
Abstract
Cell culture of human-derived neural stem cells (NSCs) is a useful tool that contributes to our understanding of human brain development and allows for the development of therapies for intractable human brain disorders. Human NSC (hNSC) cultures, however, are not commonly used, mainly because of difficulty with consistently maintaining the cells in a healthy state. In this study, we show that hNSC cultures, unlike NSCs of rodent origins, are extremely sensitive to insulin, an indispensable culture supplement, and that the previously reported difficulty in culturing hNSCs is likely because of a lack of understanding of this relationship. Like other neural cell cultures, insulin is required for hNSC growth, as withdrawal of insulin supplementation results in massive cell death and delayed cell growth. However, severe apoptotic cell death was also detected in insulin concentrations optimized to rodent NSC cultures. Thus, healthy hNSC cultures were only produced in a narrow range of relatively low insulin concentrations. Insulin-mediated cell death manifested not only in all human NSCs tested, regardless of origin, but also in differentiated human neurons. The underlying cell death mechanism at high insulin concentrations was similar to insulin resistance, where cells became less responsive to insulin, resulting in a reduction in the activation of the PI3K/Akt pathway critical to cell survival signaling.
Similar content being viewed by others


Main
Neural stem cells (NSCs) have the potential for self-renewal and differentiation toward neurons and glia, and are important in the development of the brain.1, 2 Small populations of NSCs remain in the adult mammalian brain even after development is complete;3, 4 these cells contribute to regenerative processes in response to brain damage.5, 6 NSCs can be isolated and cultured from developing and adult brains; these cultured NSCs retain the cardinal properties of NSCs in the brain, such as region-specific gene expression and neuronal subtype differentiation patterns.7 In addition, differentiation of cultured NSCs can be guided in vitro to yield specific neuronal subtypes for cell-based therapeutic approaches to neurodegenerative disorders.8, 9 Thus, NSC cultures are an invaluable experimental tool in the study of brain development and regeneration.
The culture technique for NSCs, like many other experimental systems, was initially developed using rodent cells.10 Although rodent-derived cultures remain informative and are still widely used, studies that use human cells are inherently more applicable in understanding human physiology and disease. In this regard, culturing human NSCs (hNSCs) has been attempted by direct isolation from human brain tissues.11, 12 In addition, hNSC cultures have been generated through in vitro differentiation of human pluripotent stem cells (hPSCs), which include human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs).13, 14, 15 In contrast to the limited proliferative and storage capacities displayed by rodent NSCs (roNSCs), hNSCs are extensively expandable, and can be frozen and re-cultured without altering their cardinal features.16, 17 Despite these benefits, hNSC cultures are less commonly used, mainly because of the difficulties involved in consistently maintaining healthy cell cultures. Often, culture conditions optimized for roNSCs are adopted for use in hNSC culturing. In this study, we show a clear distinction in rodent and human cells’ responses to the same culture conditions. Human NSC cultures, unlike those of rodents, are extremely sensitive to insulin concentration, a necessary supplement in the culturing of neural cell lineages.18, 19 Prolonged exposure at the concentrations optimized for roNSCs is toxic to hNSCs, which can be explained by decreased Akt intracellular signaling secondary to insulin resistance. Complete withdrawal of insulin from culture conditions was also detrimental to hNSCs, resulting in reduced cell survival and/or proliferation. As a result, healthy hNSC cultures were achievable only within a narrow range of relatively low insulin concentrations. Insulin-mediated cell death also manifested itself in human-specific neuronal cells, providing the first in vitro evidence of human neuronal cell death via insulin resistance, which provides some explanation for the high prevalence of neurodegenerative disorders among hyperinsulinemic diabetic patients.20, 21
Results
Insulin can act as an apoptotic factor for hNSCs derived from hESCs
Human pluripotent stem cells such as hESCs and hiPSCs are reliable sources for hNSCs.22 A uniform population (>90%) of hNSCs expressing the NSC-specific marker Nestin and the proliferating cell marker Ki67 was isolated by in vitro differentiation of hESCs14 (Figure 1a). hNSCs derived from hESCs (H9) were grown in the presence of basic fibroblast growth factor (bFGF), and then differentiated toward predominantly neuronal populations of central nervous system (CNS) by the withdrawal of mitogen14, 16 (Figures 1b–d). N2 is a serum-free medium optimized for roNSC cultures,23 thus our initial hNSC culture setup utilized N2.14 Surprisingly, however, hNSCs underwent massive cell death in N2, especially during the differentiation period (Figure 1e), with evidence of extensive apoptosis as indicated by cells positive for cleaved caspase-3 (Figure 1h), Annexin V, and propidium iodide (PI) (Figure 1j). Differentiation of hNSCs could not be maintained for longer than 5–7 days in N2 medium (Figure 1g). In trials testing other media, ITS, another commonly used serum-free medium,24 significantly improved cell viability of hNSCs, and showed decreased apoptosis (Figures 1f, i, and k) and prolonged viability for more than 15 days following differentiation (Figure 1g).

Human NSC (hNSC) viability versus cell death in N2 and ITS media. hNSCs were derived from hESCs (H9). The majority of hNSCs colabeled with markers specific for NSC nestin and proliferating cells Ki67 (a) entered a neuronal cell fate upon differentiation induction, as indicated by the abundance of Tuj1+ neurons compared with the few GFAP+ glia in the terminally differentiated cultures (b). Dopaminergic (TH+, c and d) and serotonergic (serotonin+, d) neurons represented the majority of the neuronal population. (e–k) Apoptotic cell death of hNSCs occurred during differentiation in N2, but not in ITS. (e and f) Phase-contrast images of the hNSC cultures 5 days after differentiation in N2 and ITS media, respectively. Greater cell viability in ITS, compared with N2, were further confirmed by MTT assays on days 1, 3, 5, 7, and 14 of differentiation (g). Values are relative to the respective values on day 1. (h–k) Images for cells expressing the apoptotic cell-specific markers cleaved caspase-3 (h and i) and Annexin-V/propidium iodide (PI) (j and k) at day 5 of differentiation in N2 (h and j) and ITS (i and k). (l) Comparison of components in N2 (Johe et al.23) and ITS (Okabe et al.24). (m–p) Cell viability (MTT, m and n) and cytotoxicity (LDH, o and p) assays carried out on hNSCs differentiated in either ITS media supplemented with N2 levels of the listed components (m and o) or N2 media with ITS levels of the listed components (n and p). In order to examine culture components responsible for the different cell viability in N2 and ITS shown in (e–g), differentiating hNSCs in (m and o) were cultured in ITS medium supplemented with N2 concentrations (colored in blue) of insulin (Ins), transferrin (Tf), putrescine (Pt), or progesterone (Pg). hNSCs were also cultured in N2 medium with ITS concentrations of the indicated components (red colored, n and p). Values are relative to the ITS controls of each respective group. *Significant difference from their respective ITS controls (P<0.001, n=8 per group, ANOVA). Scale bars=30 μm
When the components of N2 and ITS were compared (Figure 1l), the N2 constituents putrescine and progesterone were found to be absent in ITS. Insulin and transferrin, known to be necessary supplements for the survival of cultured neural cells,18, 19 were contained in both media, but in higher concentrations in N2 than ITS. The addition of putrescine and progesterone to ITS or their removal from N2 did not alter cell viability and cytotoxicity, as evidenced by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Figures 1m and n) and lactate dehydrogenase (LDH) assays (Figures 1o and p). In contrast, differentiation of hNSCs in N2 medium with the insulin concentration reduced to that of ITS greatly improved cell viability (Figures 1n and p). Accordingly, severe cell death was observed in hNSCs cultured in ITS with insulin concentrations increased to that of N2 (Figures 1m and o). These findings indicate that the higher insulin concentration in N2 is responsible for the observed hNSC cell death, and that insulin in high enough concentrations can actually be detrimental to differentiating hNSCs survival. Varying the transferrin concentration between the levels present in ITS and N2 media also affected differentiating hNSC survival (Figures 1m–p), but the effects were less substantial than those of insulin. In addition, because transferrin’s effect on cell survival may be driven indirectly by insulin-mediated signaling,25, 26 we focused on insulin-mediated hNSC cell death in this study.
Insulin concentration-dependent cell death specific to NSCs of human origin
We further examined cell viability and death of differentiating hNSCs over a range of insulin concentrations (0–4.3 μM). To minimize the effects on cell proliferation,18 cells were exposed to varying insulin concentrations 3 days after differentiation, when the majority of NSCs stop proliferating.27 Consistent with the observed differences between N2 and ITS (Figures 1e–k), severe cell death was evident at higher insulin concentrations in hNSC cultures (Figure 2a). Zero insulin supplementation or concentrations <0.09 μM also reduced cell viability (Supplementary Figure S1A and data not shown). A greater number of cells were positive for the apoptotic cell markers cleaved caspase-3 and Annexin V at the higher concentration of insulin (4.3 μM) than at the lower concentration (0.22 μM) (Figures 2d and g), indicating that apoptosis underlies the cell death induced by high concentrations of insulin. Similar patterns of insulin concentration-dependent effects were evident in all hNSCs tested, independent of their origins, which included hNSCs derived from another hESC line (HSF6), hiPSCs (IMR90-4), and human fetal ventral midbrain (VM) and cortical (Ctx) tissues (Supplementary Figures S1A–D). The optimal insulin concentrations for cell viability were lower than expected, and found to be below that of ITS (0.86 μM) in all hNSCs tested. The viability of roNSCs, on the other hand, showed very different patterns of insulin concentration dependence. Cell viability of roNSCs tended to be increased at higher insulin concentrations (Figures 2b and c), and fewer cells were positive for apoptotic cell markers (Figures 2e, f and h–i). The insulin-mediated cell survival effect was observed in all NSCs of rodent origin regardless of brain region (Ctx, VM, and ganglionic eminence (GE)) or rodent species (mouse, rat) (Supplementary Figures S1E–H). These findings collectively suggest that NSCs of human origin, unlike those of rodents, are exquisitely sensitive to insulin concentration. These cells are especially vulnerable at high insulin levels, and their survival during differentiation is ensured only within a narrow range of low insulin concentrations. Interestingly, levels of hNSC death induced by high insulin concentrations varied among the different insulin products tested, although the patterns of insulin dependency were similar (Supplementary Figure S2).

Effect of insulin concentrations on cell viability of NSCs derived from different species. Human NSCs were isolated by in vitro differentiation of hESCs (H9). Rat and mouse NSCs were cultured from embryonic forebrain (cortex) tissues at embryonic days 13 and 12, respectively. The NSCs of the different species were proliferated up to 60–80% cell confluency and then induced to differentiate for 3 days in ITS (human) or N2 (rat, mouse). From 3 days after differentiation, the NSCs were exposed to different concentrations of insulin (0–4.3 μM) for 5 days. On the last day of culture, cell viability and apoptotic cell death were assessed by MTT assays (a–c, n=8) and for the percentage of cells positive for the apoptotic cell markers cleaved caspase-3 and Annexin V (d–f). MTT values are relative to values of the none-insulin (0 μM)-supplemented samples of each group. Shown in (d–f) are representative images and (g–i) show the counts (% cells out of total DAPI+ cells) of the apoptotic cell markers in the cultures supplemented with 0.22 and 4.3 μM insulin. *Significant difference from their respective 0.22 μM insulin samples (P<0.01, n=3 coverslips per group). Scale bars=30 μm
Concentration-dependent effects of insulin on hNSCs at different developmental stages
We further examined the dose-dependent effects of insulin on other developmental stages such as proliferating NSCs and terminally differentiated neurons. As described in Figure 1a, in the presence of the mitogen bFGF, hESC-derived hNSC cultures comprise uniform populations of proliferating NSCs during the proliferation period. Insulin supplementation was proven to be important for the proliferation and survival of proliferating hNSCs, as indicated by delayed cell growth and a decreased percentage of cells positive for the proliferating cell markers Ki67 and phospho-histone H3 (pHH3) observed in cultures without insulin compared with those with insulin supplementation (Supplementary Figure S3). Similar to the insulin concentration-dependent effects observed in differentiating hNSCs (Figures 2a, d, and g), cell growth and cell proliferation indices were greater in the proliferating cell cultures treated with a low concentration of insulin (0.22 μM) than in those treated with a high dose (4.3 μM) (Supplementary Figure S3).
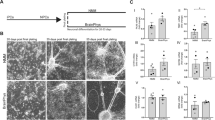
Differentiation of hNSCs is mostly complete 5–7 days following induction by withdrawal of mitogen bFGF and addition of several neurotrophic factors (see Materials and Methods). Postmitotic TuJ1+ neurons represent a major population of the cells in the differentiated cultures (Figures 1b–d, 65.47±1.27% of total cells at day 10 of differentiation). hNSCs were induced to differentiate in this condition for 10 days, after which the terminally differentiated neurons were exposed to different insulin concentrations. Similar to differentiating hNSCs, the human neuronal cells exposed to a high dose (4.3 μM) of insulin underwent apoptotic cell death, whereas cells exposed to low (0.22 μM) insulin did not (Figures 3a). In contrast, cell viability was not significantly altered by insulin concentration in primary neuron cultures derived from the mouse VM (Figures 3e) and cortex (Supplementary Figure S4). As described in Figures 1c and d, TH+ dopaminergic (DA) neurons, associated with Parkinson’s disease, are a major subtype of neurons in the hESC-derived hNSC cultures. The DA neuronal populations along with general neuronal populations in the human neuron cultures were also greatly reduced by the increased insulin concentration (Figures 3c and d), whereas cell viability of mouse DA neurons (from VM) was unaffected by the higher insulin concentration (Figures 3g and h). These findings, taken together, strongly suggest that insulin concentration is extremely important for the successful culturing of hNSCs and human neurons, regardless of their developmental stages, and that insulin concentration dependency is specific to cells of human origins.

Insulin concentration-dependent apoptotic cell death was observed in human neurons, but not in mouse neuronal cells. Human neuron-dominant cultures were obtained by 10 days of in vitro differentiation of H9 hESC-derived hNSCs. The human neurons were exposed to 0.22 or 4.3 μM of insulin for 5 days (a–d). Culture conditions and exposure to insulin was replicated in the primary culture for mouse neurons derived from embryonic VM at E14, except for supplementation of B27 (e–h). B27, a commercial supplement necessary for primary rodent neuron culture, contains insulin but its concentration is not available to the public. To take into consideration this unknown insulin amount, (e–h) represent the mouse neuron survivals at the insulin concentrations of 0.22+X and 4.3+X μM, where X denotes the insulin concentration in B27. *Significant difference from their respective 0.22 μM (or 0.22 μM+X) insulin samples (P<0.001, n=3 coverslips per group). Scale bars=30 μm
Intracellular signaling in insulin-mediated cell death
Insulin-mediated cell death was unexpected because insulin is regarded as an indispensable component of various culture media based on its role as a promoter of cell survival.19 To understand the mechanism underlying the observed insulin toxicity, we examined its intracellular signaling. The PI3K/Akt pathway is the major signaling pathway activated by the binding of insulin to cell surface insulin receptor (IR), and plays a key role in insulin-mediated cell survival.28 The hNSCs were starved of insulin overnight, and then supplemented with insulin at 0.22 or 4.3 μM. The cultures were maintained at their respective insulin concentrations for 4 days. During the culture period, fresh insulin was added to the culture at days 0, 2, and 4 along with new base media. Phosphorylated/activated Akt (pAkt) levels increased in response to the initial insulin exposure (day 0), and were greater in the hNSC cultures treated with 4.3 μM of insulin than in the cultures treated with 0.22 μM (Figure 4a). Interestingly, pAkt levels reversed after prolonged exposure at the different insulin concentrations: pAkt levels were greater in the cultures at 0.22 μM than those at 4.3 μM at 2 and 4 days after insulin exposure. This response is representative of insulin resistance, a process in which cell responsiveness to insulin becomes less effective following prolonged exposure to high levels of the molecule. Indeed, the elevated pAkt levels at 10, 30, 120 min after addition of insulin gradually decreased in cultures exposed to insulin for 0, 2, and 4 days, and the reduction in the responsiveness to insulin was more dramatic in cultures exposed to a high insulin concentration (Figures 4b and c). In contrast, a reduction in pAkt levels during culture with insulin was not observed in rat cortical NSC cultures, and pAkt protein levels were actually shown to increase following prolonged insulin exposure (Figure 4a).

Reduction of intracellular Akt signaling is responsible for hNSC death induced by long-term exposure to insulin at high concentration. (a) Human and rat NSCs exhibit different Akt signal activation patterns dependent on insulin dose and exposure time. H9 hESC-derived hNSCs at differentiation day 3 were starved with insulin overnight, and then exposed to low (0.22 μM) and high (4.3 μM) concentrations of insulin (day 0). Cultures were maintained in the same concentration of insulin for 4 days. Culture conditions and exposure to insulin was replicated with rat NSCs derived from embryonic cortices at E13. Media with freshly supplemented insulin were changed at days 0, 2, and 4. Cells were harvested 30 min after media change with the addition of freshly prepared insulin and immunoblot analyses were carried out. (b) A more drastic time-dependent reduction of pAkt levels in hNSC cultures treated with the higher concentration of insulin (4.3 μM), compared with those with the low concentration (0.22 μM). The gradual reduction of Akt signal activation in hNSC cultures during the time period of insulin exposure was further confirmed by multiple immunoblot analyses with quantification. Protein samples for immunoblot analyses were prepared 10, 30, and 120 min after media change at the indicated time points. (c) Immunoblot quantification for pAkt protein level changes in hNSCs treated with low (0.22 μM) and high (4.3 μM) concentrations of insulin. pAkt levels were quantified by measuring the band intensities of the immunoblots using a densitometer. The gray dots and lines in the graphs are arbitrary pAkt levels quantified at the indicated time points (n=4 of each), and the trend in changes of pAkt during the culture period are marked with red lines. (d) Activation of Akt signaling is critical for hNSC survival. hNSCs were differentiated with 0.22 and 4.3 μM of insulin as described. Akt activated in cultures supplemented with 0.22 μM insulin was blocked by treatment with the PI3K/Akt signal inhibitors LY294002 (LY, 20 μM) and Wortmannin (Wot, 1 μM) or by transducing with dn-Akt. Cell viability and apoptotic cell death were estimated by total viable cell counts under phase-contrast microscopy (d and e) and by the percentage of cells positive for cleaved caspase-3 (f and g). The effects of ca-Akt expression were also estimated in hNSC cultures maintained with 4.3 μM of insulin (h and i). *Significant difference from their respective controls (P<0.005, n=3 coverslips per group). Scale bars=40 μm
The timing of the decrease in pAkt levels (2 days after culturing at high insulin concentrations), which occurred 1 to 2 days before evident cell death of hNSC (3–5 days after culturing with high insulin concentrations, Figures 1 and 2), suggests that decreased Akt activation may mediate the observed cell death of hNSCs cultured at high insulin concentrations. Inhibition of PI3K/Akt signaling by the specific inhibitors LY294002 and Wortmannin and by overexpression of dominant-negative Akt (dn-Akt) resulted in severe cell death of differentiating hNSCs (Figures 4d–g). Furthermore, transduction with the constitutively active form of Akt (ca-Akt) mitigated the rates of cell death in cultures exposed to 4.3 μM insulin (Figures 4h and i), confirming that decreased activation of Akt signal underlies cell death induced by prolonged exposure to high levels of insulin.
An important mechanism of insulin resistance is the degradation of insulin receptor substrate 1 and 2 (IRS-1/2), a molecule that transduces insulin–IR binding to the intracellular PI3K/Akt pathway.29, 30 IRS-1 protein levels on the day of insulin exposure were indistinguishable between cultures treated with 0.22 and 4.3 μM of insulin (Figure 5a). However, similar to the change in pAkt levels (Figure 4a), IRS-1 protein levels in hNSC cultures decreased in cultures at days 2 and 4 of insulin exposure, and this reduction was more pronounced in the cultures treated with 4.3 μM of insulin than in those treated with 0.22 μM insulin (Figure 5a). It has been shown that IRS protein degradation in insulin resistance is mediated via mTOR activation.31, 32 Treatment with rapamycin, an mTOR signaling inhibitor, successfully blocked the apoptotic cell death of hNPCs cultured with a higher concentration (4.3 μM) of insulin (Figure 5b). Furthermore, the decrease in IRS-1 level following culture with 4.3 μM insulin at days 2 and 4 was partially rescued by the rapamycin treatment (Figure 5c). Taken together, these findings suggest that decreased PI3K/Akt signaling is mediated by the reduction of IRS-1 following prolonged exposure to high levels of insulin, and this mechanism for insulin toxicity is specific to differentiating hNSCs.

IRS-1 protein levels are decreased via mTOR signaling during insulin-supplemented hNSC culture. (a) Gradual reduction of IRS-1 protein levels over the culture period with insulin is prominent in hNSC cultures supplemented at the higher insulin concentration (left), but not in rat NSC cultures (right). hNSCs and rat NSCs were cultured in media supplemented with 0.22 and 4.3 μM of insulin as described in Figure 4. Protein samples were prepared 30 min after media change (adding freshly prepared insulin) at the time point indicated, and immunoblot analyses for IRS-1 were carried out. (b) Effect of the mTOR inhibitor rapamycin on apoptotic cell death of hNSCs at high insulin concentration. hNSCs (derived from H9 hESCs) were cultured for 4 days in the presence of 0.22 and 4.3 μM insulin, and 4.3 μM insulin+rapamycin (Rapa or R, 100 nM). Immunofluorescent staining was carried out for cleaved caspase-3 and the percentage of immunoreactive cells out of total DAPI+ cells was counted. (c) Treatment of rapamycin blocks the decrease of IRS-1 protein levels in hNSCs cultured with high concentration of insulin. *Significant difference from the high insulin sample (P<0.001, n=3 coverslips per group). Scale bars=40 μm
Gene expressions altered in high insulin concentration
In addition to IRS degradation, insulin resistance could be caused by a decrease in IR and IRS activation by insulin stimulation through various/extracellular routes.33, 34 In order to observe the dynamics of gene expression changes in insulin-resistant hNSC cultures, we conducted microarray analyses to compare gene expression profiles between hNSCs cultured with optimal (0.22 μM) and high (4.3 μM) doses of insulin. The microarray data are summarized in Supplementary Figure S5. Expression of genes associated with negative regulation of IR was increased in hNSCs cultured with insulin resistance-inducing concentrations (4.3 μM) of insulin, compared with those with the optimal insulin concentration (0.22 μM). Oxidative stress and inflammation are suggested to induce insulin resistance through decreased sensitivity as well as degradation of IR/IRS.35, 36, 37 Consistently, expressions of genes related to these events were also upregulated with high insulin dosage. In addition, genes related to cell apoptosis/programmed cell death, those associated with Wnt signaling pathway, cell–cell signaling, negative regulation of phosphorylation, and ascorbic acid binding were also upregulated with high insulin dosage. In contrast, genes related to protein–DNA complex assembly and positive regulation of cell cycles were significantly downregulated in hNSCs cultured with high insulin dosage.
Discussion
It is widely accepted that human cell culture systems are more relevant than non-human systems in obtaining information directly applicable to human physiology and pathology. However, cultures derived from human solid tissues are limited. Recent progress in stem cell biology has allowed for the establishment of human ESC/iPSCs and derivation of NSCs from these pluripotent stem cells. Disease-specific NSCs and neurons can also be cultured from hiPSCs derived from patients with neurologic diseases, and these disease-specific tissue cell cultures now offer novel in vitro models of human disease. Despite the importance of hNSC cultures, technical problems in maintaining healthy cell populations limit their widespread use. Without systematic assays for optimizing hNSC culture conditions, conditions optimized for rodent cultures have been applied in culturing hNSCs, with disappointing and inconsistent results. This study demonstrates a clear difference in optimal culture conditions between hNSCs and roNSCs. In contrast to roNSCs, hNSCs in culture were extremely sensitive to the concentration of insulin, a supplement known to be necessary in neural cell cultures, and healthy hNSC cultures were achievable only within a narrow range of relatively low insulin concentrations. Furthermore, the rate of cell death induced by insulin varied among different insulin products. It is clear that many of the past experimental failures and inconsistencies in culturing hNSCs were likely because of insufficient understanding of the insulin sensitivity specific to human neuronal cells.
In addition to the practical aspects, the findings in this study provide physiologic implications as well. The brain has long been regarded as an insulin-insensitive organ,38 but this concept has been abandoned. Insulin and its receptor are in fact highly expressed in the developing and adult brains.39 Progression of neural development through proliferation, differentiation, maturation, and survival is stimulated, at least in culture, by insulin and the insulin precursor proinsulin.19 As a result, these cytokines can now be regarded as important autocrine/paracrine signaling molecules involved in multiple steps of CNS development. In the adult brain, the elevation of circulating insulin alters brain functions such as memory,40, 41 cognition,42, 43, 44 feeding behavior,45, 46 and modulation of auditory evoked potentials.47 Furthermore, epidemiologic studies have established clinical association between hyperinsulinemia (type II diabetes) and neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease20, 21 that share a common underlying pathology of neuronal cell death. However, no studies published to date have demonstrated direct experimental data supporting a mechanistic association between hyperinsulinemia and neurodegenerative disorders. The findings reported in this study are the first evidence demonstrating that prolonged exposure of human neurons to high doses of insulin can cause apoptotic cell death in the human brain. The mechanism underlying insulin-induced cell death is insulin resistance, which has been demonstrated in extra-CNS tissues, but has not previously been reported in the neural cells of the brain. Activation of intracellular Akt in human neural cells was reduced in response to prolonged exposure of high insulin concentrations via decreased IRS-1 activation. These findings indicate that Akt activation is a crucial step in the intracellular signaling pathway regulating survival of NSCs and neuronal cells.48, 49 Accordingly, recent studies using patient brain samples have reported decreased pAkt levels in Parkinson’s disease50 and reduced insulin-mediated signaling molecules, including IRS-1/2, in brains affected by Alzheimer’s disease.51
Like NSCs, cell death manifested in human neurons by high levels of insulin was not the case in the neuronal cells primarily cultured from rodent brain tissues, indicating rodent CNS neurons physiologically require higher levels of insulin-mediated trophic supports without developing insulin resistance. Supportably, normal physiologic levels of insulin in circulating blood are greater in adult rats than those of human species (180–240 pM in rats versus 57–79 pM in humans).52, 53, 54 In contrast to rat CNS neurons requiring high levels of insulin, a recent study has demonstrated that insulin resistance was easily developed in sensory neurons derived from rat dorsal root ganglion by exposing to μM range of insulin.55 These may represent an innate difference of susceptibility to insulin resistance between central and peripheral neurons, considering that in vivo insulin levels in brain tissues are 10–100 times higher than peripheral blood.56, 57
In order to culture neural cells outside of brain, supraphysiologic levels of trophic factors should be inevitably supplemented to the cultures. Thus, findings in our study were attained at insulin concentrations far higher than those of circulating blood. Further studies must be conducted to confirm the effects of insulin dose and duration on human neural cells observed in this study in experimental systems more relevant to physiology. Considering the observed species-dependent difference in the response to insulin, it will be important that any follow-up studies use human-derived experimental systems.
Materials and Methods
NSC cultures
hNSCs were derived from hESCs (H9, University of Wisconsin, Madison, WI, USA; and HSF-6, University of California San Francisco, San Francisco, CA, USA) and hiPSCs (IMR 90-4, University of Wisconsin) using an in vitro differentiation protocol that includes neural induction on the feeder layers of MS5 stromal cells, followed by consecutive cell passages to select NSCs, as previously described.14, 15 The hNSCs underwent 2–6 NSC passages before being used in this study. NSCs derived from human embryonic cortical and VM tissues (ReN cell VM and Ctx, Millipore, Billerica, MA, USA) were also cultured and tested in this study. NSCs of rat and mouse origin were directly isolated and cultured from the embryonic Ctx, VM, and GE of embryonic day 13 (E13) rats (Sprague Dawley (SD)) and E12 mice (Imprinting Control Region (ICR)) as previously described.58 The NSCs of the different species were proliferated in the presence of bFGF (20 ng/ml; R&D Systems, Minneapolis, MN, USA) or bFGF and epidermal growth factor (EGF; 20 ng/ml; R&D Systems, for mouse culture) on poly-L-ornithine (PLO)/fibronectin (FN)-coated culture dishes. When cultures reached 80–90% cell confluency, differentiation of NSCs was induced by withdrawal of the mitogens. The cells were cultured in serum-free ITS (for hNSC culture)24 or N2 (for rat and mouse NSC culture)23 media. The components and their concentrations of N2 and ITS are summarized in Figure 1l. In order to test insulin concentration effects, the media were prepared by mixing each culture constituent without insulin, and then supplementing them with various concentrations of insulin (0–4.3 μM) later. Three different sources of insulin (12585-014, Invitrogen, Carlsbad, CA, USA; I1882 and I6634, Sigma, St. Louis, MO, USA) were tested. Except in tests to compare different insulin products, all data shown in this study were obtained with the insulin product 12585-014 from Invitrogen. hNSCs were transduced with ca-Akt or dn-Akt (kindly gifted by Dr. Incheol Shin, Hanyang University, Seoul, Korea) where necessary as described below. The following chemicals were also used: LY294002 (20 μM; Calbiochem, San Diego, CA, USA), Wortmannin (1 μM; Calbiochem), and Rapamycin (100 nM; Sigma). The medium was changed every other day, whereas the mitogens were supplemented every day. Cells were maintained at 37°C in a 5% CO2 incubator.
Neuron cultures
Cultures enriched with human neurons were derived by terminal differentiation of H9 hES-NSCs for 10 days in ITS medium (insulin concentration, 0.22 μM) and supplemented with brain-derived neurotrophic factor (BDNF; 20 ng/ml; R&D Systems), glial cell-derived neurotrophic factor (GDNF; 20 ng/ml; R&D Systems), and dibutyryl cAMP (0.5 mM; Sigma) and ascorbic acid (0.2 mM, Sigma). Neurons of mouse origin were primarily cultured from mouse (ICR) embryonic brains at E14. Cortical and VM tissues were dissected, minced, and then treated with trypsin/EDTA for 15 min at 37°C. After inactivation of trypsin by adding DMEM+10% FBS, the tissues were mechanically triturated. Dissociated cells were plated at 80 000 cells/cm2 on poly-D-lysine hydrobromide (PDL; Sigma)-coated 24-well plates and cultured for 10 days in Neurobasal (NB, Invitrogen) medium supplemented with L-Glutamine and B27 (Invitrogen).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), incubated in blocking solution (1% BSA/0.03% Triton X-100 in PBS), and then incubated with the following primary antibodies: Ki67 anti-mouse (1 : 100; Novocastra Laboratories Ltd, Newcastle, UK), nestin anti-rabbit (1 : 50; Dr. Ron McKay, NIH, Bethesda, MD, USA), GFAP anti-mouse (1 : 100; ICN Biochemicals, Costa Mesa, CA, USA), Tuj1 anti-rabbit (1 : 2000; Babco, Richmond, CA, USA), TH anti-mouse (1 : 1000; Immunostar, Hudson, WI, USA), serotonin anti-rabbit (1 : 4000; Sigma), cleaved caspase-3 anti-mouse (1 : 500; Cell Signaling Technology, Beverly, MA, USA), microtubule-associated protein 2 (MAP2; 1 : 200; Sigma), and pHH3 anti-rabbit (1 : 1000; Upstate Biotechnology, Lake Placid, NY, USA). The secondary antibodies tagged with the following fluorescent molecules were used for visualization: Alexa 488 (1 : 200, Invitrogen) and Cy3 (1 : 200; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Annexin V and PI staining were carried out using the Annexin V-FLUOS staining kit (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. The stained samples were then mounted in Vectashield with DAPI mounting medium (Vector Laboratories, Burlingame, CA, USA) and photographed using an epifluorescence microscope (Leica, Wetzlar, Germany).
Cell viability and cytotoxicity assays
Cell viability was determined by the MTT assay as previously described.59 Cytotoxicity was determined by LDH activity released from the damaged cells (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions. LDH results were expressed as a percentage of maximum LDH release obtained on complete lysis of cells with 1% Triton X-100 treatment. Media were taken as negative control (0%). Apoptotic cell death was assessed by the percent of cells positive for the apoptotic cell-specific markers cleaved Caspase-3 and Annexin V/PI after staining with the antibodies as described above.
Western blot
Whole-cell protein was extracted from cells using lysis buffer, subjected to denaturing sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, and then transferred to a nitrocellulose membrane. Transferred proteins were blocked in 5% nonfat milk in 0.001% Tween-20 with Tris-buffered saline. The blots were then incubated with the following primary antibodies: phospho-Akt (Ser473) anti-rabbit (1 : 1000; Cell Signaling Technology), Akt anti-rabbit (1 : 1000; Cell Signaling Technology), cleaved caspase-3 anti-rabbit (1 : 1000; Cell Signaling Technology), IRS anti-rabbit (1 : 1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), pmTOR anti-rabbit (1 : 1000; Cell Signaling Technology), mTOR anti-rabbit (1 : 1000; Cell Signaling Technology), and β-actin anti-mouse (1 : 5000; Abcam, Cambridge, UK). Anti-rabbit or anti-mouse IgG antibodies conjugated with peroxidase (1 : 2000; Cell Signaling Technology) were applied for visualization. Bands were visualized using an enhanced chemiluminescence (ECL) detection kit (Thermo Scientific, Rockford, IL, USA) and quantified using a densitometer (Molecular Devices, VersaMax, CA, USA) using ImageJ (v1.46r, National Institutes of Health, Bethesda, MA, USA).
Virus construction and transduction
Retroviral vectors expressing dn-Akt and ca-Akt were constructed by inserting their respective cDNAs into the pCL retroviral vector as previously described.60 The empty pCL vector was used as a negative control. Retroviruses were produced by introducing the vectors into the 293gpg retrovirus packaging cell line via transfection using Lipofectamine (Invitrogen). After 3 days, supernatant fractions were harvested and stored at −70 °C until use. For viral transduction, cultured NSCs were incubated with the viral supernatant (3–5 × 106 particles/ml) containing polybrene (hexadimethrine bromide: 1 μg/ml; Sigma) for 2 h, and then followed by media change.
Microarray analysis
Total RNA was extracted with TRIzol (Invitrogen) and purified using RNeasy columns (Qiagen) according to the manufacturer’s instructions. RNA quality was assessed by using the Agilent Model 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Microarray analysis was carried out by Macrogen Inc. (Seoul, Korea). Briefly, biotinylated complementary RNAs were amplified and purified using the Ambion Illumina RNA amplification kit (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. Labeled cRNA samples were hybridized to each human HT-12 expression v.4 bead array according to the manufacturer’s instructions (Illumina, SanDiego, CA, USA). Detection of array signal was carried out using Amersham fluoroLink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Uppsala, Sweden). Arrays were scanned with an Illumina bead array Reader confocal scanner, and the raw data were extracted using the software provided by the manufacturer (Illumina GenomeStudio v2011.1 (Gene Expression Module v1.9.0)). Gene-Enrichment and Functional Annotation analysis for significant probe list was performed using DAVID (http://david.abcc.ncifcrf.gov/home.jsp). All data analyses and visualization of differentially expressed genes were conducted using R 2.15.1 (www.r-project.org).
Cell counting and statistical analysis
Immunoreactive/DAPI-stained cells on culture coverslips were counted in at least 20 randomly selected microscopic fields using an eyepiece grid at a final magnification of × 200 or × 400. Statistical comparisons were made with SPSS software (v17.00, SPSS Inc., Chicago, IL, USA). One-way ANOVA was applied where appropriate. Data are expressed as mean±S.E.M. of at least three independent experiments and the null hypothesis was rejected on the basis of P<0.05.
Abbreviations
- NSC:
-
neural stem cell
- hESC:
-
human embryonic stem cell
- hiPSC:
-
human-induced pluripotent stem cell
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LDH:
-
lactate dehydrogenase
- Ctx:
-
cortex
- VM:
-
ventral midbrain
- GE:
-
ganglionic eminence
- pHH3:
-
phospho-histone H3
- bFGF:
-
basic fibroblast growth factor
- DA:
-
dopaminergic
- IR:
-
insulin receptor
- IRS:
-
insulin receptor substrate
- CNS:
-
central nervous system
References
Merkle FT, Alvarez-Buylla A . Neural stem cells in mammalian development. Curr Opin Cell Biol 2006; 18: 704–709.
Temple S . The development of neural stem cells. Nature 2001; 414: 112–117.
Bonfanti L, Peretto P . Radial glial origin of the adult neural stem cells in the subventricular zone. Prog Neurobiol 2007; 83: 24–36.
Gage FH . Mammalian neural stem cells. Science 2000; 287: 1433–1438.
Dietrich J, Kempermann G . Role of endogenous neural stem cells in neurological disease and brain repair. Adv Exp Med Biol 2006; 557: 191–220.
Wang YZ, Plane JM, Jiang P, Zhou CJ, Deng W . Concise review: quiescent and active states of endogenous adult neural stem cells: identification and characterization. Stem Cells 2011; 29: 907–912.
Jo AY, Park CH, Aizawa S, Lee SH . Contrasting and brain region-specific roles of neurogenin2 and mash1 in GABAergic neuron differentiation in vitro. Exp Cell Res 2007; 313: 4066–4081.
Pluchino S, Zanotti L, Deleidi M, Martino G . Neural stem cells and their use as therapeutic tool in neurological disorders. Brain Res Brain Res Rev 2005; 48: 211–219.
Martino G, Pluchino S . The therapeutic potential of neural stem cells. Nat Rev Neurosci 2006; 7: 395–406.
Temple S . Division and differentiation of isolated CNS blast cells in microculture. Nature 1989; 340: 471–473.
Kirschenbaum B, Nedergaard M, Preuss A, Barami K, Fraser RA, Goldman SA . In vitro neuronal production and differentiation by precursor cells derived from the adult human forebrain. Cereb Cortex 1994; 4: 576–589.
Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T et al. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods 1998; 85: 141–152.
Erceg S, Ronaghi M, Stojkovic M . Human embryonic stem cell differentiation toward regional specific neural precursors. Stem Cells 2009; 27: 78–87.
Park CH, Minn YK, Lee JY, Choi DH, Chang MY, Shim JW et al. In vitro and in vivo analyses of human embryonic stem cell-derived dopamine neurons. J Neurochem 2005; 92: 1265–1276.
Rhee YH, Ko JY, Chang MY, Yi SH, Kim D, Kim CH et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J Clin Invest 2011; 121: 2326–2335.
Ko JY, Park CH, Koh HC, Cho YH, Kyhm JH, Kim YS et al. Human embryonic stem cell-derived neural precursors as a continuous, stable, and on-demand source for human dopamine neurons. J Neurochem 2007; 103: 1417–1429.
Koch P, Opitz T, Steinbeck JA, Ladewig J, Brustle O . A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci USA 2009; 106: 3225–3230.
Erickson RI, Paucar AA, Jackson RL, Visnyei K, Kornblum H . Roles of insulin and transferrin in neural progenitor survival and proliferation. J Neurosci Res 2008; 86: 1884–1894.
de Pablo F, de la Rosa EJ . The developing CNS: a scenario for the action of proinsulin, insulin and insulin-like growth factors. Trends Neurosci 1995; 18: 143–150.
Craft S, Watson GS . Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004; 3: 169–178.
Watson GS, Craft S . Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer’s disease. Eur J Pharmacol 2004; 490: 97–113.
Chambers SM, Mica Y, Studer L, Tomishima MJ . Converting human pluripotent stem cells to neural tissue and neurons to model neurodegeneration. Methods Mol Biol 2011; 793: 87–97.
Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD . Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev 1996; 10: 3129–3140.
Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M, McKay RD . Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro. Mech Dev 1996; 59: 89–102.
Storch S, Kubler B, Honing S, Ackmann M, Zapf J, Blum W et al. Transferrin binds insulin-like growth factors and affects binding properties of insulin-like growth factor binding protein-3. FEBS Lett 2001; 509: 395–398.
Weinzimer SA, Gibson TB, Collett-Solberg PF, Khare A, Liu B, Cohen P . Transferrin is an insulin-like growth factor-binding protein-3 binding protein. J Clin Endocrinol Metab 2001; 86: 1806–1813.
Yi SH, Jo AY, Park CH, Koh HC, Park RH, Suh-Kim H et al. Mash1 and neurogenin 2 enhance survival and differentiation of neural precursor cells after transplantation to rat brains via distinct modes of action. Mol Ther 2008; 16: 1873–1882.
van der Heide LP, Ramakers GM, Smidt MP . Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 2006; 79: 205–221.
Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ . Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes 1999; 48: 1359–1364.
Zhande R, Mitchell JJ, Wu J, Sun XJ . Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol 2002; 22: 1016–1026.
Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes 2010; 59: 2426–2434.
Berg CE, Lavan BE, Rondinone CM . Rapamycin partially prevents insulin resistance induced by chronic insulin treatment. Biochem Biophys Res Commun 2002; 293: 1021–1027.
Zick Y . Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol 2001; 11: 437–441.
Boura-Halfon S, Zick Y . Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab 2009; 296: E581–E591.
Shoelson SE, Lee J, Goldfine AB . Inflammation and insulin resistance. J Clin Invest 2006; 116: 1793–1801.
Johnson AM, Olefsky JM . The origins and drivers of insulin resistance. Cell 2013; 152: 673–684.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM . Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003; 52: 1–8.
Rulifson EJ, Kim SK, Nusse R . Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 2002; 296: 1118–1120.
Schulingkamp RJ, Pagano TC, Hung D, Raffa RB . Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev 2000; 24: 855–872.
Greenwood CE, Winocur G . Glucose treatment reduces memory deficits in young adult rats fed high-fat diets. Neurobiol Learn Mem 2001; 75: 179–189.
Helkala EL, Niskanen L, Viinamaki H, Partanen J, Uusitupa M . Short-term and long-term memory in elderly patients with NIDDM. Diabetes Care 1995; 18: 681–685.
Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM . Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 1999; 53: 1937–1942.
Elias PK, Elias MF, D'Agostino RB, Cupples LA, Wilson PW, Silbershatz H et al. NIDDM and blood pressure as risk factors for poor cognitive performance. The Framingham Study. Diabetes Care 1997; 20: 1388–1395.
Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol 1997; 145: 301–308.
Rodin J, Wack J, Ferrannini E, DeFronzo RA . Effect of insulin and glucose on feeding behavior. Metabolism 1985; 34: 826–831.
Debons AF, Krimsky I, From A . A direct action of insulin on the hypothalamic satiety center. Am J Physiol 1970; 219: 938–943.
Kern W, Kerner W, Pietrowsky R, Fehm HL, Born J . Effects of insulin and hypoglycemia on the auditory brain stem response in humans. J Neurophysiol 1994; 72: 678–683.
Zhao Y, Xiao Z, Gao Y, Chen B, Zhang J, Dai J . Insulin rescues ES cell-derived neural progenitor cells from apoptosis by differential regulation of Akt and ERK pathways. Neurosci Lett 2007; 429: 49–54.
Ishii S, Okada Y, Kadoya T, Matsuzaki Y, Shimazaki T, Okano H . Stromal cell-secreted factors promote the survival of embryonic stem cell-derived early neural stem/progenitor cells via the activation of MAPK and PI3K-Akt pathways. J Neurosci Res 2010; 88: 722–734.
Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L et al. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest 2012; 122: 1354–1367.
Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C . Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010; 31: 224–243.
Havrankova J, Schmechel D, Roth J, Brownstein M . Identification of insulin in rat brain. Proc Natl Acad Sci USA 1978; 75: 5737–5741.
Havrankova J, Roth J, Brownstein MJ . Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest 1979; 64: 636–642.
Iwase H, Kobayashi M, Nakajima M, Takatori T . The ratio of insulin to C-peptide can be used to make a forensic diagnosis of exogenous insulin overdosage. Forensic Sci Int 2001; 115: 123–127.
Singh B, Xu Y, McLaughlin T, Singh V, Martinez JA, Krishnan A et al. Resistance to trophic neurite outgrowth of sensory neurons exposed to insulin. J Neurochem 2012; 121: 263–276.
Duarte AI, Moreira PI, Oliveira CR . Insulin in central nervous system: more than just a peripheral hormone. J Aging Res 2012; 2012: 384017.
Schechter R, Whitmire J, Holtzclaw L, George M, Harlow R, Devaskar SU . Developmental regulation of insulin in the mammalian central nervous system. Brain Res 1992; 582: 27–37.
Park CH, Kang JS, Yoon EH, Shim JW, Suh-Kim H, Lee SH . Proneural bHLH neurogenin 2 differentially regulates Nurr1-induced dopamine neuron differentiation in rat and mouse neural precursor cells in vitro. FEBS Lett 2008; 582: 537–542.
Twentyman PR, Luscombe M . A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br J Cancer 1987; 56: 279–285.
Park CH, Lim MS, Rhee YH, Yi SH, Kim BK, Shim JW et al. In vitro generation of mature dopamine neurons by decreasing and delaying the expression of exogenous Nurr1. Development 2012; 139: 2447–2451.
Acknowledgements
This work was supported by a grant from the Bio and Medical Technology Development Program (NRF-2010-0020232) and the Medical Research Center (NRF-2008-0062190) funded by the National Research Foundation of Korea (NRF) of the Ministry of Education, Science and Technology (MEST), Republic of Korea. The scholarship of Rhee, Yong-Hee was supported by the Global Ph.D. Fellowship (2011-0008831) from the National Research Foundation of Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rhee, YH., Choi, M., Lee, HS. et al. Insulin concentration is critical in culturing human neural stem cells and neurons. Cell Death Dis 4, e766 (2013). https://doi.org/10.1038/cddis.2013.295
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.295
Keywords
This article is cited by
-
Sensitivity of human pluripotent stem cells to insulin precipitation induced by peristaltic pump-based medium circulation: considerations on process development
Scientific Reports (2017)
-
Functional Activity of the Insulin Signaling System of the Brain in Health and Type 2 Diabetes Mellitus
Neuroscience and Behavioral Physiology (2017)
