
Abstract
TRAIL is a promising anticancer agent, capable of inducing apoptosis in a wide range of treatment-resistant tumor cells. In ‘type II’ cells, the death signal triggered by TRAIL requires amplification via the mitochondrial apoptosis pathway. Consequently, deregulation of the intrinsic apoptosis-signaling pathway, for example, by loss of Bax and Bak, confers TRAIL-resistance and limits its application. Here, we show that despite resistance of Bax/Bak double-deficient cells, TRAIL-treatment resulted in caspase-8 activation and complete processing of the caspase-3 proenzymes. However, active caspase-3 was degraded by the proteasome and not detectable unless the XIAP/proteasome pathway was inhibited. Direct or indirect inhibition of XIAP by RNAi, Mithramycin A or by the SMAC mimetic LBW-242 as well as inhibition of the proteasome by Bortezomib overcomes TRAIL-resistance of Bax/Bak double-deficient tumor cells. Moreover, activation and stabilization of caspase-3 becomes independent of mitochondrial death signaling, demonstrating that inhibition of the XIAP/proteasome pathway overcomes resistance by converting ‘type II’ to ‘type I’ cells. Our results further demonstrate that the E3 ubiquitin ligase XIAP is a gatekeeper critical for the ‘type II’ phenotype. Pharmacological manipulation of XIAP therefore is a promising strategy to sensitize cells for TRAIL and to overcome TRAIL-resistance in case of central defects in the intrinsic apoptosis-signaling pathway.
Similar content being viewed by others


Main
TRAIL (tumor necrosis factor-related apoptosis-inducing ligand/Apo2L) is capable of inducing cell death in a wide range of cancers resistant to conventional therapy without apparent toxic side effects to normal tissues.1 The expression of TRAIL-R1 is an independent prognostic parameter, for example, in colon carcinoma and high expression of TRAIL-R1 correlates with prolonged disease-free survival.2 Furthermore, TRAIL and the death ligands CD95L/FasL and TNFα sensitize tumor cells for ionizing radiation- and drug-induced apoptosis3, 4 albeit toxicity profiles may hamper (TNFα) or even preclude (CD95L/FasL) clinical use.
Death ligands initiate receptor oligomerization and formation of the death inducing signaling complex (DISC), resulting in activation of the initiator caspase-8. In type I cells, active caspase-8 directly mediates sufficient activation of the effector caspase-3 that triggers execution of apoptosis. In contrast, in type II cells, the death signal requires amplification via activation of the intrinsic cell death pathway5 through caspase-8-mediated cleavage and activation of Bid, a BH3-only protein of the Bcl-2 family.6
Thus, proteins of the Bcl-2 family are key regulators of both mitochondrial and death receptor-mediated apoptosis. Members of the family show homology in at least one of the four Bcl-2 homology (BH) domains. Anti-apoptotic proteins of this family (Bcl-2, Bcl-xL Bcl-w, Mcl-1 and Bfl-1/A1) are characterized by the presence of all four BH domains. Pro-apoptotic members can be subdivided into the multidomain BH123 homologs (Bax and Bak), and the proteins of the BH3-only subfamily (Bad, Bim, Puma, Noxa, Nbk/Bik, Bmf, Bnip3, Hrk and Bid).7
Activated, truncated (t)Bid triggers Bax activation to induce mitochondrial membrane permeabilization (MMP) accompanied by the release of apoptogenic factors from the mitochondrial inter-membrane space into the cytosol. One of these factors, cytochrome c, associates with APAF-1 and pro-caspase-9 to form the ‘apoptosome’, a platform that facilitates autocatalytic activation of caspase-9, which in turn triggers the effector caspases. Another pro-apoptotic factor released upon MMP is SMAC/DIABLO.8, 9 SMAC potentiates apoptosis by neutralizing cytosolic inhibitor of apoptosis proteins (IAPs). IAPs, a family of eight human analogs including cIAP1, cIAP2 and XIAP, the latter the most potent one,10 prevent inadvertent caspase activation XIAP does this by binding directly to caspases through its baculovirus IAP repeat (BIR) domain. Many human tumors express high levels of IAPs including XIAP and aberrant expression of IAPs has been linked to therapy resistance and poor prognosis.11 In addition to the upregulation of XIAP, deregulation of pro- and anti-apoptotic Bcl-2 family proteins is a central mechanism involved in TRAIL-resistance.12, 13 We have recently shown that loss of Bax, despite expression of its homolog Bak, confers resistance to TRAIL-induced apoptosis. However, inhibition of the endogenous Bak antagonist Mcl-1 enables TRAIL to kill cells via Bak.14
Here, we show that inhibition of the XIAP/proteasome pathway renders Bax/Bak double-deficient carcinoma cells sensitive to TRAIL. Because mitochondrial amplification of the death receptor signal is impossible in these cells, downregulation of XIAP converts type II into type I cells. These results show that in TRAIL-induced apoptosis, XIAP dictates the type II mode of cell death and that switching the cell death mode is a promising strategy to overcome TRAIL-resistance in cells with central defects in mitochondrial apoptosis signaling.
Results
To evaluate strategies to overcome resistance to TRAIL, we addressed the role of XIAP in a HCT116 colon cancer cell line model. We compared the impact of Mcl-1 and XIAP downregulation on TRAIL-treated isogenic HCT116 wt cells, HCT116 Bax-deficient cells (termed HCT116 Bax−) and cells devoid of Bax and Bak (termed HCT Bax−/Bak−).15 Specific loss of protein expression was achieved by knockout of bax16 and stable shRNA-mediated Bak knockdown17 and was verified by western blot analysis (Figures 1a and c can be compared because they represent the same membranes).

Silencing of XIAP by RNA interference enables TRAIL to induce Bax/Bak-independent cell death in type II cells. HCT116 wt, Bax− and Bax−/Bak− cells were transfected with control-, Mcl-1- or XIAP-siRNA. After 24 h, protein extracts were prepared and analyzed by western blot. Mcl-1 and XIAP downregulation and expression status for Bax and Bak for respective HCT116 cell lines is shown on the left of (a–c). Cells were then treated with 50 ng/ml TRAIL, cultured for additional 24 h and apoptotic cells were determined by flow cytometric measurement of the cellular DNA content. (a) HCT116 wt cells were sensitized for TRAIL-induced apoptosis upon Mcl-1 or XIAP downregulation. (b) Mcl-1 as well as XIAP downregulation overcomes TRAIL-resistance of HCT116 Bax− cells. (c) Downregulation of Mcl-1 fails to overcome TRAIL-resistance of HCT116 Bax−/Bak− cells. In contrast, knockdown of XIAP enables TRAIL to kill Bax/Bak-deficient HCT116 cells. Results are given as mean values±S.D. from three experiments for the respective cell lines in the right part of (a–c)
HCT116 cells were transfected with control-, Mcl-1- or XIAP-siRNA and downregulation of the respective proteins was confirmed by western blot analysis (figure1, left). Upon downregulation of Mcl-1 or XIAP, cells were incubated with 50 ng/ml TRAIL for 24 h and induction of apoptosis was analyzed by flow cytometric measurement of cells with a hypodiploid DNA content. TRAIL-treatment resulted in apoptotic DNA fragmentation in about 28% of the HCT116 wt cells. Sensitivity toward TRAIL was increased by knockdown of Mcl-1 and even more by XIAP knockdown, about 35 and 42% of the respective cells were apoptotic (Figure 1a, right).
Unlike HCT116 wt cells, Bax-deficient HCT116 cells were resistant to TRAIL-induced apoptosis. However, Mcl-1 or XIAP downregulation rendered these cells susceptible to TRAIL. Compared with 6% of control cells, TRAIL-induced apoptosis increased up to 32 and 41% upon Mcl-1 or XIAP downregulation, respectively (Figure 1b, right).
In contrast, TRAIL-resistance of HCT116 Bax−/Bak− cells could not be overcome by inhibition of Mcl-1. However, downregulation of XIAP strongly sensitized these double-deficient cells for TRAIL-induced apoptosis. Cells transfected with control- or Mcl-1-siRNA and treated with TRAIL showed apoptotic DNA fragmentation in <6% of the cells, which increased to 34% upon XIAP downregulation (Figure 1c, right).
Annexin V-FITC/propidium iodide (PI) staining of HCT116 wt and HCT116 Bax−/Bak− cells upon TRAIL-treatment confirmed that cell death occurs by apoptosis and that TRAIL-resistance of HCT116 Bax−/Bak− cells can be overcome by downregulation of XIAP (Supplementary Figure S1). Furthermore, XIAP downregulation overrides resistance of Bcl-2 or Bcl-xL overexpressing HCT116 cells, supporting our finding that upon TRAIL-treatment XIAP-deficient cells die independently of a Bcl-2 family protein regulated pathway (Supplementary Figure S2).
As caspase-8 and -3 are crucial for TRAIL-induced apoptosis, we next examined to which extent Bax/Bak deficiency impacts on processing of caspase-8 and -3. We treated HCT116 wt and Bax−/Bak− cells with TRAIL and analyzed the cleavage pattern of these caspases. Pro-caspase-8 and -3 levels are comparable in both cell lines, respectively (Figure 2a, left). Following TRAIL-treatment, pro-caspase-8 is cleaved in both cell lines and processed to its active subunits to a similar extent. Interestingly, in both cell lines, caspase-8 activation goes along with cleavage of the pro-caspase-3 zymogen, indicating that caspase-8 activation upon TRAIL-treatment is sufficient to cleave pro-caspase-3. Nevertheless, cleavage of pro-caspase-3 is accompanied by processing to its active p18/p16 subunits only in TRAIL-sensitive HCT116 wt cells. In contrast, TRAIL-resistant HCT116 Bax−/Bak− cells showed no processing of caspase-3 to its active subunits (Figure 2a, left). Thus, in contrast to cleavage of the pro-caspase-3 zymogen, which is independent of Bax and Bak, processing of pro-caspase-3 to its active subunits relies on an intact intrinsic mitochondrial pathway.

TRAIL-induces pro-caspase-3 processing in Bax/Bak double-deficient cells but fails to overcome inhibition by XIAP and proteasomal caspase-3 degradation. (a) HCT116 wt and Bax−/Bak− cells were treated with 50 ng/ml TRAIL and pro-caspase-8 and -3 processing were analyzed by immunoblotting. TRAIL-treatment induced cleavage of pro-caspase-8 and -3 in both cell lines. Cleavage of pro-caspase-8 is accompanied by generation of its active subunits. In contrast, processing of pro-caspase-3 to its active subunits is detectable only in HCT116 wt but not in HCT116 Bax−/Bak− cells (left). Additional downregulation of XIAP in combination with TRAIL caused full processing of pro-caspase-3 to its active subunits in HCT116 Bax−/Bak− cells (right). (b) HCT116 wt and Bax−/Bak− cells were incubated with the proteasome inhibitor MG132 prior to TRAIL-treatment. Upon inhibition of the proteasome, TRAIL-treatment resulted in full processing of pro-caspase-3 to its active subunits in both cell lines. (c, d) In addition to TRAIL, HCT116 wt and Bax−/Bak− cells were treated with 1 μM of MG132 (c) or with 1 μM of bortezomib (BZM) (d). Cells were cultured for 24 h, harvested and apoptotic cells were determined by flow cytometric measurement of cellular DNA content. HCT116 wt cells were sensitized for TRAIL-induced apoptosis by MG132 or BZM. Furthermore, inhibition of the proteasome enables TRAIL to kill Bax/Bak-deficient HCT116 cells
To analyze if the ubiquitin ligase activity of XIAP mediates degradation of the caspase-3 subunits we knocked down XIAP. In HCT116 Bax−/Bak− cells transfected with control siRNA, TRAIL-treatment resulted in pro-caspase-3 cleavage without generation of the active caspase-3. In contrast, downregulation of XIAP in combination with TRAIL caused full processing of pro-caspase-3 to its active subunits (Figure 2a, right). This indicates that caspase-8 activity triggered by TRAIL is sufficient to cleave pro-caspase-3 while caspase-3 activation is prevented by XIAP-mediated degradation of processed caspase-3.
To investigate proteasomal degradation of active caspase-3, we treated HCT116 wt and Bax−/Bak− cells with non-toxic concentrations of the proteasome inhibitor MG132. In HCT116 wt cells, TRAIL-induced caspase-3 activation is increased upon additional treatment with MG132 (Figure 2b, left). More importantly, in HCT116 Bax−/Bak− cells, TRAIL alone induced the cleavage of pro-caspase-3 without detectable levels of active subunits, whereas the combination induced full caspase-3 processing to its active subunits (Figure 2b, right).
In agreement with increased caspase-3 activation, TRAIL-induced apoptosis was increased in HCT116 wt cells upon additional inhibition of the proteasome. Moreover, MG132 treatment overcame TRAIL-resistance of Bax/Bax− deficient HCT116 cells (Figure 2c). Like MG132, bortezomib (BZM), the first therapeutic proteasome inhibitor, sensitized HCT116 wt cells for TRAIL-induced apoptosis and overcame TRAIL-resistance of HCT116 Bax−/Bak− cells (Figure 2d), indicating that sensitization towards TRAIL-induced apoptosis by inhibition of the proteasome is caused by stabilization of active caspase-3 and does not rely on the intrinsic apoptotic pathway.
To further investigate the mode of caspase-3 activation in the presence or absence of XIAP in relation to type I/II apoptosis signaling, we analyzed TRAIL-induced apoptosis in cells subjected to caspase-8 and Bid downregulation. In HCT116 wt cells, TRAIL-induced apoptosis was accompanied by pro-caspase-8 processing, Bid cleavage and processing of pro-caspase-3 to active subunits (Figure 3a). Downregulation of caspase-8 or Bid inhibited induction of apoptosis. But in contrast to caspase-8 downregulation, which blocked TRAIL-induced pro-caspase-3 cleavage, Bid knockdown did not impair pro-caspase-3 cleavage, indicating that most of the detected pro-caspase-3 processing occurs upstream of the mitochondrial amplification loop. However, although pro-caspase-3 was cleaved, subunits were decreased after Bid downregulation (Figure 3a, right), indicating that processing of pro-caspase-3 to the active subunits depends on Bid.

Upon XIAP silencing, TRAIL-induced apoptosis and caspase-3 activation become independent of Bid, Bax and Bak. (a) HCT116 wt cells were transfected with control-, caspase-8- or Bid-siRNA and subsequently incubated with TRAIL. TRAIL-induced DNA fragmentation was blocked by downregulation of either caspase-8 or Bid (left). Western blot analysis confirmed downregulation of the respective proteins (right). Furthermore, western blot analysis showed that knockdown of caspase-8 blocked pro-caspase-3 cleavage. In contrast, knockdown of Bid did not inhibit pro-caspase-3 cleavage but processing of caspase-3 to its active subunits. (b) HCT116 wt were treated as in (a). In addition, XIAP was knocked down. Upon XIAP knockdown, TRAIL-induced apoptosis was inhibited by caspase-8 downregulation but not anymore by Bid downregulation (left). In this setting, TRAIL-induced activation of pro-caspase-3, indicated by the active subunit, depends on caspase-8 but was independent of Bid. (c) HCT116 Bax−/Bak− cells were treated as in (b). Upon XIAP downregulation, TRAIL-induced apoptosis (left) is accompanied by caspase-3 activation (right). Both can be blocked by knockdown of caspase-8. In contrast, caspase-3 activation and apoptosis was not blocked by downregulation of Bid. Thus, inhibition of XIAP enables TRAIL to activate caspase-3 and to induce apoptosis despite combined loss of Bid, Bax and Bak
Additional knockdown of XIAP restored the induction of apoptosis by TRAIL in HCT116 wt cells with Bid knockdown (Figure 3b, left) and resulted in complete cleavage of pro-caspase-8 and pro-caspase-3, accompanied by high levels of the p18 caspase-3 subunit (Figure 3b, right). Knockdown of caspase-8 impaired induction of apoptosis, Bid cleavage and caspase-3 cleavage in the absence or presence of XIAP knockdown (Figures 3a and b). These results were confirmed in HCT116 Bax−/Bak− cells in which XIAP downregulation overcomes TRAIL-resistance (Figure 3c, left) and induction of apoptosis is accompanied by pro-caspase-8 and Bid cleavage. Furthermore, absence of XIAP facilitated processing of pro-caspase-3 to the active subunits (Figure 3c, right). These events were inhibited by caspase-8 knockdown but not by knockdown of Bid. Thus, inhibition of XIAP enables TRAIL to activate caspase-3 and to induce apoptosis despite combined loss of Bid, Bax and Bak, clearly demonstrating that cell death induction is independent of the intrinsic apoptotic pathway and follows a type I mode.
Next, we compared the time-dependent regulation of TRAIL-induced apoptosis in HCT116 wt and in HCT116 Bax−/Bak− cells after XIAP downregulation. Western blot analysis of TRAIL-treated HCT116 wt cells revealed early caspase-8 processing, accompanied by Bid activation, indicated by increased tBid levels detectable already after 4 h of treatment (Figure 4a, left). Furthermore, TRAIL-induced caspase-8 and Bid activation were accompanied by cytochrome c and SMAC release coinciding with caspase-3 activation and PARP cleavage (Figure 4a, left). Western blot analysis further revealed a time-dependent degradation of XIAP in response to TRAIL, which may reflect autoubiquitination and subsequent proteasomal degradation or caspase-mediated cleavage.18

Mitochondrial permeability transition precedes cell death induction in HCT 116 cells but is not involved in TRAIL-induced apoptosis after downregulation of XIAP in Bax/Bak-deficient HCT 116 cells. (a) 24 h after transfection of HCT116 wt with control siRNA (left) and HCT116 Bax−/Bak− with XIAP-siRNA (right), cells were treated with TRAIL and cultured for the indicated time. Western blot analysis revealed early processing of caspase-8 and -3 and cleavage of Bid and PARP in both cell lines. In HCT116 wt cells, cytochrome c and SMAC release into the cytosol was seen 4 h after TRAIL-treatment. In contrast, cytochrome c and SMAC release was a late event in HCT116 Bax−/Bak− cells. (b) Cells were treated as described in (a), harvested and percentages of cells with loss of ΔΨm were determined by flow cytometric measurement of JC-1 fluorescence. In addition, percentages of apoptotic cells were determined by flow cytometric measurement of hypodiploid cells. These analyses revealed that in HCT116 wt cells, loss of mitochondrial membrane potential precedes apoptotic DNA fragmentation. This is in contrast to HCT116 Bax−/Bak− cells with XIAP downregulation, where loss of mitochondrial membrane potential, release of cytochrome c and SMAC are late events. Data expressed as mean values±S.D. from three experiments
To analyze dissipation of mitochondrial membrane potential (ΔΨm) upon TRAIL-treatment, cells were incubated with the fluorochrome JC-1, which exhibits membrane potential-dependent accumulation in mitochondria. Measurement of JC-1-fluorescence intensity by flow cytometry showed a time-dependent accumulation of HCT116 wt cells with disruption of ΔΨm upon TRAIL-treatment. This was an early event detectable already after 6 h of treatment that precedes DNA fragmentation (Figure 4b, left).
In analogy, HCT116 Bax−/Bak− cells with downregulated XIAP showed caspase-8 and Bid processing as early events during TRAIL-induced apoptosis, detectable 4 h after treatment. In contrast to HCT116 wt cells, Bid cleavage did, however, not coincide with cytochrome c or SMAC release. Both occurred only late upon TRAIL-treatment, detectable after 12 h (Figure 4a, right). This indicates that tBid failed to trigger the mitochondrial pathway. Nevertheless, TRAIL-treatment in the absence of XIAP resulted in early caspase-3 activation, which coincided with PARP cleavage (Figure 4a, right). Furthermore, despite early induction of apoptosis but in line with the delayed kinetic of cytochrome c and SMAC release, MMP was also a late event in these cells. Interestingly, and in contrast to HCT116 wt cells, breakdown of ΔΨm followed DNA fragmentation upon TRAIL-treatment in HCT116 Bax−/Bak− cells (Figure 4b, right), indicating that MMP occurs late and coincides with cellular demise instead of playing an early, regulatory role. In summary, XIAP downregulation facilitates TRAIL-induced apoptosis signaling that is independent of MMP, cytochrome c and SMAC release, which all seem to be secondary effects.
Time response analysis of Annexin V-FITC/PI staining upon TRAIL-treatment confirms the different cell death mode. Early apoptotic cells were detectable already 4 h after TRAIL-treatment in HCT116 wt cells and in HCT116 Bax−/Bak− cells with downregulated XIAP. However, HCT116 wt cells tend toward early occurrence of a late apoptotic phenotype, detectable after 8 h of TRAIL-treatment. In contrast, late apoptotic HCT116 Bax−/Bak− cells were detectable after 12 h at the earliest (Supplementary Figure S3). Interestingly at this time point, HCT116 Bax−/Bak− cells also display MMP and cytochrome c release (Figures 4a and b). Taken together, the results indicate that MMP and cytochrome c release, which occur early in type II and late in type I cells, are accompanied by a late apoptotic/necrotic phenotype of the cells.
Given the therapeutic impact of our findings, we next asked if small molecules, known to downregulate or inhibit XIAP, can overcome the resistance of Bax/Bak-deficient cells. The antitumor agent Mithramycin A (Mit A) sensitizes various cancer cell lines to TRAIL-mediated apoptosis by downregulation of XIAP.19 To confirm downregulation of XIAP by Mit A, we treated HCT116 wt and HCT116 Bax−/Bak− cells with different concentrations of Mit A for 24 h. Subsequent analysis of XIAP levels confirmed reduced XIAP expression (Figure 5a). This is accompanied by an increased TRAIL sensitivity in HCT116 wt cells, mainly due to Bax/Bak-dependent, additive toxicity of Mit A. Furthermore, Mit A treatment efficiently overcame TRAIL-resistance of Bax/Bak-deficient HCT116 cells (Figure 5a, lower panel).

Downregulation or inhibition of XIAP by Mithramycin A or LBW-242, respectively, overcomes TRAIL-resistance. (a) HCT116 wt and Bax−/Bak− cells were pre-incubated with indicated concentrations of Mit A and downregulation of XIAP was observed upon immunoblotting (upper panel). Cells were then treated with TRAIL and cultured for additional 24 h. Measurement of apoptotic cells by flow cytometry revealed that Mit A sensitized Bax/Bak-deficient cells for TRAIL-induced apoptosis. Data expressed as mean values±S.D. from three experiments (lower panel). (b) To inhibit XIAP function, both cell lines were treated with 10 μM of the SMAC mimetic LBW-242 in addition to TRAIL. In HCT116 wt cells, TRAIL-induced caspase-3 processing was increased by LBW-242 (upper left). Increased caspase-3 processing was paralleled by enhanced induction of apoptosis (lower left). In HCT116 Bax−/Bak− cells, pro-caspase-3 was cleaved upon TRAIL-treatment. Proteolytic fragments, however, were detectable only upon addition of LBW-242 (upper right). Caspase-3 activation resulted in induction of apoptosis, indicating that LBW-242 can overcome TRAIL-resistance of Bax/Bak-deficient HCT116 cells (lower right). Statistical significances were determined using an unpaired Student’s t-test. Levels of statistical significance are indicated with asterisks (*P<0.05; **P<0.01)
Additionally, we targeted XIAP directly by use of the SMAC mimetic LBW-242, a synthetic small molecule that binds to and inhibits IAPs. Treatment of HCT116 cells with LBW-242 in addition to TRAIL reduced XIAP expression (Figure 5b, upper panel, lanes 2 and 4), which might be due to cleavage of XIAP by caspase-3 in a feedback loop or autoubiquitylation and proteasomal degradation. Furthermore, inhibition of XIAP resulted in an increase of TRAIL-induced apoptosis in HCT116 wt cells and, more importantly, overcame TRAIL-resistance of Bax/Bak double-deficient cells (Figure 5b, lower panel). The re-sensitization of HCT116 Bax−/Bak− cells is caused by the complete processing of pro-caspase-3 since active subunits were detectable only after the combined treatment with TRAIL and LBW-242 (Figure 5b, upper panel). Thus, like BZM, MG132 or Mit A, LBW-242 overcomes TRAIL-resistance and facilitates TRAIL-induced caspase-3 activation and induction of apoptosis in a type I manner.
To analyze if XIAP plays a similar role in cell death induced by other death receptors, we treated cells with TNFα or CD95L/FasL. In HCT116 wt cells, TNFα treatment resulted in weak induction of apoptosis; ∼10% of the cells were apoptotic (Figure 6a). Compared with TNFα, CD95L/FasL-induced DNA fragmentation was more pronounced and 24% of the cells underwent apoptosis. However, upon XIAP downregulation TNFα as well as CD95L/FasL-induced apoptosis was increased up to 16% and 37%, respectively. In contrast to HCT116 wt cells, neither TNFα nor CD95L/FasL induced apoptosis in HCT116 Bax−/Bak− cells. However, resistance was overcome by XIAP downregulation, resulting in around 17% and 25% apoptotic cells upon TNFα or CD95L/FasL treatment, respectively (Figure 6b). Altogether, these data show that downregulation of XIAP enables death receptors to kill type II cells independently of the mitochondrial death machinery by switching the apoptotic-signaling pathway from type II to type I.

XIAP mediates resistance to TNFα and CD95/FasL-induced apoptosis in Bax/Bak-deficient HCT116 cells. (a) HCT116 wt cells were transfected with control or XIAP-siRNA and cultured for 24 h. After knockdown of XIAP, cells were treated with TNFα or CD95L/FasL and cultured for additional 24 h. Measurement of apoptotic DNA fragmentation revealed that HCT116 wt cells were sensitized toward TNFα- or CD95L/FasL-induced apoptosis upon downregulation of XIAP. (b) HCT116 Bax−/Bak− cells were treated as in (a). Measurement of apoptotic cells revealed that HCT116 Bax−/Bak− cells transfected with control siRNA were resistant towards TNFα- or CD95L/FasL-induced cell death. However, XIAP downregulation enabled TNFα and CD95L/FasL (TRAIL) to induce apoptosis despite Bax/Bak deficiency. Data expressed as mean values±S.D. from three experiments are shown. Levels of statistical significance are indicated with asterisks (*P<0.05; **P<0.01)
Discussion
Strategies to overcome TRAIL-resistance comprise inhibition of pro-survival signaling, for example, the NF-κB pathway,20 inhibition of the proteasome21 or inhibition of histone deacetylases.22 The kinase inhibitors Roscovitine or Sorafenib sensitize cancer cells to TRAIL through pleiotropic mechanisms comprising facilitated DISC formation and caspase-8 activation, downregulation of cFLIP and XIAP and suppression of Mcl-1.14, 23, 24 Strategies directly targeting the Bcl-2 family include BH3 mimetics like ABT-737 that bind to the hydrophobic groove exposed at the surface of antiapoptotic proteins, block their pro-survival function and potentiate TRAIL-mediated apoptosis.25 However, cell death still relies on the presence of Bax and Bak as MMP mediators. Inhibition of IAPs is another promising approach to sensitize treatment-resistant tumor cells for TRAIL. Especially the inhibition of XIAP by small molecular SMAC mimetics has been shown to synergize with TRAIL and to potentiate TRAIL-induced apoptosis in various cancer cells.26, 27
We addressed not only the role but also the pharmacological targeting of XIAP in type I/II signaling. In contrast to type I cells, type II cells require activation of a mitochondrial amplification loop to achieve full caspase activation that depends on activation of Bid. Interestingly, a recent report showed that loss of XIAP alleviates the need for Bid in CD95/Fas-induced hepatocyte apoptosis in mice.28
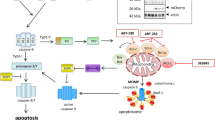
Here, we show that TRAIL-resistance of type II carcinoma cells with central mitochondrial death signaling defects was overcome by inhibition of XIAP. This enabled TRAIL to induce apoptosis in cells overexpressing the anti-apoptotic proteins Bcl-2 or Bcl-xL or in cells deficient for the pro-apoptotic proteins Bax, Bak, and Bid. Under these conditions, apoptosis is not accompanied by MMP and cytochrome c or SMAC release, indicating that cell death induction is independent of the intrinsic pathway. This is in contrast to Bax/Bak positive cells, where MMP and cytochrome c release precedes apoptosis. Thus, downregulation of XIAP bypasses a cell death blockade at the level of the mitochondria by switching apoptosis signaling from type II to type I. This observation significantly extends other studies that identified the level of caspase-8 activation upon death receptor activation as critical discriminator between type I/II apoptosis signaling.5 According to this model, type I cells display strong activation of caspase-8 upon death receptor ligation to directly activate caspase-3, thereby bypassing the mitochondria. In contrast, type II cells would generate only low amounts of active caspase-8 at the DISC and strong activation of caspase-3 occurs at a level secondary to mitochondrial events. However, in HCT116 cells caspase-8 activation induced by TRAIL is sufficient to cleave pro-caspase-3 regardless of loss of Bax/Bak expression. Nevertheless, pro-caspase-3 is processed to its active subunits only in cells with an intact mitochondrial apoptotic-signaling pathway. In the absence of MMP, caspase-3 subunits were degraded by the XIAP/proteasome pathway. Upon activation of the mitochondria through Bax/Bak, SMAC, released into the cytosol, inhibits XIAP and prevents degradation of caspase-3 subunits resulting in strong caspase-3 activation (Figure 7). Consequently, inhibition of XIAP enables caspase-8 to directly induce sufficient amounts of active caspase-3 in Bax/Bak-deficient HCT116 cells thereby making SMAC release dispensable. Taken together, we demonstrate that it is not the extent of active caspase-8 but the activity of XIAP, which makes a distinction between cell death induction via a type II or type I pathway.

Model of XIAP as a switch between type I and type II signaling. See text for details. Mit A, Mithramycin A; BZM, bortezomib
Pharmacological targeting of this type I/II switch would overcome the need for Bax/Bak proficiency. In this vein, we show that processing of pro-caspase-3 to its active subunits without activation of the mitochondria can be enforced by inhibition of the proteasome. Proteasome inhibitors may sensitize tumor cells for TRAIL by enhanced cell surface expression of TRAIL-R1 (DR4) and TRAIL-R2 (DR5), enhanced activation of the initiator caspase-8 and downregulation of cFLIP.29, 30 Furthermore, bortezomib-induced stabilization of the Bax protein as well as increased levels of Nbk/Bik and Bim were implicated in sensitization for TRAIL killing.31, 32 Our data suggest that the stabilization of active caspase-3 by blocking its proteasomal degradation, by either inhibiting proteasomal activity or preventing XIAP-mediated ubiquitylation, is the main mechanism responsible for sensitization. Interestingly, the caspase-3 fragments, which accumulate upon proteasome inhibition in addition to TRAIL-treatment, seem to be unubiquitinated (Figure 2a). Caspases have been shown to inhibit XIAP by cleavage33 and this feedback loop might be responsible for the accumulation of unubiquitinated active caspase-3 upon inhibition of the proteasome. However, inhibition of the proteasome or XIAP result in sufficient generation of active caspase-3 and mediate a switch from a type II to a type I mode of cell death. This notion is supported by experiments where we inhibited XIAP function by the use of the synthetic small molecule IAP inhibitor LBW-242 that mimics the activity of SMAC. LBW-242 is a promising compound to overcome drug resistance.34, 35 Here, we show that LBW-242 overcomes resistance of Bax/Bak-deficient cancer cells and re-sensitizes these cells to TRAIL. Loss of both Bax or Bak expression has been reported for a number of tumors.36, 37, 38, 39 Whereas loss of Bax is a frequent event in human cancer, for example, in colon cancers having a microsatellite mutator phenotype, Bak expression persists in most cancers and loss of both is a rare event. Notably, loss of Bax, despite expression of Bak, is sufficient for tumor cells to acquire TRAIL-resistance. In addition, the intrinsic apoptotic pathway of tumor cells can be blocked by other mechanisms, for example, upregulation of Bcl-xL (which efficiently inhibits both, Bax and Bak) or endogenous Bak-inhibitors Mcl-1 or VDAC2 in Bax-deficient carcinoma.14, 15, 40 The data presented in this study indicate that use of SMAC mimetics is a suitable strategy to antagonize therapy resistance caused by all these central defects in the intrinsic apoptosis machinery and delineates the combined use of TRAIL together with SMAC mimetics as a useful strategy. Altogether, these data show that targeting of XIAP enables death receptor signaling to kill type II cells via a type I pathway and define XIAP as a crucial decision point between these two cell death pathways.
Materials and Methods
Cell culture
HCT116 wild-type cells and the isogenic knockout subline HCT116-Bax−/− were kindly provided by Dr. Bert Vogelstein, Johns Hopkins Cancer Center, Baltimore, MD, USA. The stable knockdown of Bak shRNA was achieved in HCT116-Bax k.o. cells yielding Bax−/Bak− cells.17 Mock transfectants showed an identical apoptosis sensitivity as compared with HCT116 parental cells (HCT116 wt). Cells were grown in DMEM medium supplemented with 10% fetal calf serum, 100 000 U/l penicillin and 0.1 g/l streptomycin at 37 °C with 5% CO2 in a fully humidified atmosphere. Media and culture reagents were from Invitrogen (Karlsruhe, Germany).
Antibodies and reagents
SMAC/Diablo mAb (#2954), C-8 (1C12) mAb (#9746) and XIAP mAb (3B6) (#2045) were purchased from Cell Signaling Technology, Inc. (Boston, MA, USA). Bcl-2 antibody (NCL-bcl-2) was purchased from Novocastra Laboratories (Newcastle, UK). The anti-Bak Ab (clone TC102) was from Calbiochem (Darmstadt, Germany). Anti-Bax Ab (clone YTH-2D2) was purchased from Trevigen (Gaithersburg, MD, USA) and anti-Mcl-1H-260 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-Bcl-x Ab was from BD Biosciences Pharmingen (San Diego, CA, USA). Anti-caspase-8 (clone 12F5) was from Alexis (Grünberg, Germany). The anti-C-3 Ab and anti-Bid Ab were from R&D Systems GmbH (Wiesbaden-Nordenstadt, Germany), anti-actin Ab was from Sigma-Aldrich (Taufkirchen, Germany). Secondary anti-rabbit, anti-goat and anti-mouse horseradish peroxidase-conjugated antibodies were from Promega (Mannheim, Germany) or Southern Biotechnology Associates (Birmingham, AL, USA). RNase A was from Roth (Karlsruhe, Germany). The recombinant human TRAIL was from R&D Systems GmbH (Wiesbaden-Nordenstadt, Germany). TNFα and SuperFasLigand were from Alexis (Grünberg, Germany). MG132 was purchased from Sigma-Aldrich, Mit A was from Genaxxon BioScience GmbH (Ulm, Germany). The LBW-242 SMAC mimetic was kindly provided by Novartis (Basel, Switzerland).
RNA interference
On-target plus XIAP, Mcl-1, caspase-8, Bid and control siRNA were purchased from Dharmacon (Lafayette, LA, USA). Transfection of the cells was carried out by use of DharmaFECT Transfection Reagent according to the manufacturer’s instructions. Downregulation of the respective proteins was confirmed by immunoblotting 24 h after transfection.
Immunoblotting
After trypsination, cells were washed twice with ice-cold PBS and lysed in 10 mM Tris-HCl pH 7.5, 137 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 μM pepstatin, 1 μM leupeptin and 0.1 mM phenylmethyl sulfonylfluoride (PMSF). Protein concentration was determined using the bicinchoninic acid assay. Equal amounts of protein were separated by SDS-PAGE, electroblotted and visualized as described.41 For analysis of cytochrome c and SMAC release, cytosolic extracts were prepared according to a method described previously.41 Briefly, after induction of apoptosis, cells were harvested in PBS, equilibrated in hypotonic buffer (20 mM HEPES pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA) supplemented with 0.1 mM PMSF and 0.75 mg/ml digitonin (Sigma-Aldrich) and incubated on ice for 3 min. Debris was pelleted by centrifugation at 10 000 × g at 4 °C for 5 min and the supernatant was subjected to western blot analysis.
Measurement of mitochondrial permeability transition
Cells were harvested and collected by centrifugation at 300 × g at 4 °C for 5 min. Mitochondrial permeability transition was determined by staining the cells with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide; Molecular Probes, Leiden, The Netherlands), a cationic dye that exhibits membrane potential-dependent accumulation in mitochondria, as described.41 Mitochondrial permeability transition was then quantified by flow cytometric determination of cells with decreased red fluorescence, that is, with mitochondria displaying a reduced membrane potential (ΔΨm).
Measurement of apoptotic cell death by flow cytometry
DNA fragmentation was measured as described.15 Briefly, cells were collected by centrifugation at 300 × g for 5 min, washed with PBS at 4 °C, and fixed in PBS/2% (vol/vol) formaldehyde on ice for 30 min. After fixation, cells were incubated with ethanol/PBS (2 : 1, vol/vol) for 15 min, pelleted, and resuspended in PBS containing 40 μg/ml RNase A. After incubation for 30 min at 37 °C, cells were pelleted and finally resuspended in PBS containing 50 μg/ml PI. DNA fragmentation was quantified by flow cytometric determination of hypodiploid DNA. Data were collected and analyzed using a FACScan (Becton Dickinson, Heidelberg, Germany) equipped with the CELLQuest software. Data are given in % hypoploidy (subG1), which reflects the number of apoptotic cells. Alternatively, cell death was determined by staining cells with Annexin V-fluorescein isothiocyanate (FITC) and counterstaining with PI.15 Briefly, cells were washed twice with cold PBS and resuspended in 10 mM N-[2-hydroxyethyl]piperazin-N′-3[propansulfonicacid]/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2 at 1 × 106 cells/ml. Next, 5 μl of Annexin V-FITC (BD PharMingen, Heidelberg, Germany) and 10 μl PI (20 g/ml, Sigma-Aldrich) were added. Analyses were performed using a FACScan (Becton Dickinson) and CELLQuest analysis software. Results are given in % of cells.
Abbreviations
- Bak:
-
Bcl-2 homologous antagonist/killer
- Bax:
-
Bcl-2-associated x protein
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-xL:
-
long splice variant of Bcl-x
- BH3:
-
Bcl-2 homology region 3
- Bid:
-
BH3-interacting domain death agonist
- BZM:
-
bortezomib
- CD95/FasL:
-
cluster of differentiation 95/fibroblast-associated ligand
- IAP:
-
inhibitor of apoptosis proteins
- Mcl-1:
-
myeloid cell leukemia 1
- Mit A:
-
Mithramycin A
- MMP:
-
mitochondrial membrane permeabilization
- PI:
-
propidium iodide
- SMAC/DIABLO:
-
second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI
- TNF:
-
tumor necrosis factor
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- XIAP:
-
X-linked inhibitor of apoptosis
References
Gonzalvez F, Ashkenazi A . New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010; 29: 4752–4765.
Strater J, Hinz U, Walczak H, Mechtersheimer G, Koretz K, Herfarth C et al. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin Cancer Res 2002; 8: 3734–3740.
Schmelz K, Wieder T, Tamm I, Muller A, Essmann F, Geilen CC et al. Tumor necrosis factor alpha sensitizes malignant cells to chemotherapeutic drugs via the mitochondrial apoptosis pathway independently of caspase-8 and NF-kappaB. Oncogene 2004; 23: 6743–6759.
Marini P, Schmid A, Jendrossek V, Faltin H, Daniel PT, Budach W et al. Irradiation specifically sensitises solid tumour cell lines to TRAIL mediated apoptosis. BMC Cancer 2005; 5: 5.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675–1687.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
van Delft MF, Huang DC . How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res 2006; 16: 203–213.
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43–53.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102: 33–42.
Deveraux QL, Reed JC . IAP family proteins—suppressors of apoptosis. Genes Dev 1999; 13: 239–252.
LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG . IAP-targeted therapies for cancer. Oncogene 2008; 27: 6252–6275.
LeBlanc H, Lawrence D, Varfolomeev E, Totpal K, Morlan J, Schow P et al. Tumor-cell resistance to death receptor—induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med 2002; 8: 274–281.
Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res 2004; 64: 3517–3524.
Gillissen B, Wendt J, Richter A, Richter A, Muer A, Overkamp T et al. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax−deficient carcinoma. J Cell Biol 2010; 188: 851–862.
Gillissen B, Essmann F, Hemmati PG, Richter A, Richter A, Oztop I et al. Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J Cell Biol 2007; 179: 701–715.
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B . Role of BAX in the apoptotic response to anticancer agents. Science 2000; 290: 989–992.
Subramanian T, Chinnadurai G . Pro-apoptotic activity of transiently expressed BCL-2 occurs independent of BAX and BAK. J Cell Biochem 2003; 89: 1102–1114.
Hornle M, Peters N, Thayaparasingham B, Vorsmann H, Kashkar H, Kulms D . Caspase-3 cleaves XIAP in a positive feedback loop to sensitize melanoma cells to TRAIL-induced apoptosis. Oncogene 2010; 30: 575–587.
Lee TJ, Jung EM, Lee JT, Kim S, Park JW, Choi KS et al. Mithramycin A sensitizes cancer cells to TRAIL-mediated apoptosis by down-regulation of XIAP gene promoter through Sp1 sites. Mol Cancer Ther 2006; 5: 2737–2746.
Keane MM, Rubinstein Y, Cuello M, Ettenberg SA, Banerjee P, Nau MM et al. Inhibition of NF-kappaB activity enhances TRAIL mediated apoptosis in breast cancer cell lines. Breast Cancer Res Treat 2000; 64: 211–219.
Chen KF, Yeh PY, Hsu C, Hsu CH, Lu YS, Hsieh HP et al. Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem 2009; 284: 11121–11133.
Inoue S, Mai A, Dyer MJ, Cohen GM . Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res 2006; 66: 6785–6792.
Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007; 12: 66–80.
Ortiz-Ferron G, Yerbes R, Eramo A, Lopez-Perez AI, De Maria R, Lopez-Rivas A . Roscovitine sensitizes breast cancer cells to TRAIL-induced apoptosis through a pleiotropic mechanism. Cell Res 2008; 18: 664–676.
Huang S, Sinicrope FA . BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 2008; 68: 2944–2951.
Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004; 5: 25–35.
Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res 2009; 69: 2425–2434.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009; 460: 1035–1039.
He Q, Huang Y, Sheikh MS . Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax−proficient and -deficient cells. Oncogene 2004; 23: 2554–2558.
Brooks AD, Jacobsen KM, Li W, Shanker A, Sayers TJ . Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol Cancer Res 2010; 8: 729–738.
Liu FT, Agrawal SG, Gribben JG, Ye H, Du MQ, Newland AC et al. Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood 2008; 111: 2797–2805.
Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J, Kraft AS . The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim. Mol Cancer Ther 2005; 4: 443–449.
Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC . Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J 1999; 18: 5242–5251.
Hundsdoerfer P, Dietrich I, Schmelz K, Eckert C, Henze G . XIAP expression is post-transcriptionally upregulated in childhood ALL and is associated with glucocorticoid response in T-cell ALL. Pediatr Blood Cancer 2010; 55: 260–266.
Chauhan D, Neri P, Velankar M, Podar K, Hideshima T, Fulciniti M et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM). Blood 2007; 109: 1220–1227.
Sturm I, Kohne CH, Wolff G, Petrowsky H, Hillebrand T, Hauptmann S et al. Analysis of the p53/BAX pathway in colorectal cancer: low BAX is a negative prognostic factor in patients with resected liver metastases. J Clin Oncol 1999; 17: 1364–1374.
Kondo S, Shinomura Y, Miyazaki Y, Kiyohara T, Tsutsui S, Kitamura S et al. Mutations of the bak gene in human gastric and colorectal cancers. Cancer Res 2000; 60: 4328–4330.
Krajewska M, Moss SF, Krajewski S, Song K, Holt PR, Reed JC . Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res 1996; 56: 2422–2427.
Mrozek A, Petrowsky H, Sturm I, Kraus J, Hermann S, Hauptmann S et al. Combined p53/Bax mutation results in extremely poor prognosis in gastric carcinoma with low microsatellite instability. Cell Death Differ 2003; 10: 461–467.
Plötz M, Gillissen B, Hossini AM, Daniel PT, Eberle J . Disruption of the VDAC2-Bak interaction by Bcl-x(S) mediates efficient induction of apoptosis in melanoma cells. Cell Death Differ 2012; 19: 1928–1938.
von Haefen C, Wieder T, Essmann F, Schulze-Osthoff K, Dorken B, Daniel PT . Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8-driven mitochondrial amplification loop. Oncogene 2003; 22: 2236–2247.
Acknowledgements
This study was supported by the Deutsche Forschungsgemeinschaft, BMBF-MedSys and the Deutsche Krebshilfe. We thank Novartis, Basel, for kindly providing the SMAC mimetic LBW-242.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Ciliberto
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gillissen, B., Richter, A., Richter, A. et al. Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent ‘type I’ mode. Cell Death Dis 4, e643 (2013). https://doi.org/10.1038/cddis.2013.67
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.67
Keywords
This article is cited by
-
Targeting bone morphogenetic protein receptor 2 sensitizes lung cancer cells to TRAIL by increasing cytosolic Smac/DIABLO and the downregulation of X-linked inhibitor of apoptosis protein
Cell Communication and Signaling (2019)
-
The prodomain of caspase-3 regulates its own removal and caspase activation
Cell Death Discovery (2019)
-
ZBP-89 and Sp1 contribute to Bak expression in hepatocellular carcinoma cells
BMC Cancer (2018)
-
Hypertonicity-enforced BCL-2 addiction unleashes the cytotoxic potential of death receptors
Oncogene (2018)
-
MicroRNA-519a-3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells
Cell Death & Disease (2017)

{kind=link}
{kind=link}
{kind=link}