
Abstract
In the lymph node (LN) environment, chronic lymphocytic leukemia (CLL) cells display increased NF-κB activity compared with peripheral blood CLL cells, which contributes to chemoresistance. Antagonists of cellular inhibitor of apoptosis proteins (cIAPs) can induce apoptosis in various cancer cells in a tumor necrosis factor-α (TNFα)-dependent manner and are in preclinical development. Smac-mimetics promote degradation of cIAP1 and cIAP2, which results in TNFR-mediated apoptosis via formation of a ripoptosome complex, comprising RIPK1, Fas-associated protein with death domain, FLICE-like inhibitory protein and caspase-8. CD40 stimulation of CLL cells in vitro is used as a model to mimic the LN microenvironment and results in NF-κB activation and TNFα production. In this study, we investigated the response of CLL cells to smac-mimetics in the context of CD40 stimulation. We found that treatment with smac-mimetics results in cIAP1 and cIAP2 degradation, yet although TNFα is produced, this did not induce apoptosis. Despite the presence of all components, the ripoptosome complex did not form upon smac-mimetic treatment in CLL cells. Thus, CLL cells seem to possess aberrant upstream NF-κB regulation that prevents ripoptosome formation upon IAP degradation. Unraveling the exact molecular mechanisms of disturbed ripoptosome formation may offer novel targets for treatment in CLL.
Similar content being viewed by others


Main
Chronic lymphocytic leukemia (CLL) is characterized by an accumulation of CD5+ monoclonal B lymphocytes and is the most common leukemia in the western world.1 A cure for CLL is not yet available, as relapses eventually occur from chemo-resistant sites such as the lymph nodes (LNs). CD40 ligand (CD40L) presented by CD4+ T cells in the LN enhances survival of CLL cells and is an important cause of chemotherapy resistance.2 The CD40 signaling pathway activates the nuclear factor kappa B (NF-κB) pathway causing a decrease in pro-apoptotic and increase in anti-apoptotic Bcl-2 proteins rendering cells resistant to apoptosis.3, 4 From the LNs, CLL cells translocate to the peripheral blood (PB), where they arrest in G0/G1 of the cell cycle and accumulate because of a failure to undergo apoptosis.5 New therapeutic strategies are needed to overrule LN pro-survival signals and circumvent apoptosis blockades.
The inhibitor of apoptosis proteins (IAPs) are important regulators of apoptosis. The IAP family consists of eight proteins and includes X-linked IAP (XIAP) and cellular inhibitor of apoptosis protein (cIAP1) and cIAP2.6, 7, 8, 9 IAPs are characterized by the presence of one to three baculovirus inhibitor of apoptosis repeat (BIR) domains, a 70–80-amino-acid-long protein interaction domain.10, 11 XIAP is upregulated in many cancers and binds and inhibits caspases via its BIR2 and BIR3 domains. Smac/Diablo is a natural BIR-binding antagonist of IAPs that is released from the damaged mitochondria. To overcome overexpressed XIAP and render cancers more susceptible to chemotherapy, small-molecule inhibitors based on the smac/BIR interaction motif were developed and are now called smac-mimetics. In addition to inhibiting XIAP, most smac-mimetics were found to promote the proteasomal degradation of cIAP1/2.12, 13, 14, 15
cIAP1 and 2 can bind with low affinity to caspases but do not inhibit them directly. Rather, it appears IAPs indirectly prevent caspase activation.16, 17, 18 For example, during tumor necrosis factor-α (TNF)/TNF receptor 1 (TNFR1) signaling, cIAP1 and 2 are required to mediate the K63 and K11 ubiquitination of receptor-interacting protein kinase 1 (RIPK1) in the membrane-bound signaling complex. This provides docking sites for different kinases and activates the canonical NF-κB signaling pathway.15, 19, 20, 21 Canonical NF-κB drives FLICE-like inhibitory protein (cFLIP) transcription, thereby limiting caspase-8 activity. Furthermore, cIAP1/2 limits the accumulation of RIPK1 in the downstream cytosolic complex 2 by K48 ubiquitylation. In the absence of cIAPs, cFLIP transcription is reduced and RIPK1 levels are increased, thus shifting the signaling to cell death. Furthermore, smac-mimetics appear to prime cells, as intracellular RIPK1-containing complexes accumulate in the absence of an exogenous stimulus. This intracellular complex, comprising Fas-associated protein with death domain (FADD), caspase-8 and cFLIP, has been called the ripoptosome and is most likely related to the secondary cytosolic complex that assembles post-TNF treatment.22, 23, 24 cIAP1 and 2 also act as negative regulators of the alternative or non-canonical NF-κB signaling pathway, by K48 ubiquitylating the upstream regulator NF-κB-inducing kinase (NIK).15, 20 In the absence of cIAPs, NIK levels rise, promoting its auto-activation and downstream activation of IκB kinase 1, processing of NF-κB2 p100 and nuclear translocation of NF-κB2 p52. In some cells, the activation of NF-κB in this manner can promote the production of TNF.15, 20 Thus, in some cancer cells, smac-mimetics can simultaneously promote production of TNF and sensitize cells to the cytotoxic activity of TNF. In other cells where TNF is not produced, many cells are sensitized to exogenously added TNF.
In CLL, relapses have been proposed to originate from the protective LN and bone marrow microenvironment, where cells receive pro-survival signals, among others from TNF-family member CD40L. CIAP1/2 are required for NF-κB activation in response to different TNF-family members, including CD40L25 and prevent ripoptosome formation. Therefore, targeting cIAP1/2 with smac-mimetics could be an attractive way to overcome CLL chemoresistance.
The aim of this study was to investigate the various adaptor proteins of the intracellular TNFR signalosome and ripoptosome in CD40-stimulated CLL cells. We explored whether smac-mimetics could mediate the degradation of cIAP1 and 2 and induce (TNFα-dependent) cell death alone or in combination with death receptor ligands. We found that although CLL cells express all proteins required for an effective cell death response to smac-mimetics, CLL cells were resistant to smac-mimetic-induced cell death. We hypothesize that this resistance may be linked to the rapid recurrence of cIAP2.
Results
CD40 stimulation of CLL cells induces non-canonical NF-κB activation and production of TNFα and its receptors TNFR1 and TNFR2
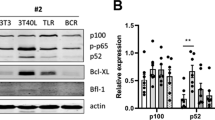
Activation of the non-canonical pathway, the subsequent production of TNFα and autocrine activation of TNFR1 and/or TNFR2 have been reported to be essential for smac mimetic-induced cell death in various cell types.14, 15 We have previously shown that prolonged CD40 stimulation induces the activation of the non-canonical NF-κB signaling pathway in CLL cells, following the activation of the canonical NF-κB signaling pathway.4 In agreement with our earlier report, we observed stabilization of NIK, phosphorylation of IκBα and the conversion of the p100 pro-form into the active NF-κB subunit p52 in patient CLL cells stimulated with CD40L (Figure 1a). The presence of cIAPs and RIPK1 in target cells is essential for the induction of cell death by smac-mimetics, because smac-mimetics target IAPs specifically while RIPK1 knockout cells are resistant to smac-mimetic-induced cell death.14, 15, 26 In line with a previous study,27 cIAP1 and 2 are both present in unstimulated PB CLL cells. Although the levels of cIAP1 and cIAP2 were unaffected by CD40 stimulation, TNF receptor-associated factor 2 (TRAF2) and RIPK1 levels were markedly enhanced (lane 3, Figure 1b). In line with previous studies,4, 14, 15 activation of non-canonical NF-κB was accompanied by the secretion of TNFα (Figure 1c). Cell-surface TNFR1 and TNFR2 expression measured by antibody staining and flow cytometry were also both significantly upregulated upon CD40 stimulation (Figure 1d).

Patient CLL cells stimulated with CD40L activate the non-canonical NF-κB pathway and the production of TNF, TNFR1 and TNFR2. (a) CLL cells were not cultured (−) or co-cultured for 48 h with 3T3 control feeder layer cells (3T3) or on CD40L-expressing 3T3 feeder layer cells (3T40) for 48 h. Lysates separated on SDS-PAGE gels and western blotted with the indicated antibodies, where actin was served as a loading control. Results of one representative CLL patient, of a total of nine analyzed, are shown. (b) CLL cells were treated as in a. Protein levels of cIAP1, cIAP2, TRAF2, TRAF6 and RIPK1 were analyzed by immunoblotting on total lysates. Blots from one representative CLL patient, of a total of three analyzed, are shown. (c) CLL cells were co-cultured with 3T3 or 3T40 cells for 72 h, after which supernatants were collected and TNFα levels were measured using ELISA. The means and S.E.M. of n=16 independent CLL samples are represented. (d) Expression levels of TNFR1 and TNFR2 were measured by flow cytometry of freshly isolated, 3T3 and 3T40 co-cultured CLL cells (n=8, independent CLL samples). Error bars represent S.E.M. Asterisks indicate statistical significance: *0.01<P<0.05 and **0.001<P<0.01
The smac-mimetic Compound A promotes proteasomal degradation of cIAP1 and cIAP2 but does not increase activation of non-canonical NF-κB in CD40-stimulated CLL cells
Our data showed that CD40 stimulation of primary patient CLL cells resulted in activation of the non-canonical NF-κB signaling pathway, TNFα secretion and upregulation of surface TNFR1 and TNFR2 expression, indicating that smac-mimetics might be effective in interfering with TNFα/NFκB signaling in CLL cells.
We therefore investigated the effect of the bivalent smac-mimetic Compound A15, 28 on cIAP1 and cIAP2 levels and on the activation of NF-κB. In agreement with earlier RNA studies,29 we observed using western blotting (Figure 2a) that CLL cells express low levels of cIAP1. The cIAP1 present rapidly disappeared with Compound A treatment. cIAP2 was expressed at readily detectable levels, which also quickly disappeared with Compound A, consistent with other studies,28, 30 and partially recovered after 4 h of treatment (Figure 2a). Bivalent smac-mimetics, such as Compound A, have been shown to induce cIAP1/2 degradation by promoting their proteasomal degradation.15, 28, 30 Alternatively, cIAPs can also reportedly be degraded via the lysosome, upon stimulation with TNF-family member TNF-related weak inducer of apoptosis (TWEAK),31 or compound A might block cIAP expression. We found that the proteasomal inhbitor MG132, but not the translation inhibitor cycloheximide (CHX) or a combination of reagents that block lysosomal activity, blocked the effects of Compound A (Figure 2b). This demonstrates that cIAP2 degradation occurs via the proteasome also in CLL cells. We have previously shown that p65 activity correlates with expression of the Bcl-2-related protein A1 (Bfl-1), whereas that of p52 correlates with B-cell lymphoma extra large (Bcl-XL) expression.4 Therefore, to assess the effect of Compound A on the activation of these two NF-κB subunits, we analyzed their DNA-binding activity on the promoters of these target genes. Consistent with the earlier studies,4 CD40L stimulation enhanced both p65 and p52 DNA-binding activity to their respective promoters (Figure 2c) and increased expression of Bfl-1 and Bcl-XL expression (Figure 2d). Compound A treatment activated both the pathways modestly in unstimulated cells, with a more marked increase in the p52/non-canonical pathway. In case of CD40-stimulated cells, compound A decreased p65- and p52-binding activity, Bcl-XL and Bfl-1 expression, and also TNFα secretion somewhat, but these effects did not reach statistical significance (Figures 2c–e).

The effect of CD40 stimulation and Compound A treatment on cIAP levels, NF-κB activity and target gene expression. (a) CLL cells were co-cultured with 3T3 cells (left) or 3T40 cells (right) in the presence of Compound A (250 nM) for 1, 4 and 24 h. Immunoblotting was performed for cIAP1 and cIAP2 on total lysates, and actin was used as a loading control. Blots from a representative CLL patient, of a total of three (left) or 4 (right), are shown. (b) CLL cells co-cultured with 3T40 cells were treated for 4 h with the indicated compounds in the following concentrations: Compound A 250 nM, CHX 20 μg/ml, MG132 500 nM, bafilomycin A 100 nM and ammonium chloride 25 μM. Immunoblotting was performed for cIAP2 on total lysates, and actin was used as a loading control. Blots are representative of two CLL patients. Patient samples used were nos. 39 and 40 from Table 1. (c) CLL cells were co-cultured with 3T3 or 3T40 cells in the presence or absence of Compound A (250 nM) for 24 h. Activity of the NF-κB subunits p65 and p52, as a measure of activity of the canonical and non-canonical NF-κB pathways respectively, was determined using ELISA with the TransAM NFκB family transcription factor assay kit (n=3). Error bars represent S.E.M. (d) CLL cells were treated as in c, but for 48 h. Bcl-XL and Bfl-1 mRNA expression levels were analyzed by RT-MLPA (n=5), and depicted as percentage of the total signal in that sample. β-Glucuronidase is depicted as a housekeeping gene not responsive to NFκB. Error bars present S.E.M. (e) CLL cells were treated as in c, but for 72 h. Supernatants were collected, and TNFα levels were measured using ELISA (n=16). Error bars represent S.E.M.
CLL cells are resistant to smac mimetic-mediated cell death
Our results suggested that the CLL cells could be sensitive to cell death induced by smac-mimetic and particularly sensitive to combined CD40L and smac-mimetic treatment, in view of the enhanced NF-κB signaling and TNFα production. Remarkably, however, not only unstimulated CLL cells but also CD40-stimulated CLL cells were insensitive to Compound A (Figures 3a and b, left panel). This was tested for >20 CLL samples in order to investigate whether (prognostic) subgroups might be sensitive, but this turned out not to be the case (see also Table 1 for patient characteristics). Only the highest dose of Compound A applied (500 nM) induced apoptosis in some CD40-stimulated CLL samples (Figure 3a). Moreover, both unstimulated and CD40-stimulated CLL cells were also unresponsive to a second bivalent smac-mimetic, smac-mimetic 83 (SM83) (Figure 3b, right panel).32 As a control, the sensitive rhabdymyosarcoma cell line Kym-115 was treated with increasing concentrations of Compound A or SM83, which resulted in high levels of apoptosis already at 1 nM (Figure 3b). The slight increase in apoptosis induced by 500 nM Compound A in CD40-stimulated CLL cells could not be blocked by anti-TNFα, suggesting that this process was TNFα independent. In addition, and consistent with the fact that TNFα is already produced by CD40L-stimulated cells, no significant increase in apoptosis of CD40-stimulated CLL cells was observed when exogenous TNFα was combined with Compound A (Figure 3c). Several studies have shown that smac-mimetics can sensitize different types of cancer cells to apoptosis induction by TNF superfamily members Fas ligand (FasL/CD95L/TNFSF6) and TNF-related apoptosis inducing ligand (TRAIL) (TNFSF10).23, 32, 33, 34, 35, 36, 37 However, we did not observe synergistic effects in CLL cells (Figure 3d). The pro-apoptotic activity of FasL and TRAIL was verified with Jurkat T cells, which readily underwent apoptosis upon exposure to TRAIL and FasL (data not shown). CD40L stimulation enhanced the expression of anti-apoptotic Bcl-2 proteins, which could contribute to Compound A resistance (Figure 2d). We therefore specifically inhibited Bcl-2 and Bcl-XL with the compound ABT-737 to assess this possibility, using concentrations of ABT-737 that induce modest apoptosis in CD40-stimulated CLL cells.2, 38 However, CD40-stimulated CLL cells could not be sensitized to Compound A with ABT-737, indicating that induction of pro-survival Bcl-2 family members by CD40 stimulation does not mediate resistance to Compound A in CLL cells (Figure 3e). In addition, no synergistic effects of Compound A with a range of cytotoxic drugs, such as fludarabine, proteasome inhibitor bortezomib, HDAC inhibitors suberohydroxamic acid (SBHA) and trichostatin A, syk inhibitors R406 and piceatannol, Src/Abl inhibitor dasatinib or NF-κB inhibitor Bay were observed for CLL cells (data not shown).

The effects of smac mimetics, alone or in combination with death receptor ligands or ABT-737, on apoptosis in unstimulated and CD40-stimulated CLL cells. (a) CLL cells were co-cultured with 3T3 (n=13) or 3T40 cells (n=26) for 72 h in the presence or absence of the indicated concentrations of Compound A. Apoptosis levels were measured with annexin V/PI staining, and averaged data are shown. Patient characteristics are summarized in Table 1. For 3T3 conditions nos. 1, 3, 5, 7–9, 13, 23–26, 28 and 29 were used, and for 3T40 conditions nos 1, 2, 4–7, 9–15 and 18–30 were used. (b) CLL cells co-cultured with 3T3 (n=4) or 3T40 cells (n=4) were treated for 48 h with the indicated concentrations of Compound A or SM83. The rhabdomyosarcoma cell line Kym-1, known to be sensitive to treatment with smac mimetics was used as a positive control and was treated with the same concentrations for 24 h. Apoptosis was analyzed by DioC6/PI staining. Error bars represent S.D. Patient samples used were nos. 31, 32, 33 and 35 from Table 1. (c) CLL cells co-cultured with 3T40 cells were treated with Compound A (250 nM) or recombinant TNFα (5 μg/ml) alone or together, or a combination of Compound A and TNFR1/2 blocking reagent Etanercept (10 μg/ml; indicated as TNFα) for 72 h (n=4). Apoptosis was measured by annexin V/PI staining. Error bars represent S.E.M. and ‘***’ indicates a statistical significance of P<0.001. Patient samples used were nos. 1,2,12 and 18 from Table 1. (d) CLL cells co-cultured with 3T40 cells were treated with a combination of Compound A (250 nM) and FasL (10 or TRAIL (100 ng/ml) for 48 h (n=3), and apoptosis was determined by annexin V/PI staining. Error bars represent S.E.M. and ‘**’ indicates a statistical significance of 0.001<P<0.01. (e) CLL cells co-cultured with 3T40 cells were treated with a combination of Compound A (1–500 nM) and ABT-737 (100 nM) for 48 h (n=3), and apoptosis was determined by DioC6/PI staining. Error bars represent S.E.M. Patient samples used were nos. 31, 39 and 40 from Table 1
Primary CLL cells are resistant to combination treatment with Compound A and TNFR1/2-specific TNFα mutants
In contrast to TNFR1, TNFR2 does not contain a death domain and can only activate NF-κB.39, 40, 41, 42 On average, we observed a strong increase in TNFR2 levels and a moderate enhancement of TNFR1 levels in CLL cells upon CD40 stimulation. Thus, potentially, TNFR2 could sequester the TNFα produced in CD40-stimulated cells and thereby antagonize pro-death TNF/TNFR1 signaling. To study this possibility, we treated CLL cells with TNFR1- and TNFR2-selective TNFα mutants (TNFα-TNFR1 and TNFα-TNFR2, respectively), in combination with Compound A. Two patient samples were studied, one with relatively high levels of TNFR2 (patient 38 from Table 1) and one with relatively high levels of TNFR1 (patient 18), in CD40-stimulated cells (Figure 4a). Pt-38 resembled the majority of patients (as depicted in Figure 1d). In both samples, combination treatment with Compound A and TNFα-TNFR1 did not induce apoptosis in unstimulated nor in CD40-stimulated CLL cells (Figures 4b and c). Decoy TNFR agent Etanercept was added to prevent TNFR2 signaling from TNFα produced by CD40-stimulated CLL cells, but again no differences in apoptosis were observed (Figure 4c). We measured whether expression of Fas receptor increased43 in response to the TNFR stimulation. Especially, in pt-18, we observed an increase of Fas expression upon addition of both TNFα variants, verifying that they were active (Figure 4d).

The effects of Compound A in combination with specific TNFR1/2 stimulation in CLL cells. CLL cells of a representative patient, which upregulate TNFR2 in response to CD40 stimulation (Pt-38 from Table 1), and cells of a patient that upregulate TNFR1 rather than TNFR2 (Pt-18 from Table 1) were selected for treatment with Compound A in combination with specific TNFR1/2 agonistic antibodies. (a) TNFR1 and TNFR2 expression levels on CLL cells of Pt-1 and Pt-2 co-cultured with control or 3T40-expressing 3T3 cells as measured with antibody staining, using flow cytometry. (b) CLL cells of Pt-1 and Pt-2 co-cultured with control 3T3 cells were treated with Compound A (50–500 nM) alone or in combination with TNFα mutants (100 ng/ml) that preferentially bind to TNFR1 (TNFα-TNFR1) or TNFR2 (TNFα-TNFR2). (c) CLL cells of Pt-1 and Pt-2 co-cultured with 3T40 cells were treated with Compound A (50–500 nM) alone or in combination with TNFα-TNFR1 (1 μg/ml) or TNFα-TNFR2 (1 μg/ml). Etanercept (10 μg/ml) was added to block endogenous TNFα activity. (d) Percentage of Fas expression in CLL cells co-cultured with 3T3 cells, in the presence and absence of TNFα-TNFR1 (1 μg/ml) and TNFα-TNFR2 (1 μg/ml), as measured with antibody staining using flow cytometry
CLL cells are unable to form the ripoptosome upon treatment with Compound A
In TNF-stimulated cells, the absence of cIAPs induces the formation of a cytosolic complex 2 with more RIPK1. Increased levels of death domain-containing RIPK1 are able to recruit and drive activation of caspase-8-causing apoptosis, or if caspase-8 activity is limiting, auto-activate and thereby phosphorylate RIPK3 and cause necroptosis. This complex is probably identical with the ripoptosome complex that can form in the absence of ligand stimulation but which can be augmented by diverse stimuli, including Fas, TRAIL, TLR ligands and DNA damage.22, 23, 24 To study whether CLL cells are able to form such a ripoptosome complex following cIAP1/2 degradation, we performed a caspase-8 immunoprecipitation (IP) with control and Compound A-treated cells and immunoblotted for RIPK1 and FADD.14 In Compound A-sensitive Kym-1 cells, we were readily able to detect such a complex 2/ripoptosome. In contrast, both in CD40L-stimulated (Figures 5a and b, right panels) and unstimulated CLL cells (Figure 5c), RIPK1 and FADD did not co-immunoprecipitate with caspase-8, indicating that CLL cells are unable to form the complex in response to Compound A. Different cFLIP isoforms have been reported to regulate the formation of the ripoptosome. The long cFLIP isoform, cFLIPL, represses the formation of the ripoptosome, whereas the short isoform, cFLIPS, promotes ripoptosome assembly.22, 23 Incorporation of cFLIPL into the ripoptosome could thus be a cause of Compound A resistance in CLL cells. However, we found that Kym-1 incorporated cFLIPL into the ripoptosome, whereas CLL cells did not incorporate either form of FLIP (Figure 5b), excluding this as a possible explanation for the lack of response to smac mimetics.

CLL cells are unable to form the ripoptosome in response to Compound A. (a–c) IP of caspase-8 in Kym-1 and CLL cells co-cultured with 3T40 cells (a and b) or not (c) (numbers refer to CLL cases in Table 1), untreated or treated with Compound A, followed by detection of ripoptosome components caspase-8, RIP-1, FADD and cFLIPL. CLL cells were co-cultured on 3T40 cells for 96 h, collected and treated with 100 nM Compound A for 4 h (a) or 24 h (b) or directly treated with 500 nM Compound A for 4 h (c). Kym-1 cells were exposed to 20 nM Compound A for 4 h. Caspase-8 IP was performed from total lysates. (a) Pre-IP, post-IP and IP samples were prepared, and western blotting was performed for caspase-8 and RIPK1. Actin serves as a loading control. (b) Pre-IP and IP samples were prepared, and western blotting was performed for caspase-8, RIPK1, FADD and cFLIPL. Actin serves as a loading control. *marks an aspecific band. (c) Pre-IP and IP samples were prepared, and western blotting was performed for caspase-8 and RIPK1. Actin serves as a loading control
Discussion
We show that CLL cells express all the components required for ripoptosome/complex 2 formation in response to smac-mimetic treatment, and that CD40 stimulation further enhances the levels of TNFR1, TRAF2 and RIPK1. Yet, despite the fact that smac-mimetics do induce the degradation of cIAP1/2 in CLL cells, they are resistant to smac-mimetic-induced cell death.
As we previously observed,4 CD40 stimulation activated the canonical and non-canonical NF-κB signaling pathways. CLL cells express cIAP1, cIAP2, TRAF2, RIPK1, caspase-8 and FADD, and CD40 stimulation further enhanced the levels of TRAF2 and RIPK1. Furthermore, CD40 stimulation mediated TNFα production and upregulation of TNFα receptors TNFR1 and TNFR2. This suggested that CD40-stimulated CLL cells might be particularly sensitive to smac mimetic treatment. However, although cIAP1 and cIAP2 are degraded via the proteasome in response to smac mimetic Compound A, CLL cells are resistant to cell death induction by Compound A15 and a second smac mimetic, SM83.32 Both agents were functional, as they could efficiently kill the rhabdymyosarcoma Kym-1 cell-line.15 Compound A was also ineffective in killing CLL cells in combination with TNFα, FasL or TRAIL. Previously, it was found that XIAP inhibitors can synergistically kill primary CLL cells when combined with FasL or TRAIL.37, 44 Surprisingly, therefore, selective smac-mimetics seem to be less effective than pan-IAP inhibitors, when combined with death receptor ligands in CLL cells.
We explored several possible explanations for the resistance of CLL cells to cell death induction by smac mimetics. First, we observed that cIAP2 levels recovered during treatment. This phenomenon has been previously described by Petersen et al.45 in lung carcinoma cell lines and by Darding et al.30 in colon carcinoma and melanoma cell lines. Cell lines that were resistant to smac mimetics showed initial degradation of cIAP2, but levels were restored after 3 h of treatment.30, 45 cIAP2 is a well-known NF-κB target, and we therefore anticipated that its recovery was the cause of smac mimetic-mediated NF-κB activation.30, 45 Yet, general NF-κB activity and TNF production was unaffected by Compound A in CD40-stimulated CLL cells. Because NF-κB activity encompasses a number of potential heteromeric transcription factors, we also examined p65- and p52-specific DNA-binding and expression of their respective target genes Bfl-1 and Bcl-XL, but these were also undisturbed by Compound A treatment. Next to NF-κB, phospho-inositide 3 kinase (PI3K) signaling can also regulate cIAP2 levels. In the study of Petersen et al.,45 cIAP2 recovery upon smac mimetic treatment could be suppressed with PI3K inhibitor LY294002, which abrogated resistance to smac mimetics. In our hands, LY294002 did not sensitize unstimulated nor CD40-stimulated CLL cells to Compound A (data not shown). Thus, NF-κB and PI3K do not appear to have a role in the cIAP2 reexpression in CLL cells.
Second, we studied whether CD40L-mediated Bcl-XL upregulation might contribute to Compound A resistance in CD40L-stimulated CLL cells. ABT-737, which specifically inhibits Bcl-XL and its close relative Bcl-2, could not sensitize to Compound A, arguing against a contribution of increased levels of Bcl-XL in resistance against smac mimetics.
Third, we investigated whether enhanced production of TNFR2, which we observed after CD40 stimulation, was the cause of resistance. As TNFR2 lacks a death domain and hence cannot induce cell death,39, 40, 41, 42 it could potentially sequester the TNFα produced in response to CD40L, and thus act as a decoy receptor. However, Compound A combined with a TNFR1-selective TNFα mutant (TNFα-TNFR1) did not induce cell death, not even in CLL cells with relatively high TNFR1 levels and low TNFR2 levels. As expected, the combination with a TNFR2-selective TNFα mutant (TNFα-TNFR2) was also not effective.
Fourth, we assessed a role for cFLIPL. CFLIP isoforms cFLIPS and cFLIPL have been reported to regulate the activity of the ripoptosome.23 Although cFLIPS promotes ripoptosome formation, cFLIPL represses the formation of the ripoptosome. cFLIPL does so by forming a heterodimer with caspase-8, which cleaves RIPK1 and RIP3.22, 46 We therefore reasoned that CLL cells might incorporate enhanced levels of cFLIPL in its ripoptosome and prevent induction of cell death. IP of caspase-8 showed that the ripoptosome formed in Kym-1 cells upon treatment with Compound A. Unexpectedly, although Kym-1 cells are exquisitely sensitive to Compound A, cFLIPL was nevertheless found to be incorporated in the complex. Kym-1 is a rather unique rhabdomyosarcoma cell line that is particularly sensitive to the cytotoxic activity of autocrine TNF induced when they are treated with TWEAK, CD40L, CD30L and even by TNFR2 stimulation alone.31, 47, 48 Thus, we speculate that this is a question of a low threshold for cell death and can therefore still be induced by a complex 2/ripoptosome in the presence of cFLIPL. In contrast, both unexposed and CD40L-exposed CLL cells failed to form the complex in response to high Compound A dosages. We could not detect any RIPK1, FADD or cFLIPL immunoprecipitating with caspase-8. Thus, cFLIPL does not have a role in CLL resistance to smac mimetics. Rather, the ripoptosome complex is not formed at all.
Finally, defective ripoptosome formation might derive from aberrant upstream NF-κB regulation. Cylindromatosis protein (CYLD) regulates ripoptosome formation by deubiquitinating enzyme RIPK1.49 In the absence of CYLD, RIPK1 is no longer deubiquitinated and thus is prevented from translocating to the ripoptosome. Lymphoid enhancer-binding factor 1 (LEF1) has been identified as a transcriptional repressor of CYLD.50 Compared with healthy B cells, CLL cells were reported to express relatively high levels of LEF1 and low levels of CYLD, causing resistance to TNFα-induced necroptosis.50 High LEF1 and low CYLD levels in CLL cells could in theory also explain CLL inability to form the ripoptosome. However, in five samples tested, we did not observe a negative correlation between LEF1 and CYLD levels in CLL (results not shown) and found no indication that this might be the cause.
In conclusion, we show that CD40 stimulation of CLL cells mediates the activation of the canonical and non-canonical NF-κB pathways, TNFα production and upregulation of both TNFR1 and TNFR2. Although unstimulated and CD40-stimulated CLL cells respond to smac mimetic treatment by degrading cIAP1 and cIAP2 via the proteasome, they are unable to form the ripoptosome and die via apoptosis or necroptosis. We have excluded decoy receptor activity of TNFR2, increased levels of Bcl-XL, cFLIPL inhibition of the ripoptosome and a possible contribution of CYLD as causal factors in smac mimetic resistance in CLL cells. Most likely, rapid recovery of cIAP2 levels during treatment contributes to the resistance. Further elucidation of the molecular mechanism of disturbed ripoptosome formation may offer novel therapeutic targets for CLL.
Materials and Methods
Patient material
After informed consent, patient material was obtained during diagnostic or follow-up procedures at the departments of Hematology and Pathology of the Academic Medical Center, Amsterdam. This study was approved by the AMC Ethical Review Board and conducted in agreement with the Helsinki Declaration of 1975, revised in 1983. PB mononuclear cells of patients with CLL obtained after Ficoll density gradient centrifugation (Pharmacia Biotech, Roosendaal, The Netherlands) were frozen and stored as described.2 Expression of CD5 and CD19 (both purchased from Beckton Dickinson (BD) Biosciences, San Jose, CA, USA) on leukemic cells was assessed by flow cytometry (FACScalibur; BD Biosciences) and analyzed with CellQuest software (BD Biosciences). Patient characteristics are reported in Table 1.
Reagents
Compound A was obtained under MTA from TetraLogic Pharmaceuticals (Malvern, PA, USA) and was dissolved in dimethyl sulfoxide (DMSO). SM8332 was kindly provided by Dr. D Lecis and Dr. D Delia (Department of Experimental Oncology, Fondazione IRCCS Instituto Nazionale Tumori, Milano, Italy). ABT-737 was obtained from Abbott (Abbott Park, Green Oaks, IL, USA).2, 38 SuperKiller TRAIL was purchased from Alexis Biochemicals (San Diego, CA, USA). TNFα-blocking reagent (Etanercept) was obtained from Pfizer (Kent, UK). Recombinant human TNFα was obtained from Invitrogen (Carlsbad, CA, USA). TNFR1 (FITC) (Lot no. LFA 015071) and TNFR2 (PE) (Lot no. LFB 046021), used for flow cytometry, were obtained from R&D systems (Minneapolis, MN, USA). Fas (CD95) antibody (BD Biosciences; Lot no. 29991) was used for detection of Fas by flow cytometry. Recombinant anti-human Fas monoclonal antibody (FAS10) was obtained from the CLB Sanquin Blood Foundation (CLB, Amsterdam, The Netherlands). The production and characterization of CysTNF32W/86T, referred to as TNFα-TNFR1, and CysTNF143N/145R, referred to as TNFα-TNFR2, has been described elsewhere.51, 52 CHX, bafilomycin A and ammonium chloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). MG132 was obtained from Calbiochem/Merck Millipore, Billerica, MA, USA). The pan-caspase inhibitor QVD was from R&D systems (Minneapolis, MN, USA).
Cell culture and stimulations
Control (3T3 fibroblast cell line (3T3)) and human CD40L stably transfected (CD40L-expressing 3T3 cell (3T40)) NIH3T3 feeder cells were cultured in Iscove’s modified Dulbecco's medium (IMDM; Gibco Life Technology, Paisley, FL, USA) supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml gentamycin and 0.00036% β-mercaptoethanol. PB lymphocytes of CLL patients were co-cultured with control 3T3 cells or CD40L-expressing 3T3 cells, as described previously.2 Briefly, DMSO-frozen CLL cells were thawed and co-cultured on irradiated (30 Gy) control or CD40L-expressing 3T3 cells. The final CLL cell concentration was 1.67 × 106 cells/ml in IMDM. Kym-1 cells were cultured in RPMI medium (Gibco Life Technology) supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml gentamycin and 0.00036% β-mercaptoethanol. Stimulations of CLL cells with Compound A, alone or in combination with other reagents, were performed simultaneously with the incubation on 3T3 or 3T40 cells. For the caspase-8 IP experiment, CLL cells were collected after co-culturing on 3T40 cells, to be treated with Compound A in a new cell culture dish.
Apoptosis assays
Apoptosis levels were determined with annexin V-APC (IQ Products, Groningen, The Netherlands) and propidium iodide (PI) staining (Sigma, St. Louis, MO, USA) or 3,3′-dihexyloxacarbocyanine iodide (DioC6; Invitrogen) and PI and flow cytometry.2
Western blot and antibodies
Western blotting was performed as described previously.2 Samples (30–60 μg protein) were separated by 7.5–13% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The caspase-8 (clone C-20, no. sc-6136) IP antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Blots were probed with polyclonal NF-κB2 p100/p52 (no. 4882; Cell Signaling, Beverly, MA, USA), monoclonal mouse antibody for p-IκBα (no. 9246, Cell Signaling), NIK (no. 4994; Cell Signaling), TRAF2 (no. sc-876; Santa Cruz Biotechnology), TRAF6 (no. 4743s; Cell Signaling), caspase-8 (clone 5F7, no. M032-3; MBL, Woburn MA, USA), RIPK1 (no. 610458; BD Biosciences), rat polyclonal antibodies against cIAP1 and cIAP2 (La Trobe University, Melbourne, Australia) and antiserum to actin (clone I-19, no. sc-1616; Santa Cruz Biotechnology). IRDye 680 donkey anti-rat IgG, IRDye 680 donkey anti-rabbit IgG, IRDye 800 donkey anti-goat IgG or IRDye 800 donkey anti-mouse IgG (Westburg, Leusden, The Netherlands) was used as secondary antibody. Western blots were scanned on the Odyssey imager (LI-COR Biosciences, Lincoln, NE, USA).
Enzyme-linked immunosorbent assay
For the determination of TNFα protein levels, PB lymphocytes of CLL patients were co-cultured with 3T40 cells (as described above), supernatant was collected and TNFα levels were measured using the PeliKine human enzyme-linked immunosorbent assay (ELISA) kit (Sanquin, Amsterdam, The Netherlands) according to the manufacturer’s recommendations. Activity of active NF-κB subunits p65 and p52 was also measured using ELISA, using the TransAM NF-κB family transcription factor assay kit (Active Motif, La Hulpe, Belgium), according to the manufacturer’s instructions.
RNA isolation and reverse transcription–multiplex ligation-dependent probe amplification assay
Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). Reverse transcription–multiplex ligation-dependent probe amplification assay (RT-MLPA) procedure was performed as described previously.29, 53 Only the mRNA levels of NF-κB target genes are shown. Equal amounts of mRNA were included per reaction, and all samples were tested in a single experiment using the same batch of reagents. Relative mRNA expression of the gene of interest is calculated by setting the total signal for each sample at 100%, and individual signals of genes of interest were calculated relative to the 100% value.
Statistical analysis
The Shapiro–Wilk normality test was performed to analyze Gaussian distributions. If there was a Gaussian distribution, a two-sided t-test was used to analyze differences between the groups. If there was no Gaussian distribution, a two-tailed Mann–Whitney U-test was used to analyze differences between the groups, and a Wilcoxon matched paired test was used to analyze differences between paired samples. Statistically significance of the data was set at P<0.05, with one asterisk (*) representing 0.01< P<0.05; two asterisks (**) representing 0.001<P<0.01; and three asterisks(***) representing P<0.001.
Abbreviations
- BD:
-
Beckton Dickinson
- DMSO:
-
dimethyl sulfoxide
- IMDM:
-
Iscove’s modified Dulbecco's medium
- ELISA:
-
enzyme-linked immunosorbent assay
- 3T3:
-
3T3 fibroblast cell line
- 3T40:
-
CD40L-expressing 3T3 cell
- Bcl-XL:
-
B-cell lymphoma extra large
- Bfl-1:
-
Bcl-2-related protein A1
- BIR:
-
baculovirus inhibitor of apoptosis repeat
- CD40L:
-
CD40 ligand
- cFLIP1/2:
-
FLICE-like inhibitory protein 1/2
- CHX:
-
cycloheximide
- cIAP1/2:
-
cellular inhibitor of apoptosis protein 1/2
- CLL:
-
chronic lymphocytic leukemia
- CYLD:
-
cylindromatosis protein
- DioC6:
-
3,3′-dihexyloxacarbocyanine iodide
- FADD:
-
Fas-associated protein with death domain
- FasL:
-
Fas ligand
- IAP:
-
inhibitor of apoptosis protein
- IP:
-
immunoprecipitation
- LEF1:
-
lymphoid enhancer-binding factor 1
- LN:
-
lymph node
- NF-κB:
-
nuclear factor kappa B
- NIK:
-
NF-κB-inducing kinase
- PB:
-
peripheral blood
- PI:
-
propidium iodide
- PI3K:
-
phospho-inositide 3 kinase
- RIPK1:
-
receptor-interacting protein kinase 1
- SBHA:
-
suberohydroxamic acid
- SM83:
-
smac-mimetic 83
- TNFα:
-
tumor necrosis factor-α
- TNFR1/2:
-
tumor necrosis factor receptor 1/2
- TRAF2:
-
TNF receptor-associated factor 2
- TRAIL:
-
TNF-related apoptosis inducing ligand
- TWEAK:
-
TNF-related weak inducer of apoptosis
- XIAP:
-
X-linked inhibitor of apoptosis protein
References
Chiorazzi N, Rai KR, Ferrarini M . Chronic lymphocytic leukemia. N Engl J Med 2005; 352: 804–815.
Hallaert DY, Jaspers A, van Noesel CJ, van Oers MH, Kater AP, Eldering E . c-Abl kinase inhibitors overcome CD40-mediated drug resistance in CLL: implications for therapeutic targeting of chemoresistant niches. Blood 2008; 112: 5141–5149.
Kater AP, Evers LM, Remmerswaal EB, Jaspers A, Oosterwijk MF, van Lier RA et al. CD40 stimulation of B-cell chronic lymphocytic leukaemia cells enhances the anti-apoptotic profile, but also Bid expression and cells remain susceptible to autologous cytotoxic T-lymphocyte attack. Br J Haematol 2004; 127: 404–415.
Tromp JM, Tonino SH, Elias JA, Jaspers A, Luijks DM, Kater AP et al. Dichotomy in NF-kappaB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene 2010; 29: 5071–5082.
Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood 1998; 91: 3379–3389.
Duckett CS, Nava VE, Gedrich RW, Clem RJ, van Dongen JL, Gilfillan MC et al. A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J 1996; 15: 2685–2694.
Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996; 379: 349–353.
Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV . The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995; 83: 1243–1252.
Uren AG, Pakusch M, Hawkins CJ, Puls KL, Vaux DL . Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc Natl Acad Sci USA 1996; 93: 4974–4978.
Hinds MG, Norton RS, Vaux DL, Day CL . Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nat Struct Biol 1999; 6: 648–651.
Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 1999; 401: 818–822.
Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 2004; 47: 4417–4426.
Sun H, Nikolovska-Coleska Z, Lu J, Qiu S, Yang CY, Gao W et al. Design, synthesis, and evaluation of a potent, cell-permeable, conformationally constrained second mitochondria derived activator of caspase (Smac) mimetic. J Med Chem 2006; 49: 7916–7920.
Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007; 12: 445–456.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131: 682–693.
Eckelman BP, Salvesen GS, Scott FL . Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 2006; 7: 988–994.
Eckelman BP, Salvesen GS . The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem 2006; 281: 3254–3260.
Huang H, Joazeiro CA, Bonfoco E, Kamada S, Leverson JD, Hunter T . The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem 2000; 275: 26661–26664.
Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 2008; 105: 11778–11783.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283: 24295–24299.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol 2009; 187: 1037–1054.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43: 432–448.
Knox PG, Davies CC, Ioannou M, Eliopoulos AG . The death domain kinase RIP1 links the immunoregulatory CD40 receptor to apoptotic signaling in carcinomas. J Cell Biol 2011; 192: 391–399.
Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J . RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ 2010; 17: 482–487.
Munzert G, Kirchner D, Stobbe H, Bergmann L, Schmid RM, Dohner H et al. Tumor necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: analysis of NF-kappa B/Rel-regulated inhibitors of apoptosis. Blood 2002; 100: 3749–3756.
Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J Biol Chem 2011; 286: 17015–17028.
Mackus WJ, Kater AP, Grummels A, Evers LM, Hooijbrink B, Kramer MH et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced Puma upregulation. Leukemia 2005; 19: 427–434.
Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J et al. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ 2011; 18: 1376–1386.
Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182: 171–184.
Lecis D, Drago C, Manzoni L, Seneci P, Scolastico C, Mastrangelo E et al. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. Br. J. Cancer 2010; 102: 1707–1716.
Abhari BA, Cristofanon S, Kappler R, von SD, Humphreys R, Fulda S . RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene 2012; 32: 3263–3273.
Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR . Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat 2013; 137: 359–371.
Basit F, Humphreys R, Fulda S . RIP1 protein-dependent assembly of a cytosolic cell death complex is required for inhibitor of apoptosis (IAP) inhibitor-mediated sensitization to lexatumumab-induced apoptosis. J Biol Chem 2012; 287: 38767–38777.
Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG . A small molecule Smac mimic potentiates T. Science 2004; 305: 1471–1474.
Loeder S, Zenz T, Schnaiter A, Mertens D, Winkler D, Dohner H et al. A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res 2009; 69: 8977–8986.
Tromp JM, Geest CR, Breij EC, Elias JA, van LJ, Luijks DM et al. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res 2012; 18: 487–498.
Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV . TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996; 4: 387–396.
Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MA Jr., Goeddel DV . The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci USA 1991; 88: 9292–9296.
Tartaglia LA, Ayres TM, Wong GH, Goeddel DV . A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993; 74: 845–853.
Tartaglia LA, Rothe M, Hu YF, Goeddel DV . Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell 1993; 73: 213–216.
Lens SM, Tesselaar K, den Drijver BF, van Oers MH, van Lier RA . A dual role for both CD40-ligand and TNF-alpha in controlling human B cell death. J Immunol 1996; 156: 507–514.
Kater AP, Dicker F, Mangiola M, Welsh K, Houghten R, Ostresh J et al. Inhibitors of XIAP sensitize CD40-activated chronic lymphocytic leukemia cells to CD95-mediated apoptosis. Blood 2005; 106: 1742–1748.
Petersen SL, Peyton M, Minna JD, Wang X . Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci USA 2010; 107: 11936–11941.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471: 363–367.
Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J 1999; 18: 3034–3043.
Schneider P, Schwenzer R, Haas E, Muhlenbeck F, Schubert G, Scheurich P et al. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur J Immunol 1999; 29: 1785–1792.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia 2011; 26: 1293–1300.
Krippner-Heidenreich A, Tubing F, Bryde S, Willi S, Zimmermann G, Scheurich P . Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J Biol Chem 2002; 277: 44155–44163.
Loetscher H, Stueber D, Banner D, Mackay F, Lesslauer W . Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J Biol Chem 1993; 268: 26350–26357.
Eldering E, Spek CA, Aberson HL, Grummels A, Derks IA, de Vos AF et al. Expression profiling via novel multiplex assay allows rapid assessment of gene regulation in defined signalling pathways. Nucleic Acids Res 2003; 31: e153.
Acknowledgements
We are grateful to Dr. D Lecis and Dr. D Delia (Department of Experimental Oncology, Fondazione IRCCS Instituto Nazionale Tumori, Milano, Italy) for providing SM83. This research was funded by the Dutch Cancer Society (grant UVA 2007-3856—JMT, JAE, EE and MHJvO) and the Stichting Hematologisch Onderzoek (SHO) (CM and JvL). JS is a member of the Scientific Advisory Board of TetraLogic Pharmaceuticals and is supported by an NHMRC Fellowship 541901.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Maas, C., Tromp, J., van Laar, J. et al. CLL cells are resistant to smac mimetics because of an inability to form a ripoptosome complex. Cell Death Dis 4, e782 (2013). https://doi.org/10.1038/cddis.2013.305
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.305
