
Abstract
Recent evidence has revealed the importance of reciprocal functional interactions between different types of mononuclear cells in coordinating the repair of injured muscles. In particular, signals released from the inflammatory infiltrate and from mesenchymal interstitial cells (also known as fibro-adipogenic progenitors (FAPs)) appear to instruct muscle stem cells (satellite cells) to break quiescence, proliferate and differentiate. Interestingly, conditions that compromise the functional integrity of this network can bias muscle repair toward pathological outcomes that are typically observed in chronic muscular disorders, that is, fibrotic and fatty muscle degeneration as well as myofiber atrophy. In this review, we will summarize the current knowledge on the regulation of this network in physiological and pathological conditions, and anticipate the potential contribution of its cellular components to relatively unexplored conditions, such as aging and physical exercise.
Similar content being viewed by others

Facts
-
In skeletal muscle regenerative disorders (e.g., muscular dystrophies) as well as age (sarcopenia)- or disease (cachexia)-related decline in muscle mass and function, there is an impairment of the regenerative potential, which correlates with a progressive replacement of contractile mass with fibrotic and adipose tissue.
-
Mesenchymal-derived cells, such as Sca1+/PDGFRα+ fibro-adipogenic progenitors (FAPs), reside in the interstitial space in skeletal muscle and can contribute either to muscle regeneration or to fibrosis and fat deposition.
-
Functional interactions between muscle stem cells (satellite cells), FAPs and cells from the inflammatory infiltrate have recently been reported and appear to determine the ability of skeletal muscle to regenerate or undergo fibro-adipogenic degeneration.
-
Ectopic adipose tissue in skeletal muscle is associated with impaired insulin sensitivity and muscle function.
Open Questions
-
Are the interactions between satellite cells, FAPs and inflammatory cells relevant in the pathogenesis of neuromuscular diseases?
-
Are the interactions between satellite cells, FAPs and inflammatory cells implicated in the functional decline of muscles during cachexia and sarcopenia?
-
Are the interactions between satellite cells, FAPs and inflammatory cells implicated in the control of muscle growth and homeostasis during exercise?
-
Does an increased ectopic fat deposition in skeletal muscle, from adipogenic differentiation of FAPs, alter the systemic metabolic profile?
-
Could the functional network between satellite cells, FAPs and inflammatory cells provide potential targets for pharmacological interventions toward promoting compensatory regeneration in muscular disease, countering age-related or cachexia-mediated muscle atrophy, or improving the response to exercise and the metabolic profile?
Reduction of muscle mass is typically observed at late stages of many neuromuscular diseases, during aging, inactivity and chronic systemic disorders (i.e., diabetes, cancer, rheumatoid arthritis (RA) and chronic obstructive pulmonary disease (COPD)) and is closely associated with impairment of metabolic control collectively worsening the recovery of the primary disease.1, 2, 3, 4
The endogenous regenerative potential of skeletal muscle provides a compensatory response against muscle loss; however, this response cannot support continuous muscle regeneration in chronic conditions. The regenerative capacity of skeletal muscle relies on muscle stem cells (satellite cells (SCs)), which proliferate in response to exercise to facilitate muscle growth and remodeling, or following myotrauma to repair the injured muscle.5, 6, 7, 8 Recent works identified mesenchymal progenitor cells termed fibro-adipogenic progenitors (FAPs) that provide functional support to SCs; however, these cells might also turn into a source of ectopic fat deposition and fibrosis in skeletal muscle.9, 10, 11, 12, 13, 14, 15 Although their role in humans is not fully understood, a cellular population phenotypically and functionally similar to mouse FAPs has been isolated from human muscle.14 FAP activity is regulated by physical and functional interactions with myofibers and SCs10 as well as cytokines released from innate immune cells.11 Moreover, key regulatory intracellular networks that control FAP lineage and function in regenerating muscles of normal and dystrophic mice have recently been reported.16
This review summarizes the current knowledge on the role played by the cellular network composed by SCs, FAPs and the inflammatory infiltrate (e.g., macrophages and eosinophils) during physiological and pathological perturbations of muscle homeostasis.
Muscle Stem Cell, SC and FAP niche, Interaction with Immune Cells and Contribution to Intramuscular Adipose Tissue
Maintenance of muscle mass depends on the integrity of the regenerative machinery, which is composed of SCs and other mononucleated cell types,17, 18, 19, 20, 21, 22, 23 although, the direct contribution of SCs to myofiber hypertrophy and maintenance remains controversial.19, 24, 25, 26
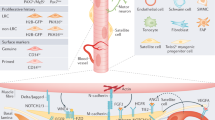
SCs are located beneath the basal lamina – the anatomical niche – and their activity is regulated by interactions with cellular components of the 'functional niche' – FAPs, immune as well as vessel-derived cells (Figures 1 and 2).

(a) Schematic illustration of localization of satellite cell, FAP, macrophage and eosinophil in relation to muscle fibers and capillaries. Compare with image in b. Sizes of individual cells are not drawn to scale. (b) Immunohistochemical staining of human muscle biopsy cross-section with antibodies against Pax7 (brown), laminin (green) and MHCI (red). Nuclei are stained blue with DAPI

Schematic illustration showing cellular interactions in skeletal muscle between immune cells, SCs, FAPs and endothelial cells with indication of some known mediators of these interactions (based on results from Heredia et al.,11 Mozzetta et al.,12 Tardif et al.,46 Moyer and Wagner60 and Snijders et al.182). For further details see text. Abbreviations: HGF, hepatocyte growth factor; IL, interleukin; IGF-1, insulin-like growth factor 1; NO, nitric oxide; TNFα, tumor necrosis factor α; TGFβ, transforming growth factor β
FAPs are non-myogenic, interstitial, mesenchymal progenitors that can be isolated by virtue of the absence of SC surface markers and by the expression of platelet-derived growth factor receptor alpha (PDGFRα)+9 or stem cell antigen 1 (Sca1)+10 (Figure 3). When isolated from regenerating muscles FAPs exhibit the remarkable property of promoting SC cell proliferation and differentiation in co-culture.9, 10 However, FAPs also possess an intrinsic adipogenic and fibrogenic potential manifested in culture by exposure to adipogenic conditions,9, 10 indicating a potential contribution of FAPs to fibrotic and adipose accumulation in diseased muscles (Figure 2). In addition to formation of ectopic fat and fibrous tissue, abnormal bone formation also occurs under some circumstances in muscle, termed heterotopic ossification,27 raising the possibility that the term FAPs does not fully cover their potential roles.28

Mononuclear cell isolation procedure from skeletal muscle composed of mechanical and enzymatic digestion, filtration, blocking, antigen labeling and finally multiple parameter FACS to sort out selected cell populations. (A) Representative plots showing FACS strategy to sort lineage-negative (Ter119− CD45− CD31−) SCs (α7 integrin+) and FAPs (Sca1+) as well as lineage-positive macrophages (MPs, CD11b+ F480+) from skeletal muscle of healthy (a) and mdx (b) mice. (B) Representative images of adipogenic (Oil Red O (a)) and fibrogenic (α-smooth muscle actin (b)) phenotype of FAPs during differentiation
Two recent studies have identified both the anatomical niche and paracrine cues from innate immune cells as signals that regulate FAP lineage and activity.9, 11 For instance, IL-4 released by eosinophils has been shown to be a key mediator of FAP fate (Figure 2).11, 15 Eosinophils provide an abundant source of IL-4/IL-13 in different conditions.11, 29, 30, 31 Chemotaxis of eosinophils to skeletal muscle and release of IL-4/IL-13 is observed in different conditions, such as muscle injury11 or exercise,29 suggesting that substantial (i.e., chemically induced muscle damage) or subtle (i.e., skeletal muscle exercise) perturbations of skeletal muscle homeostasis may both stimulate FAP-mediated activation of SCs. Inactivation of IL-4-mediated signaling or eosinophil chemotaxis stimulates the adipogenic differentiation of FAPs.11, 15 Likewise, interactions between FAPs and myofibers or SCs also regulate the FAP-mediated adipogenesis.9, 10 Finally, FAP activity is regulated by growth factors (e.g., insulin-like growth factor 1 (IGF-1), hepatocyte growth factor (HGF)), follistatin and nitric oxide (NO), which are secreted from FAPs themselves, endothelial cells and M1/M2 macrophages11, 32, 33, 34, 35, 36, 37, 38 (Figure 2).
In turn, FAPs provide a source of cytokines that regulate SC activity. For instance, FAP-derived IL-6 (LM and PLP unpublished data) activates the signal transducer and activator of transcription 3 (STAT3) in SC.10, 39, 40, 41 This pathway is involved in SC activation and its dysregulation appears implicated in age-dependent reduced regenerative capacity.42 FAPs also provide a substantial source of follistatin secretion, with a tenfold higher follistatin expression in FAPs compared to SCs.12 Follistatin is the physiological antagonist of the negative regulator of muscle mass, myostatin12, 43, 44 and besides preventing myofiber atrophy, it may influence myofiber regenerative capacity through directly targeting the SCs. Interestingly, in aged humans the reduced SC proliferation following exercise is associated with increased co-localization of myostatin in SCs of aged muscle.21 Thus, the FAP-mediated release of follistatin may play a role in SC regulation in rodent as well as in human muscle.
These results suggest that FAPs from regenerating muscles retain a functional bipotency, whose resolution is dependent on anatomical factors (e.g., interactions with myofibers, other cell types or extracellular matrix), systemic factors and local concentrations of soluble cues (such as the signals released by the niche). Dysfunctional FAP regulation by alterations of these regulatory conditions may severely deteriorate muscle health, affecting both muscle function and metabolism.11, 15, 45, 46 As for the latter, FAPs may influence the metabolic activity of the muscle since ectopic adipocytes are associated with impaired insulin sensitivity, metabolic syndrome and type 2 diabetes (T2D).47, 48, 49 In this regard, it is intriguing to speculate as to what extent systemic circulating factors may alter FAP differentiation, including high glucose conditions shown to induce adipogenesis in muscle-derived stem cells.50 Moreover, deregulated FAP activity can contribute to the increased intramuscular adipose tissue (IMAT) observed in aged muscles51 and in patients with RA,52 COPD53 and cancer cachexia (Figure 5).54 Thus, FAPs may hold a dual role in which they provide important paracrine stimuli to support SC function in healthy muscle, while contributing to ectopic adipose accumulation in pathological conditions48, 55, 56 (Figures 2 and 5), thereby leading to reduced insulin sensitivity48, 57 and decreased muscle function.58, 59
Role of SCs, FAPs and immune cells in neuromuscular disorders
Most neuromuscular disorders are initially alleviated by the regenerative potential of skeletal muscles. For instance, in Duchenne muscular dystrophy (DMD), compensatory regeneration at earlier stages of disease tends to counter the degeneration of dystrophin-deficient myofibers. While, at least in mouse models of muscular dystrophy, this reactive regeneration resembles muscle repair following injury, as the disease progresses the asynchronous waves of regeneration and the changes in niche caused by chronic regeneration eventually bias the repair toward pathogenic fibrosis and fat deposition.60, 61
Optimal regeneration entails a sequence of events that ensures temporally coordinated interactions between SCs, FAPs and cells of the immune system. An initial activation of resident immune cells and the inflammatory infiltrate,62 is typically followed by the sequential activation of FAPs and SCs.63, 64 Progressive impairment of the interplay between SCs, FAPs and immune system is emerging as a key event in switching regeneration from compensatory to pathogenic.65, 66 In particular, FAPs appear to play a central role in this switch.59, 67
Recent studies have revealed that FAPs from dystrophic muscles of mdx mice – the DMD mouse model – retain a phenotypic and functional bipotency, as they can either support compensatory regeneration at early stages of disease progression, or mediate fibrotic and fat deposition (Figure 4).12, 16 This alternative phenotype is regulated at the epigenetic level by a network formed by muscle-specific microRNA (the myomiRs miR-1.2, miR-133 and miR-206) that target key subunits of the SWI/SNF chromatin-remodeling complex. In particular, expression of myomiRs correlates with the ability of FAPs to support SC-mediated myogenesis and to adopt a myogenic phenotype at the expense of the fibro-adipogenic lineage.16 This outcome is typical of compensatory regeneration at early stages of disease progression and likely reflects the action of cues from a regeneration-conducive SC niche that resolve FAP bipotency into the pro-myogenic phenotype. Saccone et al.16 showed that myomiRs selectively target two variants of the BAF60 subunit of SWI/SNF complex (BAF60A and B), which activate chromatin remodeling at fibrotic and adipogenic loci. This leads to the selection of the alternative variant – BAF60C – which promotes chromatin remodeling at muscle loci68 and mediates FAP commitment to the myogenic lineage16 (Figure 4).

HDACs control an epigenetic network that determines FAP ability to support either regeneration or fibro-adipogenic degeneration. Inhibition of HDAC induced an upregulation of BAF60c that is engaged in the SWI/SNF complex leading to an increase of the myomiR expression and ultimately promoting a pro-myogenic phenotype in FAPs
Importantly, this intracellular network is controlled by histone deacetylases (HDACs),69 whose activity is constitutively active in DMD muscles.70 In normal conditions (i.e., physiological regeneration) reversible HDAC-mediated repression of myomiR allows the expression of BAF60A and B variants and supports maintenance of bipotency in FAPs. In dystrophic muscles, constitutive HDAC activity represses BAF60C and myomiR and favors the expression of BAF60A and B, which direct the fibro-adipogenic phenotype of FAPs. Interestingly, this dynamic HDAC-mediated regulation of phenotypic and functional bipotency of FAPs is observed at early, but not late stages of disease progression in mdx mice, and accounts for the restriction of the beneficial effects of HDAC inhibitors at early but not late stages of disease.12
Future studies should establish how FAP bipotency, and relative resolution, is regulated by cues from the regenerative microenvironment and should elucidate the signals that controls reciprocal interactions between FAPs, immune system and SCs within physiological and pathological contexts. For instance, it would be interesting to evaluate the contribution of FAPs to the excessive levels of transforming growth factor-β2 (TGFβ2), which are induced by elevated canonical Wnt signaling in dystrophic muscles and affect the behavior of SCs.71
As the human counterpart of FAPs has been identified as PDGFRα+ cells in healthy and diseased muscles,14 the functional and molecular characterization of human FAPs can provide novel interesting targets for the development of pharmacological treatment of muscular diseases.
Role of SCs, FAPs and immune cells during ageing and metabolic dysfunctions
Aging is associated with an accelerated loss of skeletal muscle mass (sarcopenia) and with a reduced regenerative capacity of the musculature, leading to a loss of strength and function.72, 73, 74 In rodents, the aging muscle and SC niche has been shown to disrupt SC function and myofiber regenerative capacity.75, 76, 77, 78 Local changes in the SC milieu include a number of events that can alter signaling in SCs. For instance, constitutively elevated activation of p38 kinase, STAT3 activity and reduced Notch signaling has been observed in aged SCs.46, 47, 79, 80, 81, 82 Likewise, increased TGFβ activity and induction of the cyclin-dependent kinase inhibitors associated to inhibition of cellular proliferation, such as p15, p16 and p21, have been reported as potential triggers of SC senescence.79, 80, 81
The events described above likely depend on extensive changes in the SC niche, including deregulated activity and number of FAPs or additional cellular components, such as fibroblasts and adipocytes,74 that originate from FAP differentiation. In a mouse model of young and old mice sharing the circulatory system (heterochronic parabiosis model) aged SCs were rejuvenated by exposure to a young systemic environment suggesting that the tissue-specific stem cells retain their proliferative potential, but that the aged systemic environment prevents full activation.76 These findings have been sparsely investigated by in vitro studies on human primary cells, leading to contradictory results.80, 82 Age-induced changes in the systemic milieu include reduced local capillary network and endothelial cell apoptosis/senescence, which can lead to reduced secretion of SC stimulatory factors, impaired chemotaxis of immune cells and collectively a more negative balance between positive and negative regulators of SC activity. Recent evidence points to the importance of systemic concentrations of the circulating proteins such as oxytocin83 or growth differentiation factor 11 (GDF11),84 although it is currently controversial whether GDF11 levels decrease or increase with aging, as well as the relative efficacy of GDF11 supplementation in countering the functional decline of aged muscle and SCs.85
Interestingly, sarcopenia in rodents is not further accelerated during conditional ablation of Pax7+ SCs.25 However, despite the lack of direct effects on muscle fiber size, ablation of Pax7+ cells during sarcopenia generated increased levels of collagen deposition, preferentially in fast muscles,25 which could derive from fibrogenic differentiation of FAPs. In human skeletal muscle in vivo the SC content in type II muscle fibers is selectively reduced with aging, whereas the number of SCs in type I fibers remains similar to young individuals, following the pattern of a selective atrophy of type II muscle fibers.86, 87 Thus, while SC content does decrease during sarcopenia in both rodent and human skeletal muscle, it is not yet entirely defined to what extent the decrease in SC content can account for muscle atrophy or vice versa. Although this selective deterioration of type II fibers and their SC content in human skeletal muscle is partly reversible by resistance training,87, 89 the responsiveness of SCs to a single bout of resistance exercise is reduced with aging.21, 88 Even lifelong (endurance) exercise does not seem to prevent the decrement in type II fiber size or SC content compared to type I fibers.90 However, the amount of adipose infiltration in the old untrained muscle was larger than in the trained groups (unpublished observation, URM). It is therefore intriguing to speculate that changes in the muscle microenvironment or systemic environment related to inactivity or ageing can condition FAP phenotype and ability to release important paracrine cues to SCs and myofibers to support regeneration and muscle growth.
In addition to muscle atrophy, inactivity and ageing are commonly associated with increased adiposity, together leading to metabolic dysfunctions such as dyslipidemia, decreased insulin sensitivity, hyperglycemia and an increased risk of developing diabetes mellitus (i.e., T2D). Since skeletal muscle is the most abundant tissue of the body for glucose disposal, muscle sensitivity to insulin action is essential in development of whole body insulin resistance and hyperglycemia.91 Moreover, patients with T2D show a greater decline in muscle mass, muscle strength and functional capacity with aging.92 A common observation in conditions associated with impaired skeletal muscle insulin sensitivity is accumulation of ectopic lipids within (intracellular) and between (extracellular) skeletal muscle fibers48, 56, 57, 93, 94 (as illustrated in Figure 1), which is linked to reduced insulin sensitivity48, 57, 94 and decreased muscle function.58 Paradoxically, endurance athletes also display an elevated level of intracellular lipid (termed athletes-paradox), presumably serving as energy source during physical activity,95 although they also exhibit increased insulin sensitivity, as compared to healthy untrained subjects.96, 97 In contrast, the IMAT (i.e., adipose tissue within a muscle but located outside the myofiber) is to our knowledge not increased in athletes and is associated with reduced insulin sensitivity in both healthy47 and obese48 subjects as well as in acromegaly patients.49 Although the origin of IMAT is not yet known,56 murine muscle-derived stem cells have been shown to undergo adipogenic differentiation upon exposure to elevated glucose levels in vitro,50 providing a potential link between adipocyte accumulation and the systemic milieu. Moreover, while previous work proposed that IMAT can originate from trans-differentiation of myogenic stem cells,50, 56, 98 increasing evidence suggests that FAPs could constitute the mesenchymal stem cells responsible for IMAT accumulation in skeletal muscle.12, 14, 99, 100 In addition, FAP-derived adipocytes may have reduced insulin sensitivity compared to conventional adipocytes, suggesting that accumulation of FAP-derived adipocytes may contribute to a compromised peripheral insulin sensitivity.101
Presently, the role of FAPs and their interplay with SCs, eosinophils or macrophages in relation to development of T2D is unknown. However, dysfunctional regulation of FAPs mediated by changes in IL-4 signaling may influence skeletal muscle homeostasis. Interestingly, IL-4 levels have been reported to be positively associated with insulin sensitivity102 and IL-4 promoter polymorphisms have been associated with T2D.103 Moreover, IL-4-mediated signaling may prevent adipogenesis in muscle and adipose tissue.102, 104 In white adipose tissue, IL-4 released from tissue-resident eosinophils regulates the presence of M2 macrophages,31 which are positively associated with insulin sensitivity.105 When genetically depleting IL-4 secreting eosinophils the content of the M2 macrophage phenotype in adipose tissue is reduced and this is associated with a substantial decrease in glucose tolerance and insulin sensitivity.31 These changes are less explored in humans, however, increased expression of the inflammatory macrophage phenotype marker; CD11c in skeletal muscle of T2D patients has been reported.106
Reduced insulin sensitivity is often associated with aging107 and in chronic diseases that cause cachexia.108, 109 Patients suffering from cachexia or patients affected by metabolic syndrome may display elevated levels of glucocorticoids110 that may influence IL-4 secretion. Interestingly, elevated glucocorticoid levels (such as those reached upon dexamethasone treatment) in mice increase the adipogenic differentiation of FAPs, which is otherwise suppressed through IL-4-mediated signaling.15 The effect of dexamethasone treatment on FAP adipogenesis may relate to suppression of eosinophil release of IL-4111 as suggested by Dong et al.15 Thus, elevated levels of glucocorticoids during disease (either as medical treatment or endocrine release) could increase adipocyte accumulation in skeletal muscle through adipogenic differentiation of FAPs and hereby negatively impact the insulin sensitivity of skeletal muscle.47, 48 Given that steroids are used in the treatment of many muscular disorders (including DMD), the interactions between glucocorticoids, FAP, SCs and cells of the immune system deserve future investigation.
Role of SCs, FAPs and immune cells in cachexia
Cachexia consists of an accelerated muscle loss that is associated with chronic diseases, complicates their recovery and is an independent predictor of morbidity and mortality.112, 113, 114 Skeletal muscle wasting is a common phenomenon in cancer patients,115, 116, 117, 118, 119 and cancer-related muscle loss affects up to 80% of patients with advanced cancer, leading to poorer prognosis, reduced treatment response and increased risk of complications during surgery and chemotherapy. Ultimately, cachexia accounts for >20% of all cancer-related deaths.120, 121, 122, 123, 124
Since both SCs and FAPs may influence muscle homeostasis and growth, their interactions can be implicated in cachexia. In rodents, cancer cachexia is associated with muscle damage and deregulation of Pax7 expression in SCs and interstitial cells, through increased NF-κB activity,125 suggesting that NF-κB may contribute to muscle wasting in cancer.125 In muscle biopsies from pancreatic cancer patients with accelerated weight loss, an increased number of Pax7+ cells was observed, indicating that cancer cachexia is associated with an expansion of the myogenic precursor pool.125 Expansion of the Sca1+ cell population that resemble mouse FAPs, was also observed in cachectic muscles from tumor-bearing mice. Interestingly, this Sca1+ population preferentially adopt the myogenic lineage under the influence of tumor environment, by expressing the SC-specific transcription factor Pax7, which is induced by serum factors from cachectic mice and patients, in an NF-κB-dependent manner; however, completion of differentiation was also inhibited by the persistent expression of Pax7.125 Restoring the myogenic potential of these cells by Pax7 downregulation or by ectopic expression of MyoD, promoted cell differentiation and fiber fusion and reversed muscle wasting.125 These results reiterate the concept that Sca1+ cells have a latent myogenic potential that can be induced by environmental signals, for example, the elevated levels of cytokines from systemic inflammation. Since, Pax7 expression is regulated by inflammation-induced signals,126 these data reveal again the important interactions between the immune system and FAPs in the control of muscle regeneration. However, it is important to note that Sca1+ cells might only account for a subpopulation of FAPs, or even a distinct population of cells induced in tumor-bearing conditions. A recent study not only identified stromal cells by expression of fibroblast activation protein α (but also uniformly expressing FAP markers such as CD90, PDGFRα and Sca1), and observed a depletion of these stromal cells to be underlying the cancer-induced cachexia.127 Specifically, these stromal cells appeared to maintain muscle size through paracrine secretion of follistatin, which in turn reduced the muscle expression of ubiquitin ligases such as atrogin-1 and muscle RING-finger 1 (MuRF1) involved in muscle protein breakdown.127 It remains to be investigated if factors released directly by the tumor, alterations in immune cell content/function or other mechanisms may underlie the depletion of the stromal cells in skeletal muscle during cancer cachexia. If these stromal cells are indeed FAPs, an alternative explanation for the cell content depletion during cachexia may relate to an increased adipogenic differentiation of the FAPs. In this regard the progression of cachexia has been associated with an increased amount of intramuscular lipid droplets,54 and although the source of these is not identified they could originate from FAPs.
Multiple chronic diseases are also associated with elevated systemic inflammation including RA.128, 129, 130, 131, 132, 133 In RA patients, loss of muscle strength is associated with RA duration rather than with chronological age, in contrast to the decline with age observed in the general population.134 This indicates a disease-related effect on muscle strength that surpasses the effect of aging. Increased IMAT, potentially originating from FAPs, is observed in the muscle of RA patients and this reduction in muscle density associates with greater joint destruction.52 Furthermore, type II fiber atrophy in RA patients has been reported,135 but compared to patients with osteoarthritis the absolute numbers of SC136 and their in vitro regenerative potential137 were not different in RA patients.
COPD is another frequent cause of disease-related cachexia and premature death worldwide.138 COPD is often accompanied by pronounced muscle wasting (atrophy of both type I and II fibers has been reported139) and metabolic dysfunction.138, 140 Notably, the loss of muscle mass is an independent predictor of mortality in COPD patients.1, 112 The alterations in immune cell function and the chronic inflammatory condition is believed to be a substantial contributor to the loss of muscle mass along with an impaired regenerative capacity and cellular apoptosis.138, 140 SC content is not altered in the muscles of COPD patients compared to controls; however, primary SCs isolated from COPD patients displayed a delayed activation in culture and decreased expression of myosin heavy chain expression during myotube formation compared to controls.141 Interestingly, the increased number of central nuclei observed in the muscles of COPD patients with preserved muscle mass, as compared to those who lost muscle mass,141, 142 suggests that the ability of the muscle to regenerate could attenuate the extent of COPD-related cachexia.
Collectively, the potential role for SC and FAPs in relation to muscle loss due to chronic inflammatory diseases is a matter of current and future investigations (Figure 5).

Schematic illustration showing potential role of FAP-SC and their interplay in muscle: atrophy (as observed with aging and disease), hypertrophy and insulin sensitivity. IL-4, interleukin-4; IMAT, intramuscular adipose tissue; T2D, type 2 diabetes
Exercise as a Strategy to Improve Muscle Health – Stimulation of SC and FAPs by Exercise
Skeletal muscle is a highly plastic tissue that adapts to stimuli, by proportionally adjusting mass and strength (resistance training) or aerobic capacity (endurance training) in response to exercise. Both acute and prolonged resistance exercise stimulates the proliferation of SCs in healthy subjects,143, 144, 145, 146, 147 and even non-hypertrophying endurance exercise can induce proliferation of at least some SC populations.7 Although the potential impact of different training modalities on the immune-FAP-SC network are described separately below, it is important to note some remarkable differences, and the relative impact on the network, between physical exercise and the pathological conditions described in the previous paragraphs. For instance, in most pathological conditions FAPs are activated by physical injury, which triggers extensive changes in the microenvironment, (e.g., myofiber degeneration in muscular dystrophies) or by elevated systemic concentration of inflammatory cytokines (e.g., cachexia); by contrast, during training most of these signals are absent – except for the case of strenuous exercise – and the predominant changes occurring in exercised muscles are of metabolic (redox alterations) or biomechanical (contraction/relaxation cycles) nature. As FAPs occupy an interstitial position they are a great candidate as cell types that sense these changes and transmit them to SCs via specific cues. In this regard, recent work reported that SIRT1, a NAD(+)-dependent HDAC known as redox and nutrient sensor, promotes the metabolic switch from fatty acid oxidation to glycolysis during the SC transition from quiescence to proliferation.148 Moreover, SIRT1 regulates autophagic flux in SCs to cope with the high bioenergetic demands during the activation process.149 Finally, SIRT1 connects changes in SC metabolism with changes in the transcriptional machinery toward myogenic commitment of the SC.148 This reprogramming of cellular metabolism decreases intracellular NAD(+) levels and the activity of the HDAC SIRT1, leading to elevated H4K16 acetylation and activation of muscle gene transcription. Future studies should establish whether SIRT1 is activated by FAP-derived signals.
Resistance and Endurance Training, Muscle Hypertrophy and Insulin Sensitivity
Although the role of SCs during myofiber regeneration has been extensively studied and reviewed,8, 22, 23, 150, 151, 152, 153 it is debated whether SCs possess a role in myofiber hypertrophy in the adult muscle.154, 155 In earlier studies, irradiation was used to ablate SC activity, whereby overload induced hypertrophy in rodents was prevented,154, 156, 157 indicating a direct linkage between SCs and myofiber hypertrophy. However, a later study opposes this contention by showing an intact hypertrophic response in myofibers of SC-depleted muscles in rodents.26 Although these findings from conditional knockout mice seem to reject the hypothesis that SCs are essential for hypertrophy, more recent results indicate that the lack of SCs can attenuate myofiber hypertrophy in the later phases of an overload period.19 The latter is supported by other studies suggesting an important role for SC in myofiber growth and myonuclei accretion.24, 158, 159 In agreement, IGF-1, which can accentuate resistance training-induced muscle hypertrophy,160 may in part act through increased proliferation and differentiation of SC to support myofiber growth.161 This is supported by the increased expression of IGF-1 splice variants in human SCs following eccentric resistance exercise.162
In several human studies, robust increases in the number of SCs has been shown both acutely21, 163, 164 and following prolonged 143, 144, 165, 166, 167, 168 resistance exercise in both young and old humans.87, 88, 89 In contrast, SC proliferation following resistance exercise may be impaired during ageing and in patients affected by chronic muscular disorders, with current evidence suggesting that this impairment originates from alterations in cues from the SC niche or the systemic environment.20, 21, 76, 83, 88 However, long-term resistance training can reverse the SC distribution in elderly muscle toward that of young muscle.169 Knowledge about the regulation of FAPs in relation to resistance training and hypertrophy is lacking, but in rodents the involvement of a SC-FAP interplay in successful muscle regeneration after muscle damage has been convincingly demonstrated.9, 11, 28 Although damage/regeneration is not a prerequisite for resistance training adaptations,170 the rodent findings combined with human SC data suggest that SC-FAP interplay may have a central role in resistance training adaptations (Figure 5). Furthermore, detraining in elderly is accompanied by an increased amount of muscle fat infiltration which can be reversed by resistance training,171 and reducing ectopic fat accumulation may enhance myofiber anabolic signaling.46 Collectively, this area awaits further investigation in humans; however, the present body of data indicates that FAPs may be regulated with resistance exercise-induced hypertrophy. One mechanism may relate to the recently identified circulating hormone Meteorin like, which is secreted from skeletal muscle upon exercise and triggers IL-4, and IL-13 production by eosinophils in adipose tissue. These cytokines cause alternative activation of M2 macrophages29, 172 as shown in Figure 2, and are also involved in the regulation of FAP activity, further providing a tentative link between exercise, immune cell activation and FAP regulation.
Endurance exercise increases insulin sensitivity and glucose tolerance,173, 174, 175, 176 for example, via increased protein expression of insulin receptor substrate-1 (IRS-1) and GLUT4176, 177 in skeletal muscle. Although increased energy expenditure through endurance training reduces accumulation of adipose tissue, insulin sensitivity and glucose tolerance are improved independent of weight loss.178 The SC response to endurance training has only been evaluated in a few human studies, and increased SC numbers are reported in most,179, 180, 181 but not all182 of these, mostly in the absence of muscle fiber hypertrophy. Recently, hypertrophy of both type I and IIa fibers was observed following 12 weeks of aerobic training, with a concomitant increase in SC number only in type I fibers.181 Furthermore, non-hypertrophying endurance exercise can induce proliferation of SC populations in hybrid fibers (type I/II) without effect on the SC content of type I or II fibers.7 Generally, these data indicate a role for SC proliferation and turnover in muscle maintenance even in the absence of fiber hypertrophy, which is in line with the suggested role for SC-FAP interplay in regulation of a healthy muscle.
Perspectives and Conclusions
FAPs are emerging as a 'cellular filter' between external perturbations (either local or systemic changes in physical, metabolic and inflammatory cues) and the effectors of the muscle regeneration machinery – the SC. Depending on the nature of the perturbations FAPs appear to adopt specific phenotypic and functional properties indicating a highly heterogeneous cell population.183 Thus, FAP heterogeneity and the dynamic transition from a physiological to compensatory or pathological subpopulation appears as a key issue to investigate in future studies. In particular, the anatomical derivation of FAPs might reveal important differences. For instance, in the presence of physical insults, the ensuing vessel injury or transient ischemia might direct a composition of FAPs that is phenotypically and functionally different from cells derived by the expansion of resident interstitial FAPs.
Overall, the elucidation of the interplay between SCs, FAPs, their niche and immune cells might have an impact not only in the discovery of interventions toward restoring muscle function and correcting metabolic dysregulation in pathological conditions, but also to improve muscle anabolism and insulin sensitivity, which commonly decreases during aging, inactivity and certain disease states.
Abbreviations
- FAP:
-
fibro-adipogenic progenitor
- SC:
-
satellite cell
- Sca1:
-
stem cell antigen 1
- PDGFRα:
-
platelet-derived growth factor receptor alpha
- FACS:
-
fluorescence-activated cell sorting
- IGF-1:
-
insulin-like growth factor 1
- HGF:
-
hepatocyte growth factor
- NO:
-
nitric oxide
- STAT:
-
signal transducer and activator of transcription
- NAD:
-
nicotinamide adenine dinucleotide
- IL-4:
-
interleukin-4
- SIRT1:
-
sirtuin 1
- COPD:
-
chronic obstructive pulmonary disease
- RA:
-
rheumatoid arthritis
- DMD:
-
Duchenne muscular dystrophy
- SWI/SNF:
-
SWItch/sucrose nonfermentable
- HDAC:
-
histone deacetylase
- TGFβ:
-
transforming growth factor beta
- GDF:
-
growth differentiation factor
- Pax7:
-
paired box transcription factor 7
- MyoD:
-
myogenic differentiation
- Wnt:
-
wingless type
- MuRF1:
-
muscle RING-finger protein 1
- T2D:
-
type 2 diabetes
- IMAT:
-
intramuscular adipose tissue
- NF-κB:
-
nuclear factor kappa-light-chain enhancer of activated B cells
References
Marquis K, Debigare R, Lacasse Y, LeBlanc P, Jobin J, Carrier G et al. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 809–813.
Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population-based study. Lancet Oncol 2008; 9: 629–635.
Langen RC, Gosker HR, Remels AH, Schols AM . Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int J Biochem Cell Biol 2013; 45: 2245–2256.
D'Souza DM, Al-Sajee D, Hawke TJ . Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front Physiol 2013; 4: 379.
Charge SB, Rudnicki MA . Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004; 84: 209–238.
Hawke TJ, Garry DJ . Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 2001; 91: 534–551.
Joanisse S, Gillen JB, Bellamy LM, McKay BR, Tarnopolsky MA, Gibala MJ et al. Evidence for the contribution of muscle stem cells to nonhypertrophic skeletal muscle remodeling in humans. FASEB J 2013; 27: 4596–4605.
Yin H, Price F, Rudnicki MA . Satellite cells and the muscle stem cell niche. Physiol Rev 2013; 93: 23–67.
Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K . Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 2010; 12: 143–152.
Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 2010; 12: 153–163.
Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013; 153: 376–388.
Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchell KJ et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med 2013; 5: 626–639.
Rodeheffer MS . Tipping the scale: muscle versus fat. Nat Cell Biol 2010; 12: 102–104.
Uezumi A, Fukada S, Yamamoto N, Ikemoto-Uezumi M, Nakatani M, Morita M et al. Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis 2014; 5: e1186.
Dong Y, Silva KA, Dong Y, Zhang L . Glucocorticoids increase adipocytes in muscle by affecting IL-4 regulated FAP activity. FASEB J 2014; 28: 4123–4132.
Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandona M et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev 2014; 28: 841–857.
Snijders T, Verdijk LB, van Loon LJ . The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Res Rev 2009; 8: 328–338.
Schultz E . Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol 1996; 175: 84–94.
Fry CS, Lee JD, Jackson JR, Kirby TJ, Stasko SA, Liu H et al. Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J 2014; 28: 1654–1665.
Suetta C, Frandsen U, Mackey AL, Jensen L, Hvid LG, Bayer ML et al. Ageing is associated with diminished muscle re-growth and myogenic precursor cell expansion early after immobility-induced atrophy in human skeletal muscle. J Physiol 2013; 591: 3789–3804.
McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G . Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J 2012; 26: 2509–2521.
Lepper C, Partridge TA, Fan CM . An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011; 138: 3639–3646.
von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA . Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci USA 2013; 110: 16474–16479.
Bruusgaard JC, Johansen IB, Egner IM, Rana ZA, Gundersen K . Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc Natl Acad Sci USA 2010; 107: 15111–15116.
Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med 2015; 21: 76–80.
McCarthy JJ, Mula J, Miyazaki M, Erfani R, Garrison K, Farooqui AB et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011; 138: 3657–3666.
Oishi T, Uezumi A, Kanaji A, Yamamoto N, Yamaguchi A, Yamada H et al. Osteogenic differentiation capacity of human skeletal muscle-derived progenitor cells. PLoS One 2013; 8: e56641.
Uezumi A, Ikemoto-Uezumi M, Tsuchida K . Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front Physiol 2014; 5: 68.
Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 2014; 157: 1279–1291.
Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014; 157: 1292–1308.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011; 332: 243–247.
Cordani N, Pisa V, Pozzi L, Sciorati C, Clementi E . Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro-adipogenic precursor differentiation. Stem Cells 2014; 32: 874–885.
Takada S, Okita K, Suga T, Omokawa M, Kadoguchi T, Sato T et al. Low-intensity exercise can increase muscle mass and strength proportionally to enhanced metabolic stress under ischemic conditions. J Appl Physiol 2012; 113: 199–205.
Laude K, Favre J, Thuillez C, Richard V . NO produced by endothelial NO synthase is a mediator of delayed preconditioning-induced endothelial protection. Am J Physiol Heart Circ Physiol 2003; 284: H2053–H2060.
Rana A, Goyal N, Ahlawat A, Jamwal S, Reddy B, Sharma S . Mechanisms involved in attenuated cardio-protective role of ischemic preconditioning in metabolic disorders. Perfusion 2014; 30: 94–105.
Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE . Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell 2002; 13: 2909–2918.
Tatsumi R, Wuollet AL, Tabata K, Nishimura S, Tabata S, Mizunoya W et al. A role for calcium-calmodulin in regulating nitric oxide production during skeletal muscle satellite cell activation. Am J Physiol Cell Physiol 2009; 296: C922–C929.
Christov C, Chretien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ et al. Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell 2007; 18: 1397–1409.
Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL . Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J 2013; 280: 4131–4148.
Begue G, Douillard A, Galbes O, Rossano B, Vernus B, Candau R et al. Early activation of rat skeletal muscle IL-6/STAT1/STAT3 dependent gene expression in resistance exercise linked to hypertrophy. PLoS One 2013; 8: e57141.
Saclier M, Yacoub-Youssef H, Mackey AL, Arnold L, Ardjoune H, Magnan M et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells 2012; 31: 384–396.
Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med 2014; 20: 1174–1181.
Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell 2004; 6: 673–684.
Lee SJ, McPherron AC . Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA 2001; 98: 9306–9311.
Akhmedov D, Berdeaux R . The effects of obesity on skeletal muscle regeneration. Front Physiol 2013; 4: 371.
Tardif N, Salles J, Guillet C, Tordjman J, Reggio S, Landrier J-F et al. Muscle ectopic fat deposition contributes to anabolic resistance in obese sarcopenic old rats through eIF2α activation. Aging Cell 2014; 13: 1001–1011.
Yim JE, Heshka S, Albu J, Heymsfield S, Kuznia P, Harris T et al. Intermuscular adipose tissue rivals visceral adipose tissue in independent associations with cardiovascular risk. Int J Obes (Lond) 2007; 31: 1400–1405.
Sinha R, Dufour S, Petersen KF, LeBon V, Enoksson S, Ma YZ et al. Assessment of skeletal muscle triglyceride content by (1)H nuclear magnetic resonance spectroscopy in lean and obese adolescents: relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes 2002; 51: 1022–1027.
Freda PU, Shen W, Heymsfield SB, Reyes-Vidal CM, Geer EB, Bruce JN et al. Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J Clin Endocrinol Metab 2008; 93: 2334–2343.
Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc Natl Acad Sci USA 2008; 105: 1226–1231.
Kent-Braun JA, Ng AV, Young K . Skeletal muscle contractile and noncontractile components in young and older women and men. J Appl Physiol 2000; 88: 662–668.
Baker JF, Von Feldt J, Mostoufi-Moab S, Noaiseh G, Taratuta E, Kim W et al. Deficits in muscle mass, muscle density, and modified associations with fat in rheumatoid arthritis. Arthritis Care Res 2014; 66: 1612–1618.
Roig M, Eng JJ, MacIntyre DL, Road JD, Reid WD . Deficits in muscle strength, mass, quality, and mobility in people with chronic obstructive pulmonary disease. J Cardiopulm Rehabil Prev 2011; 31: 120–124.
Stephens NA, Skipworth RJ, Macdonald AJ, Greig CA, Ross JA, Fearon KC . Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J Cachexia Sarcopenia Muscle 2011; 2: 111–117.
Natarajan A, Lemos DR, Rossi FM . Fibro/adipogenic progenitors: a double-edged sword in skeletal muscle regeneration. Cell Cycle 2010; 9: 2045–2046.
Vettor R, Milan G, Franzin C, Sanna M, De Coppi P, Rizzuto R et al. The origin of intermuscular adipose tissue and its pathophysiological implications. Am J Physiol Endocrinol Metab 2009; 297: E987–E998.
Goodpaster BH, Thaete FL, Kelley DE . Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr 2000; 71: 885–892.
Hilton TN, Tuttle LJ, Bohnert KL, Mueller MJ, Sinacore DR . Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: association with performance and function. Phys Ther 2008; 88: 1336–1344.
Uezumi A, Ikemoto-Uezumi M, Tsuchida K . Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front Physiol 2014; 5: 68.
Moyer AL, Wagner KR . Regeneration versus fibrosis in skeletal muscle. Curr Opin Rheumatol 2011; 23: 568–573.
Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Munoz-Canoves P . Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol 2011; 96: 167–201.
Tidball JG, Dorshkind K, Wehling-Henricks M . Shared signaling systems in myeloid cell-mediated muscle regeneration. Development 2014; 141: 1184–1196.
Paylor B, Natarajan A, Zhang RH, Rossi F . Nonmyogenic cells in skeletal muscle regeneration. Curr Top Dev Biol 2011; 96: 139–165.
Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA . Cellular dynamics in the muscle satellite cell niche. EMBO Rep 2013; 14: 1062–1072.
Madaro L, Bouche M . From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: the role of lymphocytes. Biomed Res Int 2014; 2014: 438675.
Judson RN, Zhang RH, Rossi FM . Tissue resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J 2013; 280: 4100–4108.
Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 2011; 124: 3654–3664.
Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J 2012; 31: 301–316.
Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 2010; 12: 341–351.
Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA 2008; 105: 19183–19187.
Biressi S, Miyabara EH, Gopinath SD, Carlig PM, Rando TA . A Wnt-TGFbeta2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci Transl Med 2014; 6: 267ra176.
Degens H . The role of systemic inflammation in age-related muscle weakness and wasting. Scand J Med Sci Sports 2010; 20: 28–38.
Peake J, Della GP, Cameron-Smith D . Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am J Physiol Regul Integr Comp Physiol 2010; 298: R1485–R1495.
Gopinath SD, Rando TA . Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell 2008; 7: 590–598.
Chakkalakal JV, Jones KM, Basson MA, Brack AS . The aged niche disrupts muscle stem cell quiescence. Nature 2012; 490: 355–360.
Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA . Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005; 433: 760–764.
Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 2014; 20: 255–264.
Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB . p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 2014; 20: 265–271.
Carlson ME, Hsu M, Conboy IM . Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008; 454: 528–532.
Carlson ME, Suetta C, Conboy MJ, Aagaard P, Mackey A, Kjaer M et al. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med 2009; 1: 381–391.
Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014; 20 506: 316–321.
Barberi L, Scicchitano BM, De Rossi M, Bigot A, Duguez S, Wielgosik A et al. Age-dependent alteration in muscle regeneration: the critical role of tissue niche. Biogerontology 2013; 14: 273–292.
Elabd C, Cousin W, Upadhyayula P, Chen RY, Chooljian MS, Li J et al. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat Commun 2014; 5: 4082.
Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014; 344: 649–652.
Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab 2015; 22: 164–174.
Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ . Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab 2007; 292: E151–E157.
Verdijk LB, Snijders T, Drost M, Delhaas T, Kadi F, van Loon LJ . Satellite cells in human skeletal muscle; from birth to old age. Age (Dordr) 2014; 36: 545–547.
Snijders T, Verdijk LB, Smeets JS, McKay BR, Senden JM, Hartgens F et al. The skeletal muscle satellite cell response to a single bout of resistance-type exercise is delayed with aging in men. Age 2014; 36: 9699.
Verdijk LB, Gleeson BG, Jonkers RAM, Meijer K, HHCM Savelberg, Dendale P et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J Gerontol A Biol Sci Med Sci 2009; 64A: 332–339.
Mackey AL, Karlsen A, Couppe C, Mikkelsen UR, Nielsen RH, Magnusson SP et al. Differential satellite cell density of type I and II fibres with lifelong endurance running in old men. Acta Physiol (Oxf) 2014; 210: 612–627.
Samuel VT, Shulman GI . Mechanisms for insulin resistance: common threads and missing links. Cell 2012; 148: 852–871.
Leenders M, Verdijk LB, van der Hoeven L, Adam JJ, van Kranenburg J, Nilwik R et al. Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J Am Med Dir Assoc 2013; 14: 585–592.
Goodpaster BH, Chomentowski P, Ward BK, Rossi A, Glynn NW, Delmonico MJ et al. Effects of physical activity on strength and skeletal muscle fat infiltration in older adults: a randomized controlled trial. J Appl Physiol 2008; 105: 1498–1503.
Shulman GI . Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 2014; 371: 1131–1141.
Schrauwen-Hinderling VB, Hesselink MK, Schrauwen P, Kooi ME . Intramyocellular lipid content in human skeletal muscle. Obesity 2006; 14: 357–367.
Goodpaster BH, He J, Watkins S, Kelley DE . Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 2001; 86: 5755–5761.
Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 2011; 60: 2588–2597.
Scarda A, Franzin C, Milan G, Sanna M, Dal Pra C, Pagano C et al. Increased adipogenic conversion of muscle satellite cells in obese Zucker rats. Int J Obes (Lond) 2010; 34: 1319–1327.
Agley CC, Rowlerson AM, Velloso CP, Lazarus NR, Harridge SD . Human skeletal muscle fibroblasts, but not myogenic cells, readily undergo adipogenic differentiation. J Cell Sci 2013; 126: 5610–5625.
Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature 2014; 510: 393–396.
Arrighi N, Moratal C, Clement N, Giorgetti-Peraldi S, Peraldi P, Loubat A et al. Characterization of adipocytes derived from fibro/adipogenic progenitors resident in human skeletal muscle. Cell Death Dis 2015; 6: e1733.
Chang YH, Ho KT, Lu SH, Huang CN, Shiau MY . Regulation of glucose/lipid metabolism and insulin sensitivity by interleukin-4. Int J Obes (Lond) 2012; 36: 993–998.
Ho KT, Shiau MY, Chang YH, Chen CM, Yang SC, Huang CN . Association of interleukin-4 promoter polymorphisms in Taiwanese patients with type 2 diabetes mellitus. Metabolism 2010; 59: 1717–1722.
Tsao CH, Shiau MY, Chuang PH, Chang YH, Hwang J . Interleukin-4 regulates lipid metabolism by inhibiting adipogenesis and promoting lipolysis. J Lipid Res 2014; 55: 385–397.
Sun K, Kusminski CM, Scherer PE . Adipose tissue remodeling and obesity. J Clin Invest 2011; 121: 2094–2101.
Fink LN, Oberbach A, Costford SR, Chan KL, Sams A, Bluher M et al. Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia 2013; 56: 1623–1628.
Mikkelsen UR, Couppe C, Karlsen A, Grosset JF, Schjerling P, Mackey AL et al. Life-long endurance exercise in humans: circulating levels of inflammatory markers and leg muscle size. Mech Ageing Dev 2013; 134: 531–540.
Ferraz-Amaro I, Gonzalez-Juanatey C, Lopez-Mejias R, Riancho-Zarrabeitia L, Gonzalez-Gay MA . Metabolic syndrome in rheumatoid arthritis. Mediators Inflamm 2013; 2013: 710928.
Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D et al. Cachexia: a new definition. Clin Nutr 2008; 27: 793–799.
Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP . Clinical review: the pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J Clin Endocrinol Metab 2009; 94: 2692–2701.
Lamas AM, Leon OG, Schleimer RP . Glucocorticoids inhibit eosinophil responses to granulocyte-macrophage colony-stimulating factor. J Immunol 1991; 147: 254–259.
Schols AM, Broekhuizen R, Weling-Scheepers CA, Wouters EF . Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr 2005; 82: 53–59.
Gonzalez MC, Pastore CA, Orlandi SP, Heymsfield SB . Obesity paradox in cancer: new insights provided by body composition. Am J Clin Nutr 2014; 99: 999–1005.
Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol 2013; 31: 1539–1547.
Silver HJ, Dietrich MS, Murphy BA . Changes in body mass, energy balance, physical function, and inflammatory state in patients with locally advanced head and neck cancer treated with concurrent chemoradiation after low-dose induction chemotherapy. Head Neck 2007; 29: 893–900.
Antoun S, Birdsell L, Sawyer MB, Venner P, Escudier B, Baracos VE . Association of skeletal muscle wasting with treatment with sorafenib in patients with advanced renal cell carcinoma: results from a placebo-controlled study. J Clin Oncol 2010; 28: 1054–1060.
Gilliam LA, St Clair DK . Chemotherapy-induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal 2011; 15: 2543–2563.
Haseen F, Murray LJ, Cardwell CR, O'Sullivan JM, Cantwell MM . The effect of androgen deprivation therapy on body composition in men with prostate cancer: systematic review and meta-analysis. J Cancer Surviv 2010; 4: 128–139.
Visovsky C . Muscle strength, body composition, and physical activity in women receiving chemotherapy for breast cancer. Integr Cancer Ther 2006; 5: 183–191.
Murphy KT, Lynch GS . Update on emerging drugs for cancer cachexia. Expert Opin Emerg Drugs 2009; 14: 619–632.
Antoun S, Baracos VE, Birdsell L, Escudier B, Sawyer MB . Low body mass index and sarcopenia associated with dose-limiting toxicity of sorafenib in patients with renal cell carcinoma. Ann Oncol 2010; 21: 1594–1598.
Ross PJ, Ashley S, Norton A, Priest K, Waters JS, Eisen T et al. Do patients with weight loss have a worse outcome when undergoing chemotherapy for lung cancers? Br J Cancer 2004; 90: 1905–1911.
Massicotte MH, Borget I, Broutin S, Baracos VE, Leboulleux S, Baudin E et al. Body composition variation and impact of low skeletal muscle mass in patients with advanced medullary thyroid carcinoma treated with vandetanib: results from a placebo-controlled study. J Clin Endocrinol Metab 2013; 98: 2401–2408.
Prado CM, Baracos VE, McCargar LJ, Reiman T, Mourtzakis M, Tonkin K et al. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clin Cancer Res 2009; 15: 2920–2926.
He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013; 123: 4821–4835.
Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010; 7: 455–469.
Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med 2013; 210: 1137–1151.
Mason JC, Libby P . Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J 2015; 36: 482–9c.
Ekblom B, Lovgren O, Alderin M, Fridstrom M, Satterstrom G . Physical performance in patients with rheumatoid arthritis. Scand J Rheumatol 1974; 3: 121–125.
Hsieh LF, Didenko B, Schumacher Jr HR, Torg JS . Isokinetic and isometric testing of knee musculature in patients with rheumatoid arthritis with mild knee involvement. Arch Phys Med Rehabil 1987; 68: 294–297.
Ekdahl C, Broman G . Muscle strength, endurance, and aerobic capacity in rheumatoid arthritis: a comparative study with healthy subjects. Ann Rheum Dis 1992; 51: 35–40.
Hakkinen A, Hannonen P, Hakkinen K . Muscle strength in healthy people and in patients suffering from recent-onset inflammatory arthritis. Br J Rheumatol 1995; 34: 355–360.
Nordesjo LO, Nordgren B, Wigren A, Kolstad K . Isometric strength and endurance in patients with severe rheumatoid arthritis or osteoarthrosis in the knee joints. A comparative study in healthy men and women. Scand J Rheumatol 1983; 12: 152–156.
Beenakker KG, Ling CH, Meskers CG, de Craen AJ, Stijnen T, Westendorp RG et al. Patterns of muscle strength loss with age in the general population and patients with a chronic inflammatory state. Ageing Res Rev 2010; 9: 431–436.
Magyar E, Talerman A, Mohacsy J, Wouters HW, de Bruijn WC . Muscle changes in rheumatoid arthritis. A review of the literature with a study of 100 cases. Virchows Arch A Pathol Anat Histol 1977; 373: 267–278.
Beenakker KG, Duijnisveld BJ, Van Der Linden HM, Visser CP, Westendorp RG, Butler-Brown G et al. Muscle characteristics in patients with chronic systemic inflammation. Muscle Nerve 2012; 46: 204–209.
Duijnisveld BJ, Bigot A, Beenakker KG, Portilho DM, Raz V, van der Heide HJ et al. Regenerative potential of human muscle stem cells in chronic inflammation. Arthritis Res Ther 2011; 13: R207.
Mathur S, Brooks D, Carvalho CR . Structural alterations of skeletal muscle in COPD. Front Physiol 2014; 5: 104.
Allaire J, Maltais F, LeBlanc P, Simard PM, Whittom F, Doyon JF et al. Lipofuscin accumulation in the vastus lateralis muscle in patients with chronic obstructive pulmonary disease. Muscle Nerve 2002; 25: 383–389.
Folkesundhedsrapporten (Kronisk Obstruktiv Lungesygdom), Danmark Statens Institut for Folkesundhed 2007.
Theriault ME, Pare ME, Lemire BB, Maltais F, Debigare R . Regenerative defect in vastus lateralis muscle of patients with chronic obstructive pulmonary disease. Respir Res 2014; 15: 35.
Theriault ME, Pare ME, Maltais F, Debigare R . Satellite cells senescence in limb muscle of severe patients with COPD. PLoS One 2012; 7: e39124.
Kadi F, Schjerling P, Andersen LL, Charifi N, Madsen JL, Christensen LR et al. The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. J Physiol 2004; 558: 1005–1012.
Mackey AL, Holm L, Reitelseder S, Pedersen TG, Doessing S, Kadi F et al. Myogenic response of human skeletal muscle to 12 weeks of resistance training at light loading intensity. Scand J Med Sci Sports 2010; 21: 773–782.
Mikkelsen UR, Langberg H, Helmark IC, Skovgaard D, Andersen LL, Kjaer M et al. Local NSAID infusion inhibits satellite cell proliferation in human skeletal muscle after eccentric exercise. J Appl Physiol 2009; 107: 1600–1611.
Farup J, Rahbek SK, Riis S, Vendelbo MH, Paoli F, Vissing K . Influence of exercise contraction mode and protein supplementation on human skeletal muscle satellite cell content and muscle fiber growth. J Appl Physiol 2014; 117: 898–909.
Farup J, Rahbek SK, Knudsen IS, de Paoli F, Mackey AL, Vissing K . Whey protein supplementation accelerates satellite cell proliferation during recovery from eccentric exercise. Amino Acids 2014; 46: 2503–2516.
Ryall James G, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X et al. The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015; 16: 171–183.
Tang AH, Rando TA . Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J 2014; 33: 2782–2797.
Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G . Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest 2010; 120: 11–19.
Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011; 138: 3647–3656.
Montarras D, L'Honore A, Buckingham M . Lying low but ready for action: the quiescent muscle satellite cell. FEBS J 2013; 280: 4036–4050.
Konigsberg UR, Lipton BH, Konigsberg IR . The regenerative response of single mature muscle fibers isolated in vitro. Dev Biol 1975; 45: 260–275.
O'Connor RS, Pavlath GK . Point:counterpoint: satellite cell addition is/is not obligatory for skeletal muscle hypertrophy. J Appl Physiol 2007; 103: 1099–1100.
Brack AS, Rando TA . Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 2012; 10: 504–514.
Rosenblatt JD, Parry DJ . Gamma irradiation prevents compensatory hypertrophy of overloaded mouse extensor digitorum longus muscle. J Appl Physiol 1992; 73: 2538–2543.
Adams GR, Caiozzo VJ, Haddad F, Baldwin KM . Cellular and molecular responses to increased skeletal muscle loading after irradiation. Am J Physiol Cell Physiol 2002; 283: C1182–C1195.
Blaauw B, Reggiani C . The role of satellite cells in muscle hypertrophy. J Muscle Res Cell Motil 2014; 35: 3–10.
Guerci A, Lahoute C, Hebrard S, Collard L, Graindorge D, Favier M et al. Srf-dependent paracrine signals produced by myofibers control satellite cell-mediated skeletal muscle hypertrophy. Cell Metab 2012; 15: 25–37.
Lee S, Barton ER, Sweeney HL, Farrar RP . Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol 2004; 96: 1097–1104.
Barton-Davis ER, Shoturma DI, Sweeney HL . Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand 1999; 167: 301–305.
McKay BR, O'Reilly CE, Phillips SM, Tarnopolsky MA, Parise G . Co-expression of IGF-1 family members with myogenic regulatory factors following acute damaging muscle-lengthening contractions in humans. J Physiol 2008; 586: 5549–5560.
Snijders T, Verdijk LB, McKay BR, Smeets JSJ, van Kranenburg J, Groen BBB et al. Acute dietary protein intake restriction is associated with changes in myostatin expression after a single bout of resistance exercise in healthy young men. J Nutr 2013; 144: 137–145.
Snijders T, Verdijk LB, Beelen M, McKay BR, Parise G, Kadi F et al. A single bout of exercise activates skeletal muscle satellite cells during subsequent overnight recovery. Exp Physiol 2012; 97: 762–773.
Mackey AL, Esmarck B, Kadi F, Koskinen SO, Kongsgaard M, Sylvestersen A et al. Enhanced satellite cell proliferation with resistance training in elderly men and women. Scand J Med Sci Sports 2007; 17: 34–42.
Mackey AL, Andersen LL, Frandsen U, Sjogaard G . Strength training increases the size of the satellite cell pool in type I and II fibres of chronically painful trapezius muscle in females. J Physiol 2011; 589: 5503–5515.
Olsen S, Aagaard P, Kadi F, Tufekovic G, Verney J, Olesen JL et al. Creatine supplementation augments the increase in satellite cell and myonuclei number in human skeletal muscle induced by strength training. J Physiol 2006; 573: 525–534.
Petrella JK, Kim JS, Mayhew DL, Cross JM, Bamman MM . Potent myofiber hypertrophy during resistance training in humans is associated with satellite cell-mediated myonuclear addition: a cluster analysis. J Appl Physiol 2008; 104: 1736–1742.
Verdijk LB, Gleeson BG, Jonkers RA, Meijer K, Savelberg HH, Dendale P et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J Gerontol A Biol Sci Med Sci 2009; 64: 332–339.
Paulsen G, Mikkelsen UR, Raastad T, Peake JM . Leucocytes cytokines and satellite cells: what role do they play in muscle damage and regeneration following eccentric exercise? Exerc Immunol Rev 2012; 18: 42–97.
Taaffe DR, Henwood TR, Nalls MA, Walker DG, Lang TF, Harris TB . Alterations in muscle attenuation following detraining and retraining in resistance-trained older adults. Gerontology 2009; 55: 217–223.
Lee SD, Tontonoz P . Eosinophils in fat: pink is the new brown. Cell 2014; 157: 1249–1250.
Lancaster GI, Febbraio MA . The immunomodulating role of exercise in metabolic disease. Trends Immunol 2014; 35: 262–269.
Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403.
Devlin JT, Horton ES . Effects of prior high-intensity exercise on glucose metabolism in normal and insulin-resistant men. Diabetes 1985; 34: 973–979.
Henriksen EJ . Invited review: effects of acute exercise and exercise training on insulin resistance. J Appl Physiol 2002; 93: 788–796.
Christensen B, Nellemann B, Larsen MS, Thams L, Sieljacks P, Vestergaard PF et al. Whole body metabolic effects of prolonged endurance training in combination with erythropoietin treatment in humans: a randomized placebo controlled study. Am J Physiol Endocrinol Metab 2013; 305: E879–E889.
Krogh-Madsen R, Pedersen M, Solomon TP, Knudsen SH, Hansen LS, Karstoft K et al. Normal physical activity obliterates the deleterious effects of a high-caloric intake. J Appl Physiol 2014; 116: 231–239.
Charifi N, Kadi F, Feasson L, Denis C . Effects of endurance training on satellite cell frequency in skeletal muscle of old men. Muscle Nerve 2003; 28: 87–92.
Verney J, Kadi F, Charifi N, Feasson L, Saafi MA, Castells J et al. Effects of combined lower body endurance and upper body resistance training on the satellite cell pool in elderly subjects. Muscle Nerve 2008; 38: 1147–1154.
Fry CS, Noehren B, Mula J, Ubele MF, Westgate PM, Kern PA et al. Fibre type-specific satellite cell response to aerobic training in sedentary adults. J Physiol 2014; 592: 2625–2635.
Snijders T, Verdijk LB, Hansen D, Dendale P, van Loon LJ . Continuous endurance-type exercise training does not modulate satellite cell content in obese type 2 diabetes patients. Muscle Nerve 2011; 43: 393–401.
Malecova B, Puri PL . "Mix of Mics"- phenotypic and biological heterogeneity of "Multipotent" muscle interstitial cells (MICs). J Stem Cell Res Ther 2012; e-pub ahead of print 17 October 2012.
Acknowledgements
Funding is gratefully acknowledged from The Danish Medical Research Council (grant no. 10-094021) to URM. This work was also supported by US National Institutes of Health (NIH) grants R01AR056712, R01AR052779, P30 AR061303 and EPIGEN, and Association Francaise contre le Myopathies (AFM) to PLP. We apologize to the authors of those works that have been omitted in this forum due to space limits.
Author contributions
JF, PLP and URM wrote and edited the manuscript. LM edited the manuscript. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Candi
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Farup, J., Madaro, L., Puri, P. et al. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis 6, e1830 (2015). https://doi.org/10.1038/cddis.2015.198
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.198
This article is cited by
-
Muscle fibrosis as a prognostic biomarker in facioscapulohumeral muscular dystrophy: a retrospective cohort study
Acta Neuropathologica Communications (2023)
-
Injury-experienced satellite cells retain long-term enhanced regenerative capacity
Stem Cell Research & Therapy (2023)
-
Associations of muscle and adipose tissue parameters with long-term outcomes in middle and low rectal cancer: a retrospective cohort study
Cancer Imaging (2023)
-
Interleukin 4 improved adipose-derived stem cells engraftment via interacting with fibro/adipogenic progenitors in dystrophic mice
Cellular and Molecular Life Sciences (2023)
-
Prevalence and associated factors of possible sarcopenia and sarcopenia: findings from a Chinese community-dwelling old adults cross-sectional study
BMC Geriatrics (2022)
