
Abstract
Induction of cell death and inhibition of cell survival are the main principles of cancer therapy. Resistance to chemotherapeutic agents is a major problem in oncology, which limits the effectiveness of anticancer drugs. A variety of factors contribute to drug resistance, including host factors, specific genetic or epigenetic alterations in the cancer cells and so on. Although various mechanisms by which cancer cells become resistant to anticancer drugs in the microenvironment have been well elucidated, how to circumvent this resistance to improve anticancer efficacy remains to be defined. Autophagy, an important homeostatic cellular recycling mechanism, is now emerging as a crucial player in response to metabolic and therapeutic stresses, which attempts to maintain/restore metabolic homeostasis through the catabolic lysis of excessive or unnecessary proteins and injured or aged organelles. Recently, several studies have shown that autophagy constitutes a potential target for cancer therapy and the induction of autophagy in response to therapeutics can be viewed as having a prodeath or a prosurvival role, which contributes to the anticancer efficacy of these drugs as well as drug resistance. Thus, understanding the novel function of autophagy may allow us to develop a promising therapeutic strategy to enhance the effects of chemotherapy and improve clinical outcomes in the treatment of cancer patients.
Similar content being viewed by others
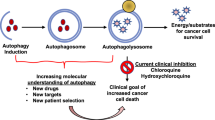

Facts
-
The induction of autophagy in response to metabolic and therapeutic stresses can have a prodeath or a prosurvival role, which contributes to the anticancer efficacy of these drugs as well as drug resistance.
-
Anticancer drugs induce different effects of autophagy on cell survival in different cancer types.
-
Autophagy as a prosurvival and resistance mechanism against chemotherapy treatment.
-
Autophagy-mediated cell death mechanism contributes to efficacy of anticancer drugs.
-
Targeting autophagy will hopefully provide a promising therapeutic strategy to circumvent resistance and enhance the effects of anticancer therapies for cancer patients.
Open Questions
-
Whether we should try to enhance or inhibit autophagy in cancer treatment?
-
Chloroquine and its derivative: just act as autophagy inhibitors?
-
Autophagy is shown to precede apoptosis or act in parallel with this cellular process in addition to be an alternative mechanism to cell death when apoptosis is inhibited. Therefore, the autophagy induction may exert other possibilities, which should be considered in the design of new treatments for the malignancies.
Resistance to anticancer drugs is a common clinical issue in the treatment of patients with cancer. Drug resistance, intrinsic or acquired, can be attributed to a wide variety of mechanisms including tumor cell heterogeneity, drug efflux and metabolism and tumor microenvironment stress-induced genetic or epigenetic alterations as a cellular response to drug exposure (Figure 1).1, 2 Among these mechanisms, the response or adaptation of cancer cell itself to anticancer drug-induced tumor microenvironment stresses is a vital cause for chemotherapy resistance.

A summary of the approaches by which cancer cells become resistant to chemotherapy and various kinds of genotoxic or metabolic stresses
Autophagy is an evolutionarily conserved catabolic process in which portions of cytosol and organelles are sequestered into a double-membrane vesicle and delivered to the lysosome for bulk degradation.3, 4, 5, 6 In this review, the term ‘autophagy’ refers to macroautophagy. The role of autophagy in regulating cancer cell death or survival remains controversial. Current evidence supports the idea that constitutive autophagy can act as a cellular housekeeper to eliminate damaged organelles and recycle macromolecules, thus protecting against cancer, particularly during malignant transformation and carcinogenesis. In established tumors, autophagy can function as a prosurvival pathway in response to metabolic stresses such as nutrient deprivation, hypoxia, absence of growth factors and the presence of chemotherapy or some targeted therapies that might mediate resistance to anticancer therapies.7, 8, 9 In this review we will summarize the possible role of autophagy as a novel target for anticancer therapies and discuss the attractive prospect of manipulating this control as a revolutionary strategy for cancer therapy.
The Regulation of Autophagy in Cancer During Response to Multiple Stresses
Autophagy is essential for not only cell survival but also organism survival in response to microenvironmental stresses. When cancer cells are subjected to stressful conditions, autophagy is rapidly upregulated to maintain metabolic homeostasis and ensure that cell growth is appropriate to its changing environmental conditions through reduced growth and increased catabolic lysis of excessive or unnecessary proteins and organelles. However, persistent or excessive autophagy is also shown to promote cell death following treatment with specific chemotherapeutic agents, either by enhancing the induction of apoptosis or mediating ‘autophagic cell death’.
Although the molecular mechanisms whereby autophagy mediates its effects on both normal and cancer cells are far from complete, various signaling pathways have been implicated in the upregulation or downregulation of autophagy.10, 11 The phosphatidylinositol 3-kinase/mammalian target of rapamycin (PI3K/mTOR) and AMP-activated protein kinase (AMPK) signaling pathways have emerged as the central conduit in the regulation of autophagy (Figure 2). mTOR can be activated by growth factors signal through the class I PI3K/Akt pathway, and inhibited by AMPK and p53.12, 13 Once activated, mTOR exerts a negative effect on autophagy by phosphorylating a complex of autophagy proteins (ULK1/2), which inhibits the downstream autophagy cascade.14, 15 In contrast, AMPK can suppress mTORC1 signaling to stimulate autophagy through TSC1/2 phosphorylation.16, 17 Several of the known tumor-suppressor genes (p53, PTEN, TSC1/TSC2) and tumor-associated genes (p21, AKT) also respectively stimulate or inhibit autophagy.10, 15

Interrelations between autophagy-related signaling and cell growth control in response to stress. Autophagy can be activated in response to multiple stresses during cancer progression, including nutrient deprivation, endoplasmic reticulum stress, hypoxia, glucose/energy depletion, chemotherapy and other diverse stresses. The AMPK/mTOR pathway functions as a central conduit for autophagic signaling pathways to promote cell survival or death
Autophagy is also induced by a variety of metabolic stresses such as endoplasmic reticulum (ER) stress, hypoxia, oxidative stress, expression of aggregate-prone proteins, glucose deprivation and so on.18 ER stress stimulates autophagy through the PERK/eukaryotic initiation factor 2α (eIF2α) and IRE1/JNK1 pathways. PERK/eIF2α phosphorylation has been shown to be essential for the transcription of key autophagy-associated genes during ER stress and may mediate the polyglutamine-induced LC3 conversion.19 The activation of IRE1/JNK promotes phosphorylation of Bcl-2 and p53, resulting in interfering with Bcl-2 binding to Beclin 1 and autophagic cell death in cancer cells.20 Depletion of nutrients or energy induces autophagy by activating the AMPK pathway or promoting upregulate transcription of certain autophagy genes.16, 17 The MEK/ERK signaling activation and Rag inactivation contribute to amino acid depletion-induced autophagy.7, 21 Many anticancer drugs including novel targeted therapies stimulate autophagy by inhibiting the PI3K/Akt/mTOR axis or altering genetic/epigenetic phenotype of cancer cells, which provides a survival advantage for struggling tumor cells.22, 23, 24 The histone deacetylase (HDAC) inhibitors are recently involved in the control of DNA damage response (DDR) and autophagy. SD118-xanthocillin X (1), a novel marine agent extracted from Penicillium commune, induces autophagy through the inhibition of the MEK/ERK pathway.25 Overall, autophagy is a cell biological process that involves diverse signals that have overlapping functions in autophagy and the control of other cellular stress responses.
Autophagy in Response to Chemotherapy
Similar to its potential to either induce cell death or promote cell survival, a growing body of evidence implicates a paradoxical role of autophagy following anticancer treatments, with response increasing or diminishing their anticancer activity. On the one hand, autophagy is activated as a protective mechanism to mediate the acquired resistance phenotype of some cancer cells during chemotherapy. Thus, the inhibition of autophagy can re-sensitize previously resistant cancer cells and augment cytotoxicity of chemotherapeutic agents. On the other hand, autophagy may also play as a death executioner to induce autophagic cell death, a form of physiological cell death which is contradictory to type I programmed cell death (apoptosis) (Figure 3). Based on current genetic and pharmacological studies, it appears that anticancer drugs induce different effects of autophagy on cell survival in different cancer types (Table 1). Here we delineate the possible role of autophagy as a novel target for anticancer therapy.

Dual role of autophagy for therapeutic purposes in cancer. On one hand, autophagy is activated as a protective mechanism to mediate the acquired resistance phenotype of some cancer cells during chemotherapy. On the other hand, autophagy may also function as a death executioner to induce autophagic cell death, a form of physiological cell death that is contradictory to apoptosis
Autophagy as a Prosurvival and Resistance Mechanism Against Chemotherapy Treatment
Recent studies have demonstrated that tumor resistance to anticancer therapies including radiation therapy, chemotherapy and targeted therapies can be enhanced through upregulation of autophagy in different tumor cell lines.26, 27 Moreover, increasing evidence suggests that autophagy inhibition augments cytotoxicity in combination with several anticancer drugs in preclinical models.28, 29, 30 Several pharmacological compounds and strategies have been reported to inhibit autophagy in vitro and in vivo.
Antimalarial Drugs
The only autophagy inhibitors whose effectiveness in vivo and safety in clinical trials have been approved by the FDA are chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) that suppress autophagy by blocking autophagosome fusion and degradation.31, 32 Both CQ and HCQ have been investigated in preclinical studies or clinical trials. In comparison with CQ, HCQ can be safely dose escalated in cancer patients.33 Currently, more than 30 phase I/II cancer clinical trials (http://clinicaltrials.gov/) involving CQ or HCQ are open around the world and many of them have evidence of preliminary antitumor activity (Table 2).
Breast cancer
The role of autophagy in breast cancer is an area of active investigation. There is evidence to suggest that epirubicin (EPI) may induce autophagy in human breast cancer MCF-7 cells, resulting in protecting MCF-7 cells from EPI-induced apoptosis.34 Autophagy is also regarded as a key mechanism of antiestrogen resistance, and blocking autophagosome can significantly reduce the emergence of antiestrogen-resistant breast cancer cells.35 A phase II clinical trial (NCT01292408) is investigating the effects of autophagy inhibition via HCQ on breast cancer patients. The current reports indicate that CQ or HCQ is often used in combination with chemotherapeutic drugs to enhance the efficacy of tumor cell killing; however, its sensitizing effects can also occur independently of autophagy inhibition, which should be considered in the ongoing clinical trials where CQ or HCQ are used in the treatment of breast cancer.36
Colorectal cancer
So far, 5-fluorouracil (5-FU), together with other drugs such as oxaliplatin, remains a widely used chemotherapeutic drug in the treatment of a variety of colorectal carcinomas. Previous researches have demonstrated that inhibition of autophagy augments anticancer effects of chemotherapy or some targeted therapies in colorectal cancer.37, 38 Recently, it has been shown that mitogen-activated protein kinase 14 (MAPK14)/p38α is involved in resistance of colon cancer cells to 5-FU and irinotecan, which triggers survival-promoting autophagy to protect tumor cells against the cytotoxic effects of these drugs.39, 40 Furthermore, autophagy inhibitor CQ significantly enhances the 5-FU-induced inhibition of tumor growth both in vitro and in vivo.41, 42 In addition, the combination of FOLFOX/bevacizumab with HCQ is currently being investigated.
Esophageal cancer
The role of autophagy in response to chemotherapy and radiotherapy has been investigated in human esophageal squamous carcinoma cells. Autophagy might play a role as a self-protective mechanism in chemotherapeutic drug-treated esophageal cancer cells, and its inhibition has the potential to improve the efficacy of chemotherapeutic agents such as cisplatin and 5-FU.43, 44, 45 However, the effect of adding HCQ to conventional therapy for esophageal cancer patients remains unclear.
Glioblastoma
Malignant cell clones resistant to chemotherapy is a key reason for treatment failure in patients with (GBM). When treated with bevacizumab alone, human GBM xenografts show increased autophagic flux and hypoxia-associated growth, which indicates that hypoxia-mediated autophagy promotes tumor cell survival and resistance to antiangiogenic therapy. However, in treatment by combining with the autophagy inhibitor CQ, tumor growth is disrupted, which elucidates a novel mechanism of overcoming resistance to antiangiogenic therapy for GMB.46 A randomized, double-blind, placebo-controlled trial examines the effect of adding CQ to conventional therapy for GBM. As a result, a median overall survival is 24 months for CQ-treated patients and 11 months for placebo-treated patients.30 Although it is not statistically different because of the small sample size, the rate of death of patients receiving CQ is prominently lower than controls. Another phase I/II trial concerning dose-limiting toxicities of HCQ with temozolomide (TMZ) and radiation for GBM patients was conducted.47 The information about the antitumor activity of this combination is underway.
Hepatocellular carcinoma
The combination of autophagy inhibitor and chemotherapy or molecular-targeted therapies has been regarded as a promising therapeutic strategy in the treatment of hepatocellular carcinoma (HCC). Autophagy is functionally activated in HCC cell lines after oxaliplatin treatment, and suppression of autophagy enhances oxaliplatin-induced cell death.48 Autophagy also contributes to HCC cell survival, and the combined treatment of an autophagy inhibitor and bevacizumab markedly inhibits the growth of HCC.49 Moreover, the combination of sorafenib with CQ can generate more ER stress-induced cell death in HCC both in vivo and in vitro.50 Therefore, autophagy inhibition may be a promising therapeutic strategy to enhance the effects of chemotherapy and improve clinical outcomes in the treatment of HCC.
Leukemia and MCL
Recently, it is reported that high-mobility group box 1 (HMGB1), a damage-associated molecular pattern (DAMP) molecule, contributes to chemotherapy resistance though the upregulation of autophagy in leukemia.51, 52 Autophagy inhibitor HCQ is also shown to decrease cell viability of B-chronic lymphocytic leukemia (B-CLL) in a dose- and time-dependent manner.53 The resistance to Akt/mTOR inhibitors such as everolimus (RAD001) is a significant clinical problem in relapsed mantle cell lymphoma (MCL) patients. Fortunately, pretreatment with HCQ can efficiently overcome this resistance, resulting in the activation of the mitochondrial apoptotic pathway.54 These data illustrate a strategy of blocking activation of adaptive autophagy pathway to improve treatment outcomes in leukemia and MCL.
Lung cancer
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) have been widely used in patients with non-small-cell lung cancer (NSCL). Unfortunately, the efficacy of these drugs is limited because of natural or acquired resistance. We report that autophagy can be activated by gefitinib or erlotinib in lung cancer cells, which contributes to the acquired drug resistance of EGFR-TKIs.55 Furthermore, the antimalaria drug CQ has been shown to enhance chemotherapy and radiation sensitivity in several preclinical models. CQ can not only potentiate the cytotoxicity of topotecan (TPT), but also substantially increase the effects of PI3K/mTOR inhibitor NVP-BEZ235 on induction of apoptosis, inhibition of colony formation and suppression of xenografts in nude mice.56, 57 A phase I study in advanced NSCL patients with prior clinical benefit from EGFR-TKIs suggests that HCQ with or without erlotinib is safe, and the recommended phase II dose for HCQ with erlotinib 150 mg is 1000 mg daily.58 Although no difference in survival was elaborated, this study explored the safety of adding HCQ to erlotinib. To assess the efficacy of the combination of HCQ with conventional chemotherapeutics, several clinical trials have been launched.
Pancreatic adenocarcinoma
The cytoprotective role of autophagy in response to chemotherapy has been confirmed in the treatment of pancreatic cancer cells.59 In agreement with this, Mirzoeva et al.60 showed that autophagy suppression with CQ promotes antitumor activity of PI3K/mTOR inhibitor for the treatment of pancreatic adenocarcinoma (PDAC) in vitro and in vivo. In addition to CQ, the efficacy and side effects of adding HCQ to conventional therapy are being investigated through several clinical trials.
Prostate cancer and renal cell carcinoma
Several recent studies have indicated that autophagy functions as a survival mechanism to promote chemoresistance in prostate and renal cancer cells.61, 62 Autophagy activation protects against ER stress-induced cell death, whereas inhibition of autophagy significantly suppresses PC-3 prostate tumor growth in vivo.63 Moreover, inhibition of autophagy by either HCQ or Beclin-1/Atg5 small interfering RNA enhances ABT-737 cytotoxicity and ursolic acid-induced apoptosis in prostate cancer cells.64, 65 Administration of high-dose interleukin-2 (HDIL-2) has durable complete and partial responses in patients with metastatic renal cell carcinoma. However, HDIL-2 treatment is often limited by side effects, because of a cytokine-induced systemic autophagic syndrome. Liang et al.66 found that the combination of IL-2 with CQ increases antitumor effects and decreases toxicity when compared with IL-2 treatment alone, which provide a novel clinical strategy to enhance the efficacy of HDIL-2 immunotherapy for patients with renal cell carcinoma. So far, two active trials (NCT 01144169 and NCT 01550367) are investigating the efficacy of adding HCQ to IL-2.
Ovarian cancer
Ovarian cancer has poor prognosis and is frequently resistant to chemotherapy. In this issue, it has been presented that autophagy may be a factor in drug resistance and poor survival in clear cell ovarian cancer patients.67 Nucleus accumbens-1 (NAC1) can mediate resistance to cisplatin in ovarian cancer cell lines because of activation of autophagy.68 FTY720, a sphingosine analog, may enhance autophagic flux when treated as a new chemotherapeutic agent for ovarian cancer, and blockade of autophagy aggravates necrotic ovarian cancer cell death in response to FTY720.69 Now, the effect of combining HCQ with sorafenib is being assessed in FIGO stage III or stage IV ovarian cancer, or extraovarian peritoneal carcinoma, or fallopian tube carcinoma failing or ineligible for first-line therapy.
Other Compounds and Strategies
In addition to antimalarial drugs, inhibition of autophagy by either pharmacological approaches or via genetic silencing of autophagy regulatory genes such as Beclin 1, ATG6, ATG5, ATG7 or ATG12 (Table 3) also results in sensitization to a variety of therapeutic agents. Different autophagy inhibitors block the autophagic process at different stages. For example, antimalarial drugs or bafilomycin A1 can inhibit autophagosome fusion with lysosomes and autophagosome degradation in the final stage of autophagy. Class III PI3K inhibitors (3-methyladenine (3-MA), LY294002 and Wortmannin) or knockdown of autophagy regulatory genes are involved in the initiation/expansion stage of autophagy.70, 71, 72, 73 So far, different autophagy inhibitors or genetic knockout of autophagy regulatory genes have been developed and used in the study of autophagy in cancer chemotherapy.74, 75, 76, 77, 78
The Molecular Mechanisms of Protective Autophagy-Mediated Chemoresistance
Although many anticancer therapeutic strategies can induce autophagic cell death, a majority of pertinent studies have been conducted to indicate that autophagy is a protective mechanism associated with increased resistance to chemotherapy. Induction of autophagy has emerged as a drug resistance mechanism that promotes cancer cell survival. There are a number of different mechanisms through which the autophagy-related functions of promoting the survival of tumor cells under the treatment of anticancer drugs (Figure 4).

The molecular mechanisms of autophagy activation in response to chemotherapeutic agents. The activation of autophagy either leads to cancer cell chemoresistance via EGFR signaling, PI3K/AKT/ mTOR pathways, p53, VEGF, MAPK14/p38α signaling and microRNA or potentiates autophagic cell death through AMPK/AKT1/mTOR axis, which depends on the tumor types and treatment characteristic
EGFR signaling
Epidermal growth factor is a key regulatory factor for cell survival. Through its binding to cell surface receptors, EGF can induce the activation of three signaling pathways which are important to the initiation and progression of cancers, Ras/MAPK, PI3K/Akt and JAK/STATs.79 In the previous study, we confirmed biochemically and morphologically that autophagy can be activated by gefitinib or erlotinib, which was accompanied by the inhibition of the PI3K/Akt/mTOR signaling pathway. Furthermore, blockage of autophagy by the pharmacological inhibitors or gene silence greatly enhanced cytotoxicity of gefitinib or erlotinib.55 PD168393, an EGFR-TKI, may induce autophagy as a cytostatic but not a cytotoxic response in malignant peripheral nerve sheath tumor (MPNST) cells that was accompanied by suppression of Akt and mTOR activation; moreover, suppression of autophagy by CQ increased caspase activation.80 These results indicate that EGFR-TKIs can induce autophagy to promote tumor cell survival in response to targeted chemotherapies, and suppression of autophagy can augment the growth inhibitory effect of these drugs through inhibition of the PI3K/Akt/mTOR signaling pathway.
PI3K/AKT/mTOR pathways
In addition to EGFR, the aberrant expression of PI3K/AKT/mTOR is also known to be a key regulator of authophagy. Genetic and pharmacologic autophagy blockade via PI3K/mTOR inhibition may reverse apoptotic resistance and result in significant cell apoptosis.81 NVP-BEZ235 (BEZ235) is a novel, orally bioavailable dual PI3K/mTOR inhibitor that has exerted a positive effect on autophagy. A recent study suggests that NVP-BEZ235 could induce apoptosis and autophagy; moreover, the combination treatment of NVP with autophagy inhibitors lead to enhanced RCC cell apoptosis.62, 82 Benzyl isothiocyanate (BITC), a dietary chemopreventive agent, is also found to induce protective autophagy in human prostate cancer cells via inhibition of mTOR signaling pathway, and inhibition of autophagy using 3-MA increased BITC-induced apoptosis.83 Taken together, targeting PI3K/AKT/mTOR-autophagy pathways displays a well-recognized contribution to overcome chemotherapy resistance and sensitize the tumor cells to anticancer therapy.
p53, VEGF and MAPK14/p38α signaling
As a well-known tumor-suppressor gene, p53 is also involved in autophagic regulation. In a mouse model of c-Myc-driven lymphoma, inhibition of autophagy with either CQ or ATG5 shRNA promotes tumor cell death by p53 activation.84 Stanton et al.85 show that the VEGF-C/NRP-2 axis is involved in the activation of autophagy, and VEGF-C or NRP-2 depletion contributes to cytotoxic drug-mediated cell death. In addition, the recent studies have provided strong evidence that the MAPK14/p38 signaling is involved in cancer cell resistance to chemotherapy treatment. Paillas et al.39 investigated the relationship between MAPK14/p38, autophagy and resistance to irinotecan and found that MAPK14/p38 was activated and triggered survival-promoting autophagy to protect tumor cells against the cytotoxic effects of irinotecan. Furthermore, p38MAPK activation is considered a key determinant in the cellular response to 5-FU by controlling the balance between apoptosis and autophagy.40
MicroRNAs
MicroRNAs (miRNAs) are a class of small, noncoding, endogenously encoded, single-stranded RNAs that regulate gene expression at the post-transcriptional level. Recently, a number of miRNAs have been reported to be deeply involved in resistance or sensitization to chemotherapy.86, 87 miR-30a, a member of miR-30 family, is a potent inhibitor of autophagy by selectively downregulating Beclin 1 and Atg5 expression. Targeting miR-30a promotes autophagy in response to imatinib treatment and enhances imatinib resistance against CML including primary stem and progenitor cells.88, 89 Moreover, the blockade of autophagy by miR-30a expression or 3-MA significantly increased cis-DDP-induced apoptosis in cancer cells.90 Downregulation of miR-199a-5p is observed in most HCC tissues of patients. More importantly, downregulation of miR-199a-5p increases cisplatin resistance by activating autophagy in HCC cells.91 Cisplatin-induced ATM-dependent phosphorylated (p)-ΔNp63α also plays an important role in chemoresistance. Further research shows that the p-ΔNp63α-induced miR-885-3p might contribute to regulation of apoptosis and/or autophagy in squamous cell carcinoma cells upon cisplatin exposure.92 Although the relationship between miRNA, autophagy and anticancer therapy resistance is quite complicated, and has not been well elucidated, miRNA may underlie key aspects of chemotherapy resistance.
Chloroquine and Its Derivative: Just Act as Autophagy Inhibitors?
CQ and its derivative are currently the only autophagy inhibitors available for clinical treatment of patients. In ongoing cancer treatment clinical trials, HCQ is often used in combination with chemotherapeutic drugs, radiotherapy, some targeted therapies and immunotherapy. The treatment of CQ or HCQ can inhibit autophagy-related survival function and exert their anticancer action. However, emerging data indicate that the ability of CQ and its derivative to inhibit the final degradative step of autophagy may not be the only mechanism by which they exert anticancer action; CQ and HCQ may also affect other pathways such as lysosomal membrane permeabilization.
Recently, Maycotte et al.36 reported that CQ could sensitize breast cancer cells to chemotherapy independent of autophagy inhibition, as sensitization was not mimicked by Atg12, Beclin 1 knockdown or bafilomycin A1 treatment, and occurred even in the absence of Atg12. In further studies, CQ and HCQ are shown to function as lysosomotropic agents to promote lysosomal membrane permeabilization (LMP), resulting in signs of apoptosis. The research from Enzenmüller et al.93 shows that PI3K/mTOR inhibitor PI103 enhances the lysosomal compartment by increasing its volume and function, whereas CQ destabilizes lysosomal membranes. Together, CQ overcomes resistance of lung carcinoma cells to PI103-induced apoptosis by cooperating with PI103 to trigger lysosome-mediated apoptosis. An additional report suggests that induction of lysosome-mediated apoptosis rather than inhibition of autophagy is critical for the CQ-mediated sensitization to BEZ235-induced apoptosis, as lysosomal enzyme inhibitors significantly decrease BEZ235- and CQ-induced drop of mitochondrial membrane potential (MMP) and caspase-dependent apoptosis.94 Similar to the potential of CQ, HCQ can also induce LMP and Bax/Bak-dependent MMP to trigger caspase activation.95
Taken together, these data indicate that the success of clinical trials using CQ or HCQ combined with other anticancer agents might not be due to CQ or HCQ effects on autophagy induced by chemotherapeutic drugs, as the effect may be mediated by mechanisms other than its inhibition of autophagy. Thus, a better knowledge of the molecular mechanisms and cellular targets of CQ or HCQ should be considered in the ongoing clinical trials where CQ or HCQ are used as autophagy inhibitors.
Autophagy-Mediated Cell Death Mechanism Contributes to Efficacy of Anticancer Drugs
Despite its a clear prosurvival role, autophagy has also been viewed as having a prodeath role under certain circumstances and following treatment with a specific set of chemotherapeutic agents, either by enhancing the induction of apoptosis or mediating ‘autophagic cell death’.
Increasing evidence supports that autophagy may mediate cell death in cancer cells which are apoptosis defective or hard to induce. Xiong et al.96 found that autophagic cell death could be induced in PUMA- or Bax-deficient human colon cancer cells after the treatment with 5-FU, consequently followed by decreased cancer proliferation. Suberoylanilide hydroxamic acid (SAHA), a prototype of the newly developed HDAC inhibitor, induces autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells and significantly reduces the tumor growth in vitro and in vivo.97 NVP-BEZ235 is demonstrated to inhibit cisplatin-resistant urothelial cancer cell proliferation by activating autophagic flux and cell cycle arrest, but not inducing apoptotic cell death.98 The antidepressants maprotiline and fluoxetine induce autophagic cell death in drug-resistant Burkitt’s lymphoma (BL), which supports a new mechanistic role for maprotiline and fluoxetine as novel proautophagic agents in the treatment of resistant BL.99 These data indicate that autophagic cell death can be induced as an alternative cell death mechanism when cells fail to undergo apoptosis.
In addition, induction of autophagic cell death is also an alternative approach to kill tumor cells without resistance to anticancer drugs. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy.100 FK-16, derived from the anticancer peptide LL-37, induces caspase-independent apoptosis and autophagic cell death in colon cancer cells.101 The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a GBM cell line.102 Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin.103 Sorafenib and SC-59, which is a novel sorafenib derivative, induce autophagic cell death in hepatocellular carcinoma cells in a dose- and time-dependent manner.104 Recently, cannabinoids have been shown to exert their anticancer activity in glioma, pancreatic cancer and hepatocellular carcinoma via stimulation of autophagy-mediated cell death.105, 106, 107, 108 Moreover, the combination of cannabinoids and TMZ strongly activates autophagy-mediated cancer cell death, resulting in a strong antitumoral action in both TMZ-sensitive and TMZ-resistant tumors.109 These studies present new insights into our understanding of the relationship between autophagy and anticancer efficacy and provide a potential therapeutic strategy for the management of some of these tumors.
Several genes and signal pathways contribute to autophagic cell death in cancer cells (Figure 4). The AMPK/AKT1/mTOR axis is critical for regulation of autophagic cell death. Tanshinone IIA induces autophagic cell death via activation of AMPK/ERK and inhibition of mTOR and p70 S6K in KBM-5 leukemia cells.110 Cannabinoids can trigger autophagic cell death in an ER stress and Akt/mTORC1-dependent manner.105, 106, 107, 108 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation.111 Diarylquinoline compounds induce a autophagic cell death by inhibiting the Akt pathway and increasing reactive oxygen species in human nasopharyngeal carcinoma cells.112 β-Catenin is also involved in activation of autophagic cell death.113 These results further strengthen the connection between autophagy and anticancer efficacy.
Recently, autophagy has been shown to precede apoptosis or act in parallel with this cellular process in addition to being an alternative mechanism to cell death when apoptosis is inhibited. It has been acknowledged that autophagy precedes caspase-dependent apoptosis.114, 115 Several studies demonstrate that autophagy precedes apoptosis and acts as a protective mechanism in cancer cells.116, 117, 118 Therefore, the autophagy induction may exert other possibilities, which should be considered in the design of new treatments for these malignancies.
Conclusions and Perspectives
Autophagy is a lysosomal degradation process usually activated in response to adverse microenvironmental stresses. Autophagy itself fulfils a dual role, having tumor-promoting and tumor-suppressing properties. As a response to anticancer treatments, whether autophagy activation leads to cell survival or cell death remains controversial. It is consensus that the outcomes of autophagy activation are highly depended on the tumor types and treatment characteristic.119, 120
Resistance to chemotherapy is a major obstacle for the success of cancer therapy. Although the controversy about the prosurvival or anticancer effect of autophagy is still heated, the data in vitro and in vivo seem more to support the idea that autophagy facilitates the cancer cells’ resistance to chemotherapy treatment, and inhibition of autophagy may potentiate the resensitization of therapeutic-resistant cancer cells to the anticancer drugs.26, 121 Several autophagy inhibitors such as CQ and its derivative HCQ have been studied in preclinical models. Although CQ or HCQ augments cytotoxicity in combination with several anticancer drugs, autophagy researchers should be careful when interpreting experiments in which CQ or HCQ treatment is used, as the effect may be mediated by mechanisms other than its inhibition of autophagy.
Currently, the combination of autophagy inhibitors with cytotoxic drugs is attracting more and more attention in cancer therapy. However, the study of the ability of autophagy inhibitors to overcome resistance to anticancer therapies and how this relates to the regulation of tumor microenvironmental stresses raises many issues. First, the question of whether we should try to enhance or inhibit autophagy in cancer treatment is not straightforward as it might vary according to cell type, the stress signal and other circumstances. What is needed to be better experimentally addressed include elucidating the impact of the tumor microenvironment on autophagy function, determining the role(s) of autophagy in the regulation of therapeutic sensitivity and defining novel mechanism by which autophagy inhibition can overcome chemotherapy resistance and sensitize the tumor cells to anticancer therapy. Second, to maximize the potential to be applied for more stringent clinical study, new and reliable methods for measuring autophagy in clinical samples need to be developed. Third, the ocular toxicities and minimal single-agent anticancer efficacy of CQ or HCQ have restricted its clinical application. New and exciting autophagy inhibitors are worthy of further investigation in the future. Thus, establishment of more effective and safe combinatorial therapeutic strategies using autophagy inhibitors will be necessary in the future. However, our increased understanding of the functional relevance of autophagy within the tumor microenvironment and ongoing dialogue between emerging laboratory and clinical research of targeting autophagy will hopefully provide a promising therapeutic strategy to circumvent resistance and enhance the effects of anticancer therapies for cancer patients.
Abbreviations
- PI3K:
-
phosphatidylinositol 3-kinase
- mTOR:
-
mammalian target of rapamycin
- AMPK:
-
AMP-activated protein kinase
- ER:
-
endoplasmic reticulum
- eIF2α:
-
eukaryotic initiation factor 2α
- HDAC:
-
histone deacetylase
- DDR:
-
DNA damage response
- CQ:
-
chloroquine
- HCQ:
-
hydroxychloroquine
- EPI:
-
epirubicin
- 5-FU:
-
5-fluorouracil
- MAPK:
-
mitogen-activated protein kinase
- GBM:
-
glioblastoma
- TMZ:
-
temozolomide
- MTD:
-
maximum tolerated dose
- HCC:
-
hepatocellular carcinoma
- MCL:
-
mantle cell lymphoma
- DAMP:
-
damage-associated molecular pattern
- B-CLL:
-
B-chronic lymphocytic leukemia
- EGF:
-
epidermal growth factor
- EGFR-TKI:
-
epidermal growth factor receptor tyrosine kinase inhibitor
- TPT:
-
topotecan
- PDAC:
-
pancreatic adenocarcinoma
- HDIL-2:
-
high-dose interleukin-2
- NAC1:
-
nucleus accumbens-1
- 3-MA:
-
3-methyladenine
- MPNST:
-
malignant peripheral nerve sheath tumor
- BITC:
-
benzyl isothiocyanate
- LMP:
-
lysosomal membrane permeabilization
- MMP:
-
mitochondrial membrane potential
- SAHA:
-
suberoylanilide hydroxamic acid
- BL:
-
Burkitt’s lymphoma
References
Ringborg U, Platz A . Chemotherapy resistance mechanisms. Acta Oncol 1996; 35: 76–80.
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM . Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5: 219–234.
Yang Z, Klionsky DJ . Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12: 814–822.
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544.
Sui X, Jin L, Huang X, Geng S, He C, Hu X . p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment. Autophagy 2011; 7: 565–571.
Yang Z, Klionsky DJ . An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335: 1–32.
Kondo Y, Kanzawa T, Sawaya R, Kondo S . The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726–734.
Maycotte P, Thorburn A . Autophagy and cancer therapy. Cancer Biol Ther 2011; 11: 127–137.
Janku F, McConkey DJ, Hong DS, Kurzrock R . Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8: 528–539.
Glick D, Barth S, Macleod KF . Autophagy: cellular and molecular mechanisms. J Pathol 2010; 221: 3–12.
Cecconi F, Levine B . The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008; 15: 344–357.
Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012; 142: 1504–1515.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465: 942–946.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93.
Yang Z, Klionsky DJ . Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22: 124–131.
Tsuchihara K, Fujii S, Esumi H . Autophagy and cancer: dynamism of the metabolism of tumor cells and tissues. Cancer Lett 2009; 278: 130–138.
Hardie DG . AMPK and Raptor: matching cell growth to energy supply. Mol Cell 2008; 30: 263–265.
Mahoney E, Lucas DM, Gupta SV, Wagner AJ, Herman SE, Smith LL et al. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood 2012; 120: 1262–1273.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–239.
Park KJ, Lee SH, Lee CH, Jang JY, Chung J, Kwon MH et al. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun 2009; 382: 726–729.
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL . Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10: 935–945.
Ciuffreda L, Di Sanza C, Incani UC, Milella M . The mTOR pathway: a new target in cancer therapy. Curr Cancer Drug Targets 2010; 10: 484–495.
Botrugno OA, Robert T, Vanoli F, Foiani M, Minucci S . Molecular pathways: old drugs define new pathways: non-histone acetylation at the crossroads of the DNA damage response and autophagy. Clin Cancer Res 2012; 18: 2436–2442.
Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M . Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res 2012; 72: 1332–1335.
Zhao Y, Chen H, Shang Z, Jiao B, Yuan B, Sun W et al. SD118-xanthocillin X (1), a novel marine agent extracted from Penicillium commune, induces autophagy through the inhibition of the MEK/ERK pathway. Mar Drugs 2012; 10: 1345–1359.
Hu YL, Jahangiri A, Delay M, Aghi MK . Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res 2012; 72: 4294–4299.
Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012; 8: 1798–1810.
Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G . Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote γ-irradiation-induced cell death in primary stem-like glioma cells. PLoS One 2012; 7: e47357.
Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007; 110: 313–322.
Sotelo J, Briceno E, Lopez-Gonzalez MA . Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2006; 144: 337–343.
Poole B, Ohkuma S . Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 1981; 90: 665–669.
Nilsson JR . Does chloroquine, an antimalarial drug, affect autophagy in Tetrahymena pyriformis? J Protozool 1992; 39: 9–16.
Gunja N, Roberts D, McCoubrie D, Lamberth P, Jan A, Simes DC et al. Survival after massive hydroxychloroquine overdose. Anaesth Intensive Care 2009; 37: 130–133.
Sun WL, Chen J, Wang YP, Zheng H . Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011; 7: 1035–1044.
Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT . Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy 2009; 5: 400–403.
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A . Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012; 8: 200–212.
Li J, Hou N, Faried A, Tsutsumi S, Kuwano H . Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer 2010; 46: 1900–1909.
Yang PM, Liu YL, Lin YC, Shun CT, Wu MS, Chen CC . Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res 2010; 70: 7699–7709.
Paillas S, Causse A, Marzi L, de Medina P, Poirot M, Denis V et al. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012; 8: 1098–1112.
de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya EM et al. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene 2012; 31: 1073–1085.
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10: 370.
Sasaki K, Tsuno NH, Sunami E, Kawai K, Hongo K, Hiyoshi M et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer Drugs 2012; 23: 675–682.
Liu D, Yang Y, Liu Q, Wang J . Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 2011; 28: 105–111.
O'Donovan TR, O'Sullivan GC, McKenna SL . Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7: 509–524.
Chen YS, Song HX, Lu Y, Li X, Chen T, Zhang Y et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus 2011; 24: 437–443.
Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 2012; 72: 1773–1783.
Rosenfeld MRGS, Brem S, Mikkelson T, Wang D, Piao S, Davis L et al. Pharmacokinetic analysis and pharmacodynamic evidence of autophagy inhibition in patients with newly diagnosed glioblastoma treated on a phase I trial of hydroxychloroquine in combination with adjuvant temozolomide and radiation (ABTC 0603). J Clin Oncol 2010; 28: 3086.
Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, Gu CY et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res 2011; 17: 6229–6238.
Guo XL, Li D, Sun K, Wang J, Liu Y, Song JR et al. Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J Mol Med (Berl) 2013; 91: 473–483.
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7: 1159–1172.
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y X et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011; 7: 112–114.
Zhao M, Yang M, Yang L, Yu Y, Xie M, Zhu S et al. HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. BMB Rep 2011; 44: 601–606.
Lagneaux L, Delforge A, Carlier S, Massy M, Bernier M, Bron D . Early induction of apoptosis in B-chronic lymphocytic leukaemia cells by hydroxychloroquine: activation of caspase-3 and no protection by survival factors. Br J Haematol 2001; 112: 344–352.
Rosich L, Xargay-Torrent S, López-Guerra M, Campo E, Colomer D, Roué G . Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin Cancer Res 2012; 18: 5278–5289.
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 2011; 6: e18691.
Wang Y, Peng RQ, Li DD, Ding Y, Wu XQ, Zeng YX et al. Chloroquine enhances the cytotoxicity of topotecan by inhibiting autophagy in lung cancer cells. Chin J Cancer 2011; 30: 690–700.
Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK et al. Augmentation of NVP-BEZ235’s anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol Ther 2011; 12: 549–555.
Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol 2012; 7: 1602–1608.
Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010; 17: 666–676.
Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K et al. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl) 2011; 89: 877–889.
Kaini RR, Sillerud LO, Zhaorigetu S, Hu CA . Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate 2012; 72: 1412–1422.
Li H, Jin X, Zhang Z, Xing Y, Kong X . Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTor inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem Funct 2013; 31: 427–433.
Kumano M, Furukawa J, Shiota M, Zardan A, Zhang F, Beraldi E et al. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol Cancer Ther 2012; 11: 1661–1671.
Shin SW, Kim SY, Park JW . Autophagy inhibition enhances ursolic acid-induced apoptosis in PC3 cells. Biochim Biophys Acta 2012; 1823: 451–457.
Saleem A, Dvorzhinski D, Santanam U, Mathew R, Bray K, Stein M et al. Effect of dual inhibition of apoptosis and autophagy in prostate cancer. Prostate 2012; 72: 1374–1381.
Liang X, De Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer Stolz D et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res 2012; 72: 2791–2801.
Gibson SB . Autophagy in clear cell ovarian cancer, a potential marker for hypoxia and poor prognosis? J Pathol 2012; 228: 434–436.
Zhang Y, Cheng Y, Ren X, Zhang L, Yap KL, Wu H et al. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene 2012; 31: 1055–1064.
Zhang N, Qi Y, Wadham C, Wang L, Warren A, Di W et al. FTY720 induces necrotic cell death and autophagy in ovarian cancer cells: a protective role of autophagy. Autophagy 2010; 6: 1157–1167.
Zhao S, Ma CM, Liu CX, Wei W, Sun Y, Yan H et al. Autophagy inhibition enhances isobavachalcone-induced cell death in multiple myeloma cells. Int J Mol Med 2012; 30: 939–944.
Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N et al. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int J Cancer 2009; 124: 1060–1071.
Liu F, Liu D, Yang Y, Zhao S . Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol Lett 2013; 5: 1261–1265.
Cao X, Liu B, Cao W, Zhang W, Zhang F, Zhao H et al. Autophagy inhibition enhances apigenin-induced apoptosis in human breast cancer cells. Chin J Cancer Res 2013; 25: 212–222.
Gao P, Bauvy C, Souquère S, Tonelli G, Liu L, Zhu Y et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem 2010; 285: 25570–25581.
Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V . AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med 2009; 13: 3644–3654.
Ren Y, Huang F, Liu Y, Yang Y, Jiang Q, Xu C . Autophagy inhibition through PI3K/Akt increases apoptosis by sodium selenite in NB4 cells. BMB Rep 2009; 42: 599–604.
Filomeni G, Desideri E, Cardaci S, Graziani I, Piccirillo S, Rotilio G et al. Carcinoma cells activate AMP-activated protein kinase-dependent autophagy as survival response to kaempferol-mediated energetic impairment. Autophagy 2010; 6: 202–216.
Deng L, Lei Y, Liu R, Li J, Yuan K, Li Y et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis 2013; 4: e614.
Henson ES, Gibson SB . Surviving cell death through epidermal growth factor (EGF) signal transduction pathway: implications for cancer therapy. Cell Signal 2006; 18: 2089–2097.
Kohli L, Kaza N, Lavalley NJ, Turner KL, Byer S, Carroll SL et al. The pan erbB inhibitor PD168393 enhances lysosomal dysfunction-induced apoptotic death in malignant peripheral nerve sheath tumor cells. Neuro Oncol 2012; 14: 266–277.
Ghadimi MP, Lopez G, Torres KE, Belousov R, Young ED, Liu J et al. Targeting the PI3K/mTOR axis, alone and in combination with autophagy blockade, for the treatment of malignant peripheral nerve sheath tumors. Mol Cancer Ther 2012; 11: 1758–1769.
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 2010; 70: 8097–8107.
Lin JF, Tsai TF, Liao PC, Lin YH, Lin YC, Chen HE et al. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis 2013; 34: 406–414.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–336.
Stanton MJ, Dutta S, Zhang H, Polavaram NS, Leontovich AA, Hönscheid P et al. Autophagy control by the VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer Res 2013; 73: 160–171.
Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res 2007; 67: 11111–11116.
Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P et al. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep 2010; 23: 997–1003.
Yu Y, Cao L, Yang L, Kang R, Lotze M, Tang D . microRNA 30A promotes autophagy in response to cancer therapy. Autophagy 2012; 8: 853–855.
Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P et al. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012; 26: 1752–1760.
Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X et al. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem 2012; 287: 4148–4156.
Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F et al. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun 2012; 423: 826–831.
Huang Y, Chuang AY, Ratovitski EA . Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle 2011; 10: 3938–3947.
Enzenmüller S, Gonzalez P, Debatin KM, Fulda S . Chloroquine overcomes resistance of lung carcinoma cells to the dual PI3K/mTOR inhibitor PI103 by lysosome-mediated apoptosis. Anticancer Drugs 2013; 24: 14–19.
Seitz C, Hugle M, Cristofanon S, Tchoghandjian A, Fulda S . The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk. Int J Cancer 2013; 132: 2682–2693.
Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 2003; 22: 3927–3936.
Xiong HY, Guo XL, Bu XX, Zhang SS, Ma NN, Song JR et al. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Cancer Lett 2010; 288: 68–74.
Lee YJ, Won AJ, Lee J, Jung JH, Yoon S, Lee BM et al. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int J Med Sci 2012; 9: 881–893.
Li JR, Cheng CL, Yang CR, Ou YC, Wu MJ, Ko JL . Dual inhibitor of phosphoinositide 3-kinase/mammalian target of rapamycin NVP-BEZ235 effectively inhibits cisplatin-resistant urothelial cancer cell growth through autophagic flux. Toxicol Lett 2013; 220: 267–276.
Cloonan SM, Williams DC . The antidepressants maprotiline and fluoxetine induce Type II autophagic cell death in drug-resistant Burkitt's lymphoma. Int J Cancer 2011; 128: 1712–1723.
Leng S, Hao Y, Du D, Xie S, Hong L, Gu H et al. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int J Cancer 2013 e-pub ahead of print 16 July 2013; doi:10.1002/ijc.28301.
Ren SX, Shen J, Cheng AS, Lu L, Chan RL, Li ZJ et al. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One 2013; 8: e63641.
Josset E, Burckel H, Noël G, Bischoff P . The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a glioblastoma cell line. Anticancer Res 2013; 33: 1845–1851.
Guo WJ, Zhang YM, Zhang L, Huang B, Tao FF, Chen W et al. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013; 9: 996–1008.
Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 2013; 4: e485.
Salazar M, Carracedo A, Salanueva IJ, Hernández-Tiedra S, Lorente M, Egia A et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119: 1359–1372.
Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis 2011; 2: e152.
Dando I, Donadelli M, Costanzo C, Dalla Pozza E, D'Alessandro A, Zolla L et al. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis 2013; 4: e664.
Vara D, Salazar M, Olea-Herrero N, Guzmán M, Velasco G, Díaz-Laviada I . Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ 2011; 18: 1099–1111.
Torres S, Lorente M, Rodríguez-Fornés F, Hernández-Tiedra S, Salazar M, García-Taboada E et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther 2011; 10: 90–103.
Yun SM, Jung JH, Jeong SJ, Sohn EJ, Kim B, Kim SH et al. Tanshinone IIA induces autophagic cell death via activation of AMPK and ERK and inhibition of mTOR and p70 S6K in KBM-5 leukemia cells. Phytother Res 2013 e-pub ahead of print 27 June 2013; doi:10.1002/ptr.5015.
Kohli L, Kaza N, Coric T, Byer SJ, Brossier NM, Klocke BJ et al. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res 2013; 73: 4395–4405.
Cai Y, Wan Z, Sun T, Shi Y, Sun Y, Huang P et al. Diarylquinoline compounds induce autophagy-associated cell death by inhibiting the Akt pathway and increasing reactive oxygen species in human nasopharyngeal carcinoma cells. Oncol Rep 2013; 29: 983–992.
Chang HW, Lee YS, Nam HY, Han MW, Kim HJ, Moon SY et al. Knockdown of β-catenin controls both apoptotic and autophagic cell death through LKB1/AMPK signaling in head and neck squamous cell carcinoma cell lines. Cell Signal 2013; 25: 839–847.
Franzetti E, Huang ZJ, Shi YX, Xie K, Deng XJ, Li JP et al. Autophagy precedes apoptosis during the remodeling of silkworm larval midgut. Apoptosis 2012; 17: 305–324.
Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z et al. PARP and RIP 1 are required for autophagy induced by 11'-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011; 7: 598–612.
Abe A, Yamada H, Moriya S, Miyazawa K . The β-carboline alkaloid harmol induces cell death via autophagy but not apoptosis in human non-small cell lung cancer A549 cells. Biol Pharm Bull 2011; 34: 1264–1272.
Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem 2012; 287: 31104–31115.
Francisco R, Pérez-Perarnau A, Cortés C, Gil J, Tauler A, Ambrosio S . Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett 2012; 318: 42–52.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17: 654–666.
Choi KS . Autophagy and cancer. Exp Mol Med 2012; 44: 109–120.
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM, Lotze MT . Cell-mediated autophagy promotes cancer cell survival. Cancer Res 2012; 72: 2970–2979.
Acknowledgements
This study is supported by grants from the National Natural Science Foundation of China (grants 81301891, 81272593, 81071651 and 81071963) and the Zhejiang Provincial Natural Science Foundation of China (grant LQ13H160008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Sui, X., Chen, R., Wang, Z. et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 4, e838 (2013). https://doi.org/10.1038/cddis.2013.350
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.350
Keywords
This article is cited by
-
LAMP2A regulates cisplatin resistance in colorectal cancer through mediating autophagy
Journal of Cancer Research and Clinical Oncology (2024)
-
SIRT4 is an independent prognostic factor in bladder cancer and inhibits bladder cancer growth by suppressing autophagy
Cell Division (2023)
-
Nanomedicine for autophagy modulation in cancer therapy: a clinical perspective
Cell & Bioscience (2023)
-
Autophagy dictates sensitivity to PRMT5 inhibitor in breast cancer
Scientific Reports (2023)
-
Actionable cancer vulnerability due to translational arrest, p53 aggregation and ribosome biogenesis stress evoked by the disulfiram metabolite CuET
Cell Death & Differentiation (2023)
