.43, 44 The cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (Hyclone, Beijing, China), 100 mg/ml penicillin and 100 mg/ml streptomycin at 37 °C in the presence of 5% CO2. For synchronization, the cells were cultured in DMEM without FBS for 22 h. After that FBS was added to the culture to a final concentration of 10%, and the culture was incubated for 2 h before the addition of nocodazole (Sigma, St. Louis, MO, USA) to a final concentration of 50 ng/ml. After 16 h of incubation, the cells were washed twice with complete DMEM medium.45 For ubiquitination assay, the cells were treated with 10 μM MG132 (Sigma) for 8 h before harvest. For protein half-life assay, the cells were treated with 50 μM CHX alone or together with MG132 after transfection. Transfection of the cells was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction.46
Plasmids, siRNA and antibodies
pTB701-HA/cyclin E was provided by Mikiko Takahashi (Biosignal Research Center, Kobe University, Kobe, Japan). pCS2+-Myc/cyclin E harboring wild-type and mutant forms (T380A and T380S mutants) of cyclin E were acquired from Clurman Lab (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). p3xFlag-CMV7.1/Fbw7α was provided by Dr. Deanna M Koepp (University of Minnesota-Twin Cities, Minneapolis and St. Paul, MN, USA). HA-Cdk2 was kindly given by Dr. Greg H. Enders (Fox Chase Cancer Center, Philadelphia, PA, USA). pCMV-Myc/cyclin A was provided by Dr. Liang Zhu (Department of Developmental and Molecular Biology, Jack and Pearl Resnick Campus, Bronx, NY, USA). The plasmids p3xFlag-CMV-10/DEC1, p3xFlag-CMV-10, pcDNA3.1-Myc/Ub, pcDNA3.1(−) and pcDNA3.1-HA/DEC1 were acquired as previously described.47, 48 PCR primers for constructing siRNAs, pRNAT-U6.1/shDEC1, pACT/DEC1, pACT/cyclin E, pBIND/cyclin E and pBIND/Cdk2 are presented in Supplementary Table S1.
Antibodies used in this study included anti-Myc (9E10), -Flag (M2; Sigma); anti-DEC1 (A300-649A; Bethyl Laboratories, Montgomery, TX, USA); anti-Ub (10201-2-AP; Proteintech, Wuhan, China); anti-Fbw7 (A301-720A-T; Bethyl Laboratories); anti-Cdk2 (ab6538; Abcam, Cambridge, UK); anti-cyclin B1 (AB60234a; Sango, Shanghai, China); anti-HA (sc-7392), -cyclin E (sc-198), -cyclin A (sc-751), -mouse and -rabbit secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA); mouse monoclonal (SB62a) antibody against rabbit IgG light chain (HRP, ab99697; Abcam).
Immunoblotting and protein-protein interaction
Cells were lysed in 200 μl lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40 and 0.5% sodium deoxycholate), and centrifuged at 12 000 × g/4 °C for 10 min to obtain the cell extract.49, 50 The protein concentration of the cell extract was determined before subjecting to western blot or immunoprecipitation analysis. For immunoblotting, aliquot of this cell extract was first resolved in 10% SDS-PAGE gel, and the protein bands were then transferred to Millipore (Billerica, MA, USA) PVDF membranes.
For CoIP assay, the cell extract was incubated with the appropriate primary antibody and protein A-Sepharose (Amersham Biosciences, Piscataway, NJ, USA) at 4 °C for 24 h, followed by centrifugation at 5000 × g/4 °C for 10 min. The pellet was washed twice with wash buffer I (50 mM Tris-HCl pH 7.5, 500 mM sodium chloride, 0.1% NP-40 and 0.05% sodium deoxycholate) and once with wash buffer II (50 mM Tris-HCl pH 7.5, 0.1% NP-40 and 0.05% sodium deoxycholate), and then subjected to SDS-PAGE using 10% gel followed by western blot.51 Immunoblot data were quantified by scanning the appropriate bands of interest and plotted as relative density of gray scale. The CheckMate mammalian two-hybrid system (Promega) was used according to manufacturer’s protocol. The primers used are shown in Supplementary Table S1.
GST pull-down assay
GST and GST-tagged cyclin E-fusion proteins were expressed in Escherichia coli BL21(DE3) and purified with the Pierce GST spin purification kit (Thermo scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The identity of the purified protein was confirmed by western blot with an anti-GST antibody. The purified GST-tagged fusion protein (BAIT) was immobilized on the Pierce spin column. MCF-7 cells were lysed in pull-down lysis buffer containing DNase (Takara, Dalian, China) and centrifuged at 12 000 × g/4 °C for 15 min to obtain the cell extract. The supernatant was loaded onto the Pierce spin column, and then incubated at 4 °C for at least 6 h with gentle agitation. After that the column was centrifuged at 700 × g for 1 min and the flow through was discarded. The column was then washed five times with wash solution and centrifuged as before with each wash. After the last wash, elution buffer was added to the column and the column was incubated for 5 min with gentle agitation, before it was centrifuged at 700 × g for 1 min. The resulting eluent was subjected to western blot assay.
Cell cycle analysis
MCF-7 were trypsinized, fixed in 70% ethanol at 4 °C, and washed according to previously described protocol.52 The washed cells were incubated in PBS containing propidium iodide and RNase A at 37 °C for 30 min and then subjected to cell cycle analysis performed with a Becton Dickinson FACScan run by the FlowJo 7.6 software (BD Biosciences, San Jose, CA, USA).53
Cell proliferation assays
Cell proliferation was determined by MTT assay and colony formation assay. For MTT assay, cells (5 × 104 per well) were seeded into 96-well plate and incubated with appropriate selective medium for several days. After that, MTT assay was performed according to the manufacturer’s protocol (KeyGen, Nanjing, China).
For colony formation assays, the cells were transfected with the appropriate plasmids and then transferred to 35-mm plate containing the appropriate selective medium and incubated for different times. After that, the cells were fixed in cold 75% ethanol for 10 min and dyed with 30% crystal violet (Sigma) for 15 min at room temperature. The cells were then washed with water to remove the residual crystal violet, and the number of individual colonies was counted.
Soft-agar colony culture
Anchorage-independent growth of MCF-7 cells was estimated using soft agar colony formation assay as described previously.54 The cells were transfected with the appropriate plasmids and the same number of cells from each transfection were then cultured in selective medium containing 1 × DMEM, 10% FBS and 800 μg/ml G418 (Sigma). After 15 days of culturing, the cell were collected and 6 × 104 cells were suspended in 1 ml of 0.35% soft agar medium (top agar pre-warmed to 37 °C), which was then added to the top of a 0.6% agar base layer (prepared in 1 ml medium) in a 35-mm-diameter dish. Fresh medium (200 μl, containing 200 μg/ml G418) was added to the plate 3 days later and on every successive 3 days over a 30-day period. The colonies that appeared in the plate were counted and photographed using a phase-contrast microscope, and the diameters of the colonies was measured the with the software Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).55
Immunofluorescence staining
MCF-7 cells were grown on cover slips for overnight, and then transfected with the desired plasmids. Twenty-four hours after transfection the cells were washed three times with PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS solution at −20 °C, and finally blocked with 0.8% BSA for 1 h at 4 °C. The cells were then incubated with the corresponding antibodies and examined according to the manufacturer’s instructions.
Kinase activity assay
The kinase activity of cyclin E/Cdk2 complex was measured with a kinase activity assay kit (Genmed Scientifics, Arlington, MA, USA), which is a complete assay system designed to measure the activity of cyclin E/Cdk2 by coupling the formation of ADP to the reaction catalyzed by PK and LDH in the presence of phosphoenolpyruvate, which would result in the oxidation of NADH. The disappearance of NADH was detected by measuring the decrease in absorbance at 340 nm at every 1-min interval over a 5-min period. The assay was repeated three times for each sample.
Human breast cancer xenograft model
Five- to six-week-old athymic BALB/c nude male mice were purchased from the Animal Experiment Center of Dalian Medical University and maintained under specific pathogen-free (SPF) conditions (NO.SCXK2013-0003). All experiments were carried out according to the regulation set by the Ethics Committee for Biology and Medical Science of Dalian University of Technology. About 6 × 105 MCF-7 cells stably transfected with Flag-DEC1 or empty plasmid (as selected by G418) were resuspended in a final volume of 100 μl containing 50% Matrigel (BD Matrigel, BD Biosciences) and then injected into the right flanks of the animals (n=5 mice per group). Tumor growth rates were analyzed by caliper measurements every 8 days post injection using the formula: tumor volume (V)=length (L) × width (W)2 × 0.5. After 48 days, the animals were killed in a humane manner and the tumors were weighed, photographed and subjected to further analysis (paraffin embedding and digestion for subsequent extraction of protein) as previously described.56
Luciferase reporter assays
Cells were seeded in 24-well plates at a density of 2 × 105 per well and cultured for 24 h before they were transfected with the appropriate plasmids. Twenty-four hours after transfection, the cells were harvested and Luc reporter assay was performed according to the manufacturer’s instructions (Promega).
Statistical analysis
All statistical analyses of data were performed with ANOVA with LSD method. Data were expressed as means±S.D., and significance was considered at either P-value <0.05 or 0.01 level.54
Abbreviations
- DEC1:
-
differentiated embryo-chondrocyte expressed gene 1
- bHLH:
-
basic helix-loop-helix
- HDAC:
-
histone deacetylase
- MIC-1:
-
macrophage inhibitory cytokine 1
- TGF:
-
transforming growth factor
- E2F:
-
adenovirus E2 factor
- Cdk:
-
cyclin-dependent kinase
- Cul:
-
cullin
- SCF:
-
Skp1-Cul1-F-box protein complex
- BCR:
-
BTB-Cul3-Rbx1 complex
- FACS:
-
fluorescence-activated cell sorting
- miRNA:
-
microRNA
- shRNA:
-
short hairpin RNA
- MCM:
-
minichromsome maintenance
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FBS:
-
fetal bovine serum
- MTT:
-
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
- CHX:
-
cycloheximide
References
Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002; 419: 841–844.
Kawamoto T, Noshiro M, Sato F, Maemura K, Takeda N, Nagai R et al. A novel autofeedback loop of Dec1 transcription involved in circadian rhythm regulation. Biochem Biophys Res Commun 2004; 313: 117–124.
Liu Y, Sato F, Kawamoto T, Fujimoto K, Morohashi S, Akasaka H et al. Anti-apoptotic effect of the basic helix-loop-helix (bHLH) transcription factor DEC2 in human breast cancer cells. Genes Cells 2010; 15: 315–325.
Grechez-Cassiau A, Panda S, Lacoche S, Teboul M, Azmi S, Laudet V et al. The transcriptional repressor STRA13 regulates a subset of peripheral circadian outputs. J Biol Chem 2004; 279: 1141–1150.
Sato F, Kawamoto T, Fujimoto K, Noshiro M, Honda KK, Honma S et al. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. Interaction with BMAL1. Eur J Biochem 2004; 271: 4409–4419.
Qian Y, Zhang J, Yan B, Chen X . DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J Biol Chem 2008; 283: 2896–2905.
Qian Y, Chen X . ID1, inhibitor of differentiation/DNA binding, is an effector of the p53-dependent DNA damage response pathway. J Biol Chem 2008; 283: 22410–22416.
Sun H, Taneja R . Stra13 expression is associated with growth arrest and represses transcription through histone deacetylase (HDAC)-dependent and HDAC-independent mechanisms. Proc Natl Acad Sci USA 2000; 97: 4058–4063.
Ehata S, Hanyu A, Hayashi M, Aburatani H, Kato Y, Fujime M et al. Transforming growth factor-beta promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res 2007; 67: 9694–9703.
Bek MJ, Wahle S, Muller B, Benzing T, Huber TB, Kretzler M et al. Stra13, a prostaglandin E2-induced gene, regulates the cellular redox state of podocytes. FASEB J 2003; 17: 682–684.
Qian Y, Jung YS, Chen X . DEC1 and MIC-1: new players of p53-dependent cell fate decision. Cell Cycle 2012; 11: 3525–3526.
Dulic V, Lees E, Reed SI . Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992; 257: 1958–1961.
Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA . Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992; 70: 993–1006.
Welcker M, Clurman BE . FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8: 83–93.
Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI . Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001; 413: 316–322.
Kossatz U, Breuhahn K, Wolf B, Hardtke-Wolenski M, Wilkens L, Steinemann D et al. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest 2010; 120: 3820–3833.
Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP . Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 2007; 26: 131–143.
Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J et al. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. J Biol Chem 2004; 279: 50110–50119.
Moore JD, Kornbluth S, Hunt T . Identification of the nuclear localization signal in Xenopus cyclin E and analysis of its role in replication and mitosis. Mol Biol Cell 2002; 13: 4388–4400.
Donnellan R, Chetty R . Cyclin E in human cancers. FASEB J 1999; 13: 773–780.
Jones RM, Mortusewicz O, Afzal I, Lorvellec M, Garcia P, Helleday T et al. Increased replication initiation and conflicts with transcription underlie cyclin E-induced replication stress. Oncogene 2013; 32: 3744–3753.
Minella AC, Loeb KR, Knecht A, Welcker M, Varnum-Finney BJ, Bernstein ID et al. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev 2008; 22: 1677–1689.
Abbas T, Dutta A . p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9: 400–414.
Le Francois BG, Maroun JA, Birnboim HC . Expression of thymidylate synthase in human cells is an early G(1) event regulated by CDK4 and p16INK4A but not E2F. Br J Cancer 2007; 97: 1242–1250.
Ge JN, Huang D, Xiao T, Wang Z, Li XL, Xiao H et al. Effect of starvation-induced autophagy on cell cycle of tumor cells. Ai Zheng 2008; 27: 788–794.
Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM . Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev 1996; 10: 1979–1990.
Lu X, Liu J, Legerski RJ . Cyclin E is stabilized in response to replication fork barriers leading to prolonged S phase arrest. J Biol Chem 2009; 284: 35325–35337.
Wang H, Zhang X, Geng L, Teng L, Legerski RJ . Artemis regulates cell cycle recovery from the S phase checkpoint by promoting degradation of cyclin E. J Biol Chem 2009; 284: 18236–18243.
Wimmel A, Lucibello FC, Sewing A, Adolph S, Muller R . Inducible acceleration of G1 progression through tetracycline-regulated expression of human cyclin E. Oncogene 1994; 9: 995–997.
Delk NA, Hunt KK, Keyomarsi K . Altered subcellular localization of tumor-specific cyclin E isoforms affects cyclin-dependent kinase 2 complex formation and proteasomal regulation. Cancer Res 2009; 69: 2817–2825.
Turley H, Wykoff CC, Troup S, Watson PH, Gatter KC, Harris AL . The hypoxia-regulated transcription factor DEC1 (Stra13, SHARP-2) and its expression in human tissues and tumours. J Pathol 2004; 203: 808–813.
Shen M, Yoshida E, Yan W, Kawamoto T, Suardita K, Koyano Y et al. Basic helix-loop-helix protein DEC1 promotes chondrocyte differentiation at the early and terminal stages. J Biol Chem 2002; 277: 50112–50120.
Zawel L, Yu J, Torrance CJ, Markowitz S, Kinzler KW, Vogelstein B et al. DEC1 is a downstream target of TGF-beta with sequence-specific transcriptional repressor activities. Proc Natl Acad Sci USA 2002; 99: 2848–2853.
Bhawal UK, Sato F, Arakawa Y, Fujimoto K, Kawamoto T, Tanimoto K et al. Basic helix-loop-helix transcription factor DEC1 negatively regulates cyclin D1. J Pathol 2011; 224: 420–429.
Hong Y, Xing X, Li S, Bi H, Yang C, Zhao F et al. SUMOylation of DEC1 protein regulates its transcriptional activity and enhances its stability. PLoS One 2011; 6: e23046.
Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE . p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol 2002; 12: 1817–1827.
Huang M, Yang S, Liao S, Zhang B, You J . The effects of cyclin E on the growth and other cell cycle related genes of breast carcinoma cells MCF-7. Zhonghua Bing Li Xue Za Zhi 2000; 29: 192–195.
Pillay K, McCleod H, Chetty R, Hall P . A study to investigate the role of p27 and cyclin E immunoexpression as a prognostic factor in early breast carcinoma. World J Surg Oncol 2011; 9: 31.
Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci USA 2011; 108: 3761–3766.
Wang WL, Huang HC, Kao SH, Hsu YC, Wang YT, Li KC et al. Slug is temporally regulated by cyclin E in cell cycle and controls genome stability. Oncogene 2014; 34: 1116–1125.
Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI . Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 2004; 165: 789–800.
Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD et al. Kinase-independent function of cyclin E. Mol Cell 2007; 25: 127–139.
Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y et al. Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 2005; 438: 981–987.
Li S, Wang M, Ao X, Chang AK, Yang C, Zhao F et al. CLOCK is a substrate of SUMO and sumoylation of CLOCK upregulates the transcriptional activity of estrogen receptor-alpha. Oncogene 2013; 32: 4883–4891.
Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y . MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J 2009; 28: 3005–3014.
Wu H, Sun L, Zhang Y, Chen Y, Shi B, Li R et al. Coordinated regulation of AIB1 transcriptional activity by sumoylation and phosphorylation. J Biol Chem 2006; 281: 21848–21856.
Yang C, Li S, Wang M, Chang AK, Liu Y, Zhao F et al. PTEN suppresses the oncogenic function of AIB1 through decreasing its protein stability via mechanism involving Fbw7 alpha. Mol Cancer 2013; 12: 21.
Bi H, Li S, Wang M, Jia Z, Chang AK, Pang P et al. SUMOylation of GPS2 protein regulates its transcription-suppressing function. Mol Biol Cell 2014; 25: 2499–2508.
Li S, Yang C, Hong Y, Bi H, Zhao F, Liu Y et al. The transcriptional activity of co-activator AIB1 is regulated by the SUMO E3 ligase PIAS1. Biol Cell 2012; 104: 287–296.
Xing X, Bi H, Chang AK, Zang MX, Wang M, Ao X et al. SUMOylation of AhR modulates its activity and stability through inhibiting its ubiquitination. J Cell Physiol 2012; 227: 3812–3819.
Liu Y, Ao X, Jia Z, Bai XY, Xu Z, Hu G et al. FOXK2 transcription factor suppresses ERalpha-positive breast cancer cell growth through down-regulating the stability of ERalpha via mechanism involving BRCA1/BARD1. Sci Rep 2015; 5: 8796.
Lai MC, Chang WC, Shieh SY, Tarn WY . DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol 2010; 30: 5444–5453.
Zhao F, Wang M, Li S, Bai X, Bi H, Liu Y et al. DACH1 inhibits SNAI1-mediated epithelial-mesenchymal transition and represses breast carcinoma metastasis. Oncogenesis 2015; 4: e143.
Xiao L, Chang AK, Zang MX, Bi H, Li S, Wang M et al. Induction of the CLOCK gene by E2-ERalpha signaling promotes the proliferation of breast cancer cells. PLoS One 2014; 9: e95878.
Li YY, Bao YL, Song ZB, Sun LG, Wu P, Zhang Y et al. The threonine protease activity of testes-specific protease 50 (TSP50) is essential for its function in cell proliferation. PLoS One 2012; 7: e35030.
Lombardo Y, Filipovic A, Molyneux G, Periyasamy M, Giamas G, Hu Y et al. Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci USA 2012; 109: 16558–16563.
Acknowledgements
We thank Dr. Alan K Chang for valuable discussion and for revising the language of the manuscript. We also thank Dr. Hongming Pan (Qiqihar Medical University) for providing the breast cancer tissue samples. This work was supported by grants (31171353 and 31271500 to HW, 31301159 to SL and 81301504 to MW) from National Natural Science Foundation of China (http://www.nsfc.gov.cn/Portal0/default106.htm), Program for Liaoning Innovative Research Team in University (LT2015008 to HW) from the Department of education of Liaoning Province (http://www.lnen.cn/) and grants (973 Program 2011CB504201 to HW) from the Ministry of Science and Technology of China (http://www.most.gov.cn/eng/).
Author contributions
HW conceived and designed the experiments; HB and SL performed the experiments; HB, SL, XQ and MW analyzed the data; HB, SL, XQ, MW, XB, ZX, XA, ZJ, XJ and YY contributed reagents, materials and analysis tools; HW, HB and SL wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Chipuk
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bi, H., Li, S., Qu, X. et al. DEC1 regulates breast cancer cell proliferation by stabilizing cyclin E protein and delays the progression of cell cycle S phase. Cell Death Dis 6, e1891 (2015). https://doi.org/10.1038/cddis.2015.247
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.247
This article is cited by
-
The matrisome landscape controlling in vivo germ cell fates
Nature Communications (2024)
-
KLF12 promotes the proliferation of breast cancer cells by reducing the transcription of p21 in a p53-dependent and p53-independent manner
Cell Death & Disease (2023)
-
USP13 promotes breast cancer metastasis through FBXL14-induced Twist1 ubiquitination
Cellular Oncology (2023)
-
ELF5 inhibits the proliferation and invasion of breast cancer cells by regulating CD24
Molecular Biology Reports (2021)
-
The bHLH transcription factor DEC1 promotes thyroid cancer aggressiveness by the interplay with NOTCH1
Cell Death & Disease (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}