
Abstract
Lon protease is a multifunction protein and operates in protein quality control and stress response pathways in mitochondria. Human Lon is upregulated under oxidative and hypoxic stresses that represent the stress phenotypes of cancer. However, little literature undertakes comprehensive and detailed investigations on the tumorigenic role of Lon. Overexpression of Lon promotes cell proliferation, apoptotic resistance to stresses, and transformation. Furthermore, Lon overexpression induces the production of mitochondrial reactive oxygen species (ROS) that result from Lon-mediated upregulation of NDUFS8, a mitochondrial Fe-S protein in complex I of electron transport chain. Increased level of mitochondrial ROS promotes cell proliferation, cell survival, cell migration, and epithelial–mesenchymal transition through mitogen-activated protein kinase (MAPK) and Ras-ERK activation. Overall, the present report for the first time demonstrates the role of Lon overexpression in tumorigenesis. Lon overexpression gives an apoptotic resistance to stresses and induces mitochondrial ROS production through Complex I as signaling molecules to activate Ras and MAPK signaling, giving the survival advantages and adaptation to cancer cells. Finally, in silico and immunohistochemistry analysis showed that Lon is overexpressed specifically in various types of cancer tissue including oral cancer.
Similar content being viewed by others

Main
Several cancer hallmarks, for example, disabled apoptosis, invasion/metastasis, and oxidative stress, are directly or indirectly linked to deregulated mitochondria.1, 2, 3 Mitochondria have an important role in carcinogenesis partly because of their roles in mediating apoptosis4 and as a major source of endogenous reactive oxygen species (ROS) that escape from the electron transport chain (ETC) during oxidative phosphorylation (OXPHOS) process.5, 6 The OXPHOS system consists of five multi-protein complexes (I–V), and there are two main sites of superoxide generation in the inner mitochondrial membrane: NADH dehydrogenase at complex I, and the interface between ubiquinone and complex III.5 ROS act as a double-edged sword in carcinogenesis.7, 8 First, ROS make both nuclear DNA and mitochondrial DNA susceptible to damage, and mutations in these two DNA pools are reported to lead to carcinogenesis.9 Second, ROS are crucial intermediates of cellular signaling that promote and suppress tumorigenesis simultaneously.7, 10, 11 It is well known that ROS activate mitogen-activated protein kinase (MAPK) family comprising of JNK, p38, and ERK.12 MAPK family members function in cell context-specific manner to integrate signals that regulate proliferation, survival, apoptosis, and invasiveness.13, 14, 15 In addition, Ras is activated by ROS and drives the activation of MAPKs that are required for transformation phenotypes including epithelial–mesenchymal transition (EMT).2, 7, 16
Mitochondrial Lon operates in protein quality control and stress response pathways in mitochondria by its multiple functions.17, 18, 19, 20 In addition to its proteolytic activity, Lon has been found to show chaperone properties.17, 21 Lon homeostasis is important to cell fate, as its downregulation leads to reduced cell proliferation and apoptosis.22, 23 Lon is a stress protein and induced by a number of stresses such as oxidative stress and hypoxia.21, 24, 25 During hypoxia, Lon is upregulated by the hypoxia inducible factor-1α (HIF-1α) and involved in a mechanism to response low oxygen availability and adapt cancer cells to a hypoxic environment.25 Thus, Lon upregulation may be critical for cancer cell survival by regulating stress responses induced by oxidative and hypoxic condition, which are common stress phenotypes of cancer cells. However, no report has been described how upregulated Lon promotes cancer cell survival and tumorigenesis.
We previously found that human Lon is upregulated in non-small-cell lung cancer cell lines and its downregulation triggers caspase-3-mediated apoptosis.23 The purpose of this present study was to investigate the detailed mechanisms of Lon-mediated tumorigenesis.
Results
Lon is upregulated in various types of cancer
To understand whether Lon overexpression has a crucial role in tumorigenesis, we first performed a comprehensive in silico analysis on several types of cancer by using several publicly available gene expression data sets of cancer-versus-normal, including Oncomine26 and Gene Expression Omnibus (GEO) at the NCBI. The expression profiles complied in Oncomine showed that Lon is upregulated in most of cancer types, including lung, colorectal, and head-and-neck cancer (Supplementary Figure S1A). Consistently, the transcript level of Lon is significantly overexpressed in lung adenocarcinoma, GSE767027 (P=3.1 × 10−4) and GSE10072 data set28 (non-paired, P=1.39 × 10−6; matched pairs, P=2.9 × 10−6) (Supplementary Figure S1B), breast cancer (Supplementary Figure S1C), and malignant melanoma (Supplementary Figure S1D). These in silico data indicate the overexpression pattern of Lon in various cancers, which strongly suggests that Lon overexpression may have a role during tumorigenesis.
Lon is a stress protein and its upregulated level is associated with cell survival
Next we attempted to unveil the molecular mechanism of Lon upregulation in tumorigenesis. Lon protein is largely induced by hydrogen peroxide (H2O2), hypoxia, CoCl2, and ultra-violet (UV) irradiation (Figure 1A). Stress proteins induce their expression to enhance cell survival during stress and are involved in the regulation of gene expression, signal transduction, and apoptosis.29 We first performed an in silico analysis to show that Lon is selectively overexpressed in the transformed cells that bypasses oncogene-induced senescence triggered by Ras-MEK activation (Supplementary Figure S2). As hypoxia regulates survival and apoptotic cell death,30 we examined whether the expression of Lon correlates to cell survival under hypoxia. The expression pattern of anti-apoptotic protein Bcl-2 and phosphorylated MEK1/2 paralleled to the one of Lon (Figure 1B, left panel). Moreover, the pattern of Lon expression was able to reflect on the extent of cell viability under different periods of exposure of hypoxia (Figure 1B, right). Similarly, the level of Lon expression was correlated to a decrease in cleaved-caspase 3 after H2O2 treatment (Figure 1C, left), suggesting that upregulation of Lon protects cells from apoptosis under recovery of oxidative stress. To prove that Lon is important for cell survival, we knocked-down Lon expression by short hairpin RNA (shRNA) technique to examine the effect on apoptosis under H2O2 treatment or none (Figure 1C, right and Figure 1D). In the Lon-compromised cells, they were proceeding to apoptosis during recovery from H2O2 treatment or even without treatment (Figure 1C, right and Figure 1D). Likewise, cleaved-caspase 3 and PARP were observed in the cells without Lon overexpression after UV treatment, whereas almost no signal was found in the cells overexpressing Lon (Figure 1E). We consistently observed that apoptosis is attenuated in Lon-overexpressed cells after UV treatment (Figure 1F); DNA fragmentation positive cells (Figure 1G) and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells (e in Figure 1H) were not detected in cells overexpressing Lon. Interestingly, we found that the protein level of Lon is reversely correlated to that of p53 (Figures 1C, D), proposing that the mechanism of cell protection by increased Lon under stress is associated with and through regulating the protein level of p53. Together, these results indicate that overexpression of Lon protects cells from apoptosis and promotes cell survival upon environmental stress.

Lon is a stress protein and its level is associated with cell survival. (A) Lon expression is induced with various stresses. Lon protein analysis of 293T cells treated with nothing (No), 200 μM hydrogen peroxide (H2O2) for 24 h, hypoxia (1% O2) for 24 h, CoCl2 (100 μM) for 24 h, or UV (50 J/m2, recovery for 3 h). The cell extracts were immunoblotted with an antibody to Lon and antibody to actin as a loading control. Result is presented as a representative of three independent experiments. (B) The level of Lon is associated with cell survival under hypoxia 293T cells (3 × 104 cells) were seeded and collected at indicated time point after exposure to hypoxia (1% O2). The protein expression was detected by western blotting using the indicated antibodies. An antibody to Actin was used as a loading control. The viable cells determined by trypan blue dye exclusion assay were shown in right panel. Data are presented as mean±S.D. of at least three independent experiments. (C) The level of Lon is associated with cell survival under oxidative stress 293 cells were retrovirally infected with pMKO vector or pMKO expressing shRNAs for human Lon. Subsequently, these cells were treated with 200 μM H2O2 for 4 h and then incubated for indicated recovery time after the treatment. Immunoblots were probed with indicated antibodies and anti-actin antibody acts as a loading control. The expression of Lon with or without infection of shRNAs was shown. (D) Downregulation of Lon is associated with apoptotic activation FADU cells were retrovirally infected with pMKO vector, Lon-pBABE-puro, or pMKO expressing shRNAs for human Lon. Immunoblots were probed with indicated antibodies and anti-actin antibody acts as a loading control. (E) Overexpression of Lon protects the cells from apoptosis under UV treatment. 293T cells were infected with pcDNA3-Lon, pIRES2-hLon, or vector and treated with UV (50 J/m2, recovery for 3 h). The protein expression was detected by western blotting using the indicated antibodies. An antibody to Actin was used as a loading control (left panel). Exogenous expression of Lon in cells was verified by western blotting with anti-Lon antibody (right panel). The result is a representative of those obtained in at least three individual experiments. (F) Overexpression of Lon protects the cells from UV-induced apoptosis. 293T cells were infected with pcDNA3-Lon or not and treated with UV (50 J/m2). The protein expression was detected by western blotting using the indicated antibodies. An antibody to Actin was used as a loading control. The result is a representative of those obtained in at least three individual experiments. (G) Overexpression of Lon inhibits UV-induced apoptosis shown by DNA fragmentation analysis. DNA fragmentation analysis demonstrated that Lon overexpression prevents UV-induced apoptosis (50 J/m2). M: DNA marker; C: control; V: Vector only; L: Lon overexpression. (H) Overexpression of Lon inhibits UV-induced apoptosis shown by TUNEL assay. 293T cells were transfected with pcDNA3-Lon or not and treated with UV (50 J/m2). TUNEL assay was applied to the transfected cells and the effect of Lon overexpression on UV-induced apoptosis was examined. TUNEL-positive cells (green fluorescence, arrow indicated) were observed in the cells with UV treatment (a) or without Lon overexpression (c), however, no TUNEL-positive cells were detected in the cells with Lon overexpression (e). DAPI was used for nuclear staining. The error bars shown in the right panel represent the S.D. from three different experiments
Overexpression of Lon facilitates cell proliferation and transformation
To delineate the role of increased Lon expression in tumorigenesis, Lon was stably expressed in 293, FADU and OEC-M1 cells to establish Lon-overexpressing cells (293/Lon, FADU/Lon and OEC-M1/Lon) and their expression and localization were identified (Figure 2A and Supplementary Figure S3). The proliferation of Lon-overexpressing cells was significantly faster than that of their wild-type counterparts after 48 h (Figure 2B), thus indicating that upregulation of Lon is associated with cell proliferation. Colony formation assay showed the significantly increased colony formation activity in the Lon-overexpressing cells compared with the control cells (Figure 2c and Supplementary Figure S4A). The representative picture of FADU/Lon versus FADU cells was shown (Figure 2c, bottom). Consistently, knocking-down Lon by shRNA significantly reversed the increased colony formation activity in FADU/Lon and SCC-15/Lon cells, confirming that Lon is important for cell transformation (Figure 2d and Supplementary Figure S4B). These data indicate that Lon overexpression increases the activity of cell proliferation and transformation.

Lon overexpression increases cell proliferation and transformation activity. (a) Lon-myc is successfully overexpressed in 293/Lon cells. 293 cells were infected by the retrovirus expressing myc-Lon for 48 h. After 24 h for recovery, puromycin (2 μg/ml) was added to select successfully infected 293 cells stably overexpressing Lon (293/Lon). The expression of Lon and myc-Lon were detected by western blotting using an antibody to Lon and myc, respectively. An antibody to actin was used as a loading control. (b) Overexpression of Lon increases cell growth. 106 Of 293 and 293/Lon cells were seeded. After 24 and 48 h, cell number was analyzed by counting. *P<0.05. (c) Overexpression of Lon increases cell transformation. The long-term effects of Lon overexpression on cancer cell transformation were determined by colony formation assay in FaDu and FaDu/Lon cells. **P<0.01. Bottom, a representative picture of clonogenicity assay using 1 × 103 cells in each well. (d) Downregulation of Lon decreases cell transformation. The long-term effects of Lon downregulation on cancer cell transformation were determined by colony formation assay in FaDu/Lon and FaDu/Lon+shLon cells. ***P<0.001. Bottom, a representative picture of clonogenicity assay using 1 × 103 cells in each well
Overexpression of Lon causes mitochondrial ROS generation
To seek molecular mechanisms of Lon upregulation-mediated tumorigenesis, we surveyed literatures and tried to find a linkage. We and others showed that Lon is induced by hypoxia (Figure 1A), and it regulates ROS generation of hypoxic cells by degrading COX 4–1.25 We thus proposed that Lon overexpression triggers ROS generation to activate cell signaling-linked cell survival and proliferation. ROS could be detected by staining cells with the H2O2 indicator dichlorodihydrofluorescein diacetate (DCFDA) when cells were treated with H2O2 and UV (Supplementary Figure S5). We first showed that ROS level was dramatically increased under hypoxia and further diminished by the addition of ROS scavenger, N-acetylcysteine (NAC) (Figure 3a). We then overexpressed Lon by an increasing amount in 293T cells and found that the ROS level was significantly increased in a dose-dependent manner (Figure 3b) and inhibited by the addition of NAC (Figure 3c, left). Consistently, similar results were observed in SCC-15 and H1299 cells (Figure 3c). To test whether ROS elicited by Lon overexpression are derived from mitochondria, we stained 293/Lon cells with a mitochondrial superoxide indicator MitoSox. We found that 293/Lon cells exhibit a dramatic increase in mitochondrial superoxide production according to the staining (Figure 3d), suggesting that Lon overexpression increases generation of mitochondrial ROS including H2O2 and anion superoxide. To confirm this, we knocked-down Lon expression by shRNA in cancer cells to examine the effect on mitochondrial ROS production. SCC-15 or FADU/Lon expressing Lon-shRNA cells exhibited a decrease in mitochondrial superoxide production (Figure 3e and Supplementary Figure S6). These studies suggest that Lon overexpression triggers mitochondrial ROS generation, which may be linked to cell survival and proliferation.

Upregulation of Lon increases the level of mitochondrial ROS. (a) ROS generation in 293T cells exposed to hypoxia (1% O2 for 24 h) were assessed using DCFDA staining and fluorescence intensity of the cells was measured in the presence or absence of NAC. (b) ROS generation in 293T cells overexpressing different amount of Lon as indicated were assessed using DCFDA staining and fluorescence intensity of the cells was measured. (c) Intracellular ROS generation by the cells overexpressing Lon was assessed using DCFDA staining in the presence or absence of NAC. The cell lines are as indicated. ROS level is presented as a ratio of fluorescence intensity of the stained cells and the buffer-only control. The error bars in graph indicate the S.E. from three independent experiments. *P<0.05, **P<0.01, and ***P<0.001. (d) Mitochondrial superoxide generation in 293 cells expressing Lon was assessed using MitoSox staining and fluorescence intensity of the cells was measured. (e) Mitochondrial superoxide generation in SCC-15 cells expressing vector or Lon-shRNA was assessed using MitoSox staining and fluorescence intensity of the cells was measured
Overexpressed Lon-induced mitochondrial ROS generation is due to the upregulation of NDUFS8 in Complex I by Lon
As Complex I is one of major sources of mitochondrial ROS,5 we wondered whether the function of Complex I is required for production of ROS induced by Lon. To prove this, we tested the effects of rotenone, a specific inhibitor of Complex I, on Lon-induced ROS generation using MitoSox as a probe. Rotenone reduces Lon overexpression-induced superoxide in 293/Lon cells (Figure 4a), which is consistent with the fact that rotenone reduces high NaCl-induced ROS.31 Similar results were observed in OEC-M1/Lon (Supplementary Figure S7) and FADU/Lon (data not shown). These results point to a mitochondrial complex I source of the Lon overexpression-induced increase in ROS.

Upregulation of Lon increases mitochondrial ROS through the stabilization and upregulation of NDUFS8 of complex I by Lon. (a) Mitochondrial superoxide generation in 293/Lon cells was inhibited by rotenone (2 μM, 3 h). MitoSox staining and fluorescence intensity of the cells was measured. (b) Upregulation of Lon increases the protein level of NDUFS3 and NDUFS8. 293T, OEC-M1, or FADU cells were stably transfected with plasmid encoding Lon. The cell extracts were immunoblotted with indicated antibodies and antibody to actin as a loading control. (c) Expression level of Lon and NDUFS8 in 293 cells overexpressing Lon and NDUFS8-shRNA. 293/Lon cells were retrovirally infected with pMKO vector or pMKO expressing shRNAs for human NDUFS8. Immunoblots were probed with indicated antibodies and anti-actin antibody acts as a loading control. The expression of NDUFS8 with or without infection of shRNAs was shown. (d) Mitochondrial superoxide generation in 293 cells overexpressing Lon was inhibited by NDUFS8-shRNA. MitoSox staining and fluorescence intensity of the cells was measured. (e) Lon interacts with NDUFS8 shown by Co-IP. OEC-M1 or FADU cells were stably transfected with plasmid encoding Myc-Lon followed by Co-IP with anti-Lon and anti-NDUFS8, respectively. The immunoprecipitation complex was analyzed by western blotting using indicated antibodies. (f) The complex formation between mitochondrial Lon and NDUFS8 was verified by confocal immunofluorescence OEC-M1/Lon cells were fixed and immunostained by anti-NDUFS8 (green) and anti-Lon (red) antibodies. DNA was stained with DAPI (blue). (g) Downregulation of Lon decreases the protein level of NDUFS8. 293 and OEC-M1 cells were retrovirally infected with pMKO vector or pMKO expressing shRNAs for GFP or Lon. Immunoblots were probed with indicated antibodies and anti-actin antibody acts as a loading control. The expression of NDUFS8 with or without infection of Lon-shRNA was shown. (h) The proteolytic activity of Lon is not a major factor to the stabilization of NDUFS8 by Lon. 293T cells were transfected with wild-type pcDNA3-Lon (WT), a proteolytic mutant (LonS855 A) or pcDNA3 vector and treated with H2O2 (200 μM for 4 h) or not. Exogenous overexpression of Lon was verified by western blotting with anti-Lon antibody. Western blotting was performed by using indicated antibodies. An antibody to actin was used as a loading control. The number below Aconitase and TFAM blot represents the band intensity normalized against actin. The numbers shown in the bottom represent Lane 1 to 6
Five Fe-S proteins (NDUFS1, 2, 3, 7, and 8) are important functional components in the catalytic core of Complex I, which are responsible for electron transfer to ubiquinone.32 Thus, we wondered whether these molecules are required for the Lon-induced ROS through Complex I. Unexpectedly, we found that Lon overexpression increases the protein level of NDUFS3 and NDUFS8 in 293 T, OEC-M1, and FADU cells (Figure 4b). This observation allowed us to understand a novel mechanism that how Lon controls production of ROS through upregulating these Fe-S proteins. To confirm the Fe-S protein is required for the Lon-induced ROS through Complex I, the expression of NDUFS8 was suppressed by shRNA in 293 cells overexpressing Lon (Figure 4c). Figure 4d shows that the suppression of NDUFS8 in the cells nearly abolished the Lon-induced ROS formation, confirming that NDUFS8 is required for the Lon-induced ROS through Complex I. We observed that Lon may interact with NDUFS8 according to a proteomic analysis (unpublished results). In addition, recent studies showed that Lon is attached to prohibitin complex and recruited to the inner membrane of mitochondria,33, 34 suggesting that Lon could be recruited to the inner membrane where NDUFS8 is located. Therefore, we hypothesized that Lon interacts with NDUFS8 and regulates the stability or level of the latter. The interaction between Lon and NDUFS8 was confirmed by co-immunoprecipitation (Co-IP) experiments (Figure 4e). In addition, confocal immunofluorescence analysis showed Lon’s colocalization with NDUFS8, suggesting that they are in the same protein complex (Figure 4f). The Lon depletion by shRNA decreased NDUFS8 level in 293 and OEC-M1 cells (Figure 4g), confirming that NDUFS8 level is regulated by Lon. To exclude the possibility that the proteolytic activity of overexpressed Lon is defective, we overexpressed the proteolytic mutant of Lon (LonS855A) to examine the status of NDUFS8. Overexpression of LonS855A caused the accumulation of aconitase and TFAM that are physiological substrates of Lon protease19, 20 (Figure 4h), confirming the efficacy of LonS855A mutant. We found that level of NDUFS8 is increased under not only Lon overexpression but also H2O2 treatment (Figure 4h, lane 1, 3, and 5). However, the level of NDUFS8 showed no substantial difference between samples from Lon WT and LonS855A overexpression regardless H2O2 treatment or not (Figure 4h, lane 3–6). Taken together, these results suggest that overexpressed Lon-induced mitochondrial ROS is due to the upregulation of NDUFS8 protein by Lon. In addition, this upregulation may need the interaction with Lon rather than the proteolytic activity of Lon.
Lon overexpression-induced ROS promotes cell proliferation through Ras-ERK signaling
We further pursued molecular mechanisms of Lon-promoted cell proliferation and tumorigenesis through ROS production. As Ras acts as an upstream activator of ROS-associated signal pathways,35 we tested whether Lon overexpression activates Ras signaling pathway. The data showed that Lon overexpression upregulates Ras activity and activates phosphorylations of c-Raf, MEK1/2, and ERK1/2, which are dependent on ROS generation (Figure 5a). This result is consistent with that ROS is required for Kras-induced anchorage-independent growth through activation of ERK MAPK signaling pathway.16 In addition, other p38 and JNK MAPK concurrently were activated by ROS derived from Lon overexpression (Figure 5a). To verify this, we knocked-down Lon expression by shRNA in cancer cells to examine the effect on Ras-MAPK activation. FADU expressing Lon-shRNA cells exhibited a decrease in phosphorylations of MEK1/2, ERK1/2, and p38 (Figure 5b). To confirm Ras-MEK-ERK signaling is involved in Lon-promoted cell proliferation, we treated 293T/Lon cells with specific MAPK inhibitors to evaluate the status of cell cycle progression by the level of cyclin D1, cyclin E (G1/S), and cyclin A (S/G2). The treatment with MEK inhibitor PD98059 significantly reduced the expression of cyclin proteins but not with p38 inhibitor SB203580 and JNK inhibitor SP600125 (Figure 5c). Consistently, the treatment with PD98059 significantly reduced the Lon-induced cell proliferation but not with SB203580 and SP600125 (Figure 5d). These findings are supported by that Lon is selectively overexpressed in the transformed cells that bypasses senescence triggered by Ras-MEK activation (Supplementary Figure S2). These results indicate that three members of MAPK are activated by ROS generation caused by Lon overexpression, but only the ERK pathway promotes cell proliferation.

Overexpression of Lon enhances cell proliferation via ROS-dependent Ras-ERK1/2 signaling. (a) Overexpression of Lon activates ROS-dependent MAPK and Ras-c-Raf-MEK1/2-ERK1/2 signaling. Ectopic expression of Lon in 293 T cells is performed by transfected with pcDNA3-Lon. The expression of Lon, phosphorylated signaling kinases were detected by western blotting using indicated specific antibodies. The ROS production was inhibited by addition of NAC (1, 5, or 10 mM) for 24 h. Result is presented as a representative of three independent experiments. (b) Downregulation of Lon diminishes the MAPK activation triggered by increased Lon FADU cells were retrovirally infected with pMKO vector, Lon-pBABE-puro, or pMKO expressing shRNAs for human Lon. Immunoblots were probed with indicated antibodies and anti-GAPDH antibody acts as a loading control. Result is presented as a representative of three independent experiments. (c and d) Overexpression of Lon enhances cell proliferation via ERK signaling. 293T cells overexpressing Lon were treated with 20 μM PD98059 (PD, MEK inhibitor), 5 μM SB203580 (SB, p38 inhibitor), and 10 μM SP600125 (SP, JNK inhibitor) for 24 h. The protein levels of cyclin A, D1, and E were analyzed by western blotting using indicated antibodies (c) Result is presented as a representative of three independent experiments. For cell growth curves, 293T cells overexpressing Lon were seeded onto 24-well plates (2 × 105 cells/well). After 20 h, the cells were left non-treated or treated with the inhibitors and the cell proliferation was measured using the MTT assay at the indicated time intervals (d)
Overexpression of Lon promotes cell migration/invasion and EMT through ROS-mediated ERK and p38 MAPK pathways
ROS is required for hypoxia-dependent EMT that has been reported to be associated with tumor cell migration, invasion, and metastasis.36, 37, 38 We asked whether Lon overexpression also has a role during cancer metastasis. The in silico analysis revealed that Lon is selectively overexpressed in metastatic prostate cancer39 (Figure 6a), suggesting that Lon overexpression may have a role during tumor metastasis. Lon-overexpressed cells induce the expression of MMP-2 (Figure 6b) and significantly increase their ability of cell migration (Figure 6c and Supplementary Figure S8), suggesting that Lon overexpression facilitates cell invasion/migration. To confirm this, we knocked-down Lon expression by shRNA in cancer cells to examine the effect on cell migration. FADU/Lon or SCC-15 expressing Lon-shRNA cells exhibited a decrease in ability of cell migration (Figure 6d). In addition, 293/Lon cells show a progressive change into a cell morphology closer to that of fibroblasts (Figure 6e). We thus tested whether EMT process is induced by Lon overexpression. Lon overexpression promotes the activation of EMT markers, for example, E-cadherin, N-cadherin, Vimentin, and Snail (Figure 6f). More importantly, the changes in EMT markers were inhibited by the addition of NAC (Figure 6f), suggesting that Lon-induced EMT process is dependent on ROS generation. Moreover, the treatment with MEK inhibitor and p38 inhibitor inactivated EMT markers but not with PI3K inhibitor (Figure 6g). These results indicate that Lon overexpression-induced EMT process is activated by ERK and P38 signaling, for which ROS generation is required.

Overexpression of Lon induces EMT through ROS-dependent MAPK signaling. (a) Lon expression is upregulated in metastatic prostate tumor cells. Box plots showed relative Lon mRNA level of benign, primary, and metastatic tumor cells in the public data sets GDS1439. (b) Upregulation of Lon increases the protein level of MMP-2. OEC-M1 or FADU cells were stably transfected with plasmid encoding Lon. The cell extracts were immunoblotted with indicated antibodies and antibody to GAPDH as a loading control. (c) Overexpression of Lon induces cell migration. 293 and 293/Lon cells were seeded in Oris 96-well, and removed the stopper after 16 h. Cell migration was analyzed by their motility ability after 24 and 48 h. The diameter of cell-free area was measured (Supplementary Figure S8), and data were collected and quantitated from three independent experiments. **P<0.01. (d) Downregulation of Lon reduces cell migration. FADU or SCC-15 cells were retrovirally infected with pMKO vector, Lon-pBABE-puro, and/or pMKO expressing shRNAs for human Lon. Cells were seeded in Oris 96-well, and removed the stopper after 16 h. Cell migration was analyzed by their motility ability after 24 and 48 h. The diameter of cell-free area was measured as described in Figure 6c, and data were collected and quantitated from three independent experiments. *P<0.05; **P<0.01. (e) EMT-like cell morphology was observed in 293 cells overexpressing Lon (293/Lon). Scale bar was shown. (f) Overexpression of Lon induces ROS-dependent EMT. Ectopic expression of Lon in 293T cells is performed by transfected with pcDNA3-Lon. The protein expression was detected by western blotting using the indicated antibodies. The ROS production was inhibited by addition of NAC (1, 5, or 10 mM) for 24 h. Result is presented as a representative of three independent experiments. (g) Overexpression of Lon induces EMT via ERK and p38 signaling. 293T cells overexpressing Lon were treated with 20 μM PD98059 (PD, MEK inhibitor), 5 μM SB203580 (SB, p38 inhibitor) and 10 μM LY294002 (LY, PI3K inhibitor) for 24 h. The protein levels were analyzed by western blotting using indicated antibodies. The result is a representative of those obtained in at least three individual experiments
Clinical significance of Lon overexpression in oral squamous cell carcinoma (OSCC)
To examine our findings in human tumors, we studied the expression of Lon in OSCC cell lines and specimens because Lon overexpression has never been documented in oral cancer to date and our in silico analysis showed Lon overexpression in head-and-neck ranks at top 1% (Supplementary Figure S1A). We found that most of oral cancer-derived cells showed a higher level of Lon than the cells derived from pre-cancer lesion (DOK) and normal primary cells (CGHNK2 and CGK6) (Figure 7a). Consistent with Figure 7a, Lon mRNA was upregulated in oral cancer-derived cells, OEC-M1, HSC-3, and OC3, compared with the normal cells (Figure 7b). To link the clinical significance of increased Lon level in cancer tissues, 99 samples of tumor tissues and 5 benign tissues from OSCC patients were used to determine Lon expression pattern by immunohistochemistry (IHC) analysis. The positive staining was observed in the majority of OSCC tumor tissues (86/99, 86.9%), whereas absent staining was observed in benign tissue (Figure 7c and Supplementary Table S1). These results indicate that Lon is overexpressed specifically in OSCC patients.

Lon is upregulated in OSCC. (a) Lon is overexpressed in oral cancer cell lines. The extracts of oral cancer cell lines were immunoblotted with an antibody to Lon and antibody to actin as a loading control. Densiometric analysis of Lon correlated to actin level is shown at the bottom. (b) mRNA expression profile of Lon in oral cancer cell lines. Quantitative real-time Quantitative real-time PCR analysis showed increased Lon expression in samples from three of four oral cancer cell lines. The error bars shown in the graph represent the S.D. from three different experiments. The results were presented as fold increase relative to CGK6 cells (deliberately set to 1). (c) Immunohistochemical analysis of Lon expression in OSCC patients. Representative immunohistochemical analysis of Lon was performed by using paraffin-embedded sections of OSCC and normal epithelium. The representative results shown here are negative staining intensity of normal oral epithelium and moderate staining intensity of oral cancer tissues. Microscopic magnification was × 200 (left) and × 400 (right)
Discussion
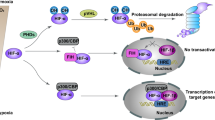
To our knowledge, the present report for the first time demonstrates how Lon contributes cancer cell survival and tumor progression. The Lon has essential roles in tumorigenesis by both inhibiting apoptosis and promoting cell proliferation, transformation, invasion/migration, and EMT. Lon is upregulated in cancer cells, resulting in increased concentration of mitochondrial ROS. They are produced from Complex I and used to promote cell survival and aggressive phenotype of cancer through activation of MAPK and RAS-ERK1/2 signaling (Figure 8).

The role of Lon overexpression in tumorigenesis and malignancy progression. Several stresses in tumor microenvironment, hypoxia, oxidative stress, genetic instability, upregulate the level of mitochondrial Lon. Upregulation of Lon increases the protein level of NDUFS8 in mitochondrial complex I, leading to the increased concentration of mitochondrial ROS from complex I. Increased ROS levels in cancer cells promote cell survival, transformation, and malignancy progression, for example, cell migration and EMT, through activation of MAPK and/or Ras-ERK1/2 signaling
Here we confirm that Lon acts as a stress protein and its expression is induced by oxidative stress, hypoxia, and UV irradiation (Figure 1a). We also show that these stresses including hypoxia induce Lon expression whose level is important for cell survival. The findings connect Lon and hypoxic adaptations that enable cancer cell survival, including apoptosis repression and activated angiogenesis.25, 40 In addition to degrading damaged proteins, Lon may also be a protein chaperone to assist cells to survive and adapt to various stresses that are linked to oncogenesis. Molecular chaperones of heat shock protein (HSP) family have important roles in promoting tumor growth and survival.41, 42 In present work, indeed, we found that increased Lon enhances cell survival and malignant phenotypes through a putative chaperone activity but not a proteolytic activity. These results are supported by the fact that HSP70 and HSP60 are overexpressed in cancer cells and have crucial roles in modulating the apoptotic pathways and in cancer development.41 However, as very few associated proteins of Lon have been identified in human, the mechanism of how Lon employs its chaperone activity to regulate mitochondrial functions linked to tumorigenesis still remains obscure.
The present studies show that overexpression of Lon exhibits a significant increase in the level of mitochondrial ROS that come from complex I of ETC. These results are in agreement with previous reports clearly showing that the MAPK pathway can be activated by ROS generated in complex I or III in mitochondria.12 Increased Lon interacts with NDUFS8 of complex I and causes an increased level of this protein, which is similar to the finding that overexpression of Rieske Fe-S protein in complex III enhanced the hypoxic ROS formation.43 In addition, mitochondrial Fe-S proteins in complexes II and III, for example, SDHB and Rieske, and COX 4-1 are potential substrates of Lon.25, 44, 45 These reports strongly support that Lon overexpression leads to ROS generation owing to dysfunction of mitochondrial complex I to III of ETC.
Our results show that ROS induced by Lon overexpression activate MAPK (p38, JNK, and ERK1/2) and Ras-ERK signaling. These findings are in good agreement with that ROS are shown to exhibit oncogenic effects at low concentrations and act as second messengers that activate various signaling pathways controlling cell proliferation, transformation, and survival.10, 46 In this work, ERK1/2 is involved in the regulation of cell survival (anti-apoptosis), proliferation, and cell migration/EMT, whereas p38 and JNK are shown to mainly activate EMT in response to oxidative stress caused by Lon overexpression (Figure 8), suggesting that ERK is important for overall cell survival, and p38/JNK are involved in stress response and adaptation. Consistently, ERK1/2 is mostly known for their stimulation on cell survival and proliferation; p38 and JNK are deemed stress responsive.12, 13, 47 In addition, our finding and other lines of evidence suggest that p38 and JNK have emerged as a key signaling molecule in the regulation of cancer invasion/metastasis by modulating the activity of molecules governing EMT.14, 48 EMT and metastasis are proposed to be an integrated strategy for cancer cells to avoid oxidative damage and escape excess ROS in the primary tumor site.36 Indeed, our studies indicate that the activation of MAPK signaling due to Lon overexpression induces EMT, suggesting that MAPKs are involved in determining cell fates in oxidative stress response and giving survival advantages to the cancer cells overexpressing Lon.
ROS positively contribute to carcinogenesis and to malignant progression of tumor cells through driving genomic damage, and as signaling intermediates in cellular signal transduction for various cellular processes in tumor microenvironment.10, 46 On the other hand, excessive ROS can damage to cellular proteins, DNA, or lipids and trigger the signaling to senescence or apoptosis. The paradoxical roles of ROS imply that cancer cells die by the same mechanism that promotes their survival. Thus, the big question is how cells integrate conflicting signals to make cell fate decisions for survival or death? A current accepted model is that the role of ROS depends on their level.10, 49 Low levels and local distribution of mitochondrial ROS mediates its role as a second messenger and are required for cellular survival and adaptation, whereas high levels of ROS mediates its role as a death signal. This regulation could be explained by the term “mitohormesis”, which is used to describe that nonlethal stress doses that induce oxidative stress give an adaptive, beneficial effect on organismal fitness.50, 51 The relationship between Lon and the regulation of ROS level will be the subject of increased study.
Materials and Methods
Cell culture
293, 293T, H1299, HSC-3, FADU, and KB cells were cultured in medium containing Dulbecco’s modified Eagle’s essential medium (DMEM), supplemented with 5% fetal bovine serum (FBS) and 5% super calf serum. The all medium contain penicillin 100 Units/ml, streptomycin 100 μg/ml except mentioned. SCC-4, SCC-9, SCC-15, SCC-25, and SAS cells were cultured in medium containing a 1 : 1 mixture of DMEM/F12 medium, supplemented with 10% FBS. OC3 cells was cultured in medium containing a 1 : 1 mixture of DMEM/K-SFM medium, supplemented with 10% FBS, and OEC-M1 and YD-15 cells were cultured in medium containing RPMI 1640 medium, supplemented with 10% FBS. Oral pre-cancer lesion cell DOK (dysplastic oral keratinocyte) was cultured in medium containing DMEM, supplemented with 10% FBS.52 Oral keratinocyte normal cell lines, CGHNK2 and CGK6,53 were grown in a culture medium containing K-SFM medium, supplements for K-SFM, supplemented with G418 (400 μg/ml).
Cell treatment
Cultured cells were treated with hydrogen peroxide (H2O2, Sigma-Aldrich, St. Louis, MO, USA) for 200 μM 1 or 24 h at 37 °C and the medium was changed to recover for the indicated period. For hypoxic treatment, cells were grown in a hypoxia chamber and equilibrating for 30 min with humidified gas containing 1% oxygen, 5% CO2, and 94% nitrogen. The cells were exposed under hypoxic conditions for various time courses as indicated.
Plasmid construction
Human mitochondrial Lon protease with Myc tag was cut by EcoRV (New England Biolabs, Hitchin, UK) from hLon-Myc-pcDNA3β and cloned into pBabe-puro vector, which was cut by the same restriction enzyme. The sequence of Lon-Myc-pBABE-puro plasmid was verified by DNA sequencing (Genomics BioSci&Tech. Co., Taipei, Taiwan). For construction of shRNA expression vectors, the retrovirus vector pMKO-puro54 was used. Sequences of the shRNA target sites were shown as below.55 Lon-shRNA: 5′-GAAAGUUCGUCUCGCCCAGCC-3′ or 5′-AGGAGCAGCUAAAGAUCAUCA-3′; NDUFS8-shRNA: 5′-CAUCAACUACCCGUUCGAGAA-3′.
Cell transfection and retroviral infection
The cells were grown in 10 cm dish until they reached 70–80% confluence before introducing the expression of individual constructs by transfection with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Stable cell lines expressing tagged Lon or Lon-shRNA were generated by retroviral infection using pBabe or pMKO vector, respectively. The plasmids, hLon-Myc-pBABE-puro for stable expression and Lon-shRNA-pMKO-puro for stable knocked-down cell, along with packaging plasmid gag-pol and envelope plasmid VSV-G, were transfected into 293T cells by TurboFect. The cells were incubated for 24 h and the medium was changed to remove remaining transfection reagent. Retroviral supernatant was collected at 24 and 48 h post-transfection and used to infect the target cells 293 for 48 h. Polybrene (hexadimethrine bromide) was added to the medium for improving infection efficacy. The following day, medium was changed and the infected cells were incubated for recovery for 24 h. Puromycin (Sigma-Aldrich) was used to select the successfully infected cells at a final concentration of 2 μg/ml and the survived cells were collected to check the expression of human Lon protease by western blotting.
Reagents
ERK inhibitor (PD98059), p38 inhibitor (SB203580), JNK inhibitor (SP600125), and PI3K inhibitor (LY294002) were purchased from Calbiochem (San Diego, CA, USA). NAC and rotenone were obtained from Sigma Chemical Co. (St. Louis, MO, USA). For mitochondrial location, MitoTracker Red FM (Invitrogen) was added in DMEM at a final concentration of 500 nM for 30 min at 37 °C.
Antibodies
Antibodies to human Lon was produced as described previously.23 Antibodies used in this study were purchased as indicated: HIF-1α, cleaved-caspase 3, cleaved-PARP, phsopho-p53(Ser46), TFAM (no. 7495), phospho-c-Raf (Ser289/296/301), phospho-MEK1/2 (Ser217/221), phospho-ERK1/2 (Thr202/Tyr204), phospho-p38 (Thr180/Tyr182), phospho-JNK (Thr183/Tyr185), MMP-2 (no. 4022), and Snail antibodies were from Cell Signaling Technology (Beverly, MA, USA); p53, Bax, Bcl-2, cyclin A, and cyclin D1 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); Myc (9E10), Actin from Millipore (Temecula, CA, USA); cyclin E, N-cadherin and Vimentin were obtained from BD Transduction Laboratories (Franklin Lakes, NJ, USA); FLAG (M2) from Sigma-Aldrich; Aconitase (ab71452), VDAC2 (ab37985), α-tubulin (ab4074) from Abcam (Cambridge, MA, USA); caspase 3 from IMGNEX (San Diego, CA, USA); E-cadherin, GAPDH, and beta-actin from GeneTex (Hsinchu, Taiwan) NDUFS3 (1D6) and NDUFS8 (B01P) from Abnova (Taipei, Taiwan).
Western blot analysis
The cells were harvested by trypsinization and lysed with NETN buffer (20 mM Tris (pH 8.0), 1 mM EDTA, 150 mM NaCl, 0.5% nonidet P-40 (NP-40)) containing protease inhibitor cocktail (Roche, Mannheim, Germany). The cell lysates were then centrifuged at 10 000 g at 4 °C to obtain solubilized cellular proteins. Protein was quantified with a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Proteins were separated by 8 or 10 or 12% SDS-PAGE and electrotransfered to a polyvinylidene fluoride membrane. Blots were probed with primary antibodies, followed by HRP-conjugated goat anti-rabbit IgG (1 : 5000, v/v) (Zymed, South San Francisco, CA, USA) or HRP-conjugated goat anti-mouse IgG (1 : 5000, v/v) (Zymed). After washing with phosphate buffer saline (PBS) containing 0.5% Tween-20, peroxidase activity was assessed using enhanced chemiluminescence (PerkinElmer Life Science, Boston, MA, USA). For an internal control, the same membrane was re-probed with a monoclonal antibody directed against β-actin (1 : 10 000, v/v) or GAPDH (1 : 5000). The intensities of the reaction bands were analyzed with the Image Gauge System (Fuji, Tokyo, Japan).
Cell viability analysis
Cell viability analysis was performed by either cell number counting or MTT assay. For cell number counting, cells were seeded on six-well plastic dishes at a concentration of 3 × 104 per well. Before the experiment, 60–70% confluence cells were washed twice with PBS and treated with hypoxia (1% O2) or not. Cells were then stained with trypan blue and cell numbers were counted under an inverted microscope. Data are presented as the mean±SD of three replicates from six separate experiments. For 293/Lon cell growth assay, 106 cells were seeded in the 10 cm dishes and incubate in 5% CO2 incubator at 37 °C. After 0, 24, and 48 h, trypan blue assay were used for testing cell viability. Cell suspensions were diluted with 0.4% trypen blue solution. Cells were counted under the microscope; the viable cells would not be stained, nonviable cells would be blue. Cell numbers were calculated after normalized with dilution rate. For MTT assay, 293T cells were seeded into 24-well plates at a density of 2 × 105 cells/well and incubated overnight. Cells were transfected with pcDNA3-Lon plasmid and treated with PD98059, SB203580, and SP600125 after transfection for 6 h. After treatment for 24 or 48 h, cells were stained with MTT reagent (5 mg/ml). After 2 h incubation with MTT reagent, the crystals were produced in wells and then dissolved with DMSO. Two-hundred microlitter of the final solutions were transferred to 96-well plate and measured at 562 nm by spectrophotometer.
DNA fragmentation analysis
After treatment, both detached and attached cells were collected and washed with PBS. The analysis was performed using the Tissue and Cell Genomic kit (GeneMark, Taichung, Taiwan), according to the manufacturer’s instructions. Fragmented DNA was extracted and electrophoresed on a 2.5% agarose gel containing 0.1 μg/ml ethidium bromide.
TUNEL staining
Cell apoptosis was detected by TUNEL assay according to the manufacturer’s instructions (TaKaRa BIO, Shiga, Japan) and was performed as described previously.23 Briefly, the cells grown on a cover slip in a six-well plate were transfected with Lon plasmid or vector only for 24 h. After transfection, cells were exposed to UV and recovered for 24 h. The cells were then chilled on ice, washed three times with cold 0.3% Triton-X-100-PBS, and fixed in 2% paraformaldehyde for 30 min at room temperature. After fixation, the cells were washed three times with cold 0.3% Triton-X-100-PBS, and cover slip cultures were incubated with a TUNEL reaction mixture in a humidified atmosphere for 60 min at 37 °C in the dark. After incubation, the cells were washed three times with cold 0.3% Triton-X-100-PBS, and then cover slip cultures were counterstained with 4′,6 diamidino-2-phynylindole (DAPI) and examined by a Leica microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Colony formation assay
To determine long-term effects, 1 × 103 of FaDu and FaDu/Lon cells were seeded in six-well plate. Cells were culture with DMEM containing 5% FBS in 37 °C incubator. After 15 days, cell colonies were stained by crystal violet and calculated.
Evaluation of ROS production
The production of hydrogen peroxide (H2O2) was measured with DCFDA (Sigma, St. Louis, MO, USA) staining as described according to the manufacturer’s instructions. Briefly, cells were washed three times with PBS and incubated with 10 μM DCFDA for 1 h at 37 °C, 5% CO2. After incubation, cells were washed and fluorescence was measured using a Berthold Technologies LB941 TriStar Multimode Reader (BERTHOLD TECHNOLOGIES GmbH & Co. KG, Bad Wildbad, Germany), with excitation at 485 nm and emission at 535 nm. To detect mitochondrial superoxide levels, cells were stained with 5 μM MitoSOX (Molecular Probes, Eugene, OR, USA). After incubation 37 °C for 10 min, cells were washed three times with PBS. The mitochondrial ROS were visualized by using fluorescence microscope or analyzed by using FACS (Ex/Em: 510/580 nm).
Co-IP assay
Cells were lysed in NETN (150 mM NaCl, 1 mM EDTA, 20 mM Tris-Cl (pH 8.0), 0.5% NP-40) containing protease and phosphatase inhibitors (1.0 mM sodium orthovanadate, 50 μM sodium fluoride). Immunoprecipitation was performed by incubating primary antibodies with cell lysates at 4 °C for 4 h, followed by the addition of secondary antibody and protein A/G-agarose (Calbiochem) for one additional hour. For western blot analysis, proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA), and overnight incubated in primary antibodies followed by 1 h of incubation in horseradish peroxidase-conjugated secondary antibodies.
Confocal immunofluorescence staining
The immunofluorescence staining was determined as described previously56 with minor modifications. Cells were fixed in 3.7% formaldehyde in PBS for 10 min at room temperature and permeabilized with 0.1% Triton X-100 for 5 min. After blocked in 3% BSA, the cells were incubated with primary antibody 1 h at 37 °C and then with the anti-mouse fluorescence-labeled secondary antibody 1 h at room temperature. DNA was stained with DAPI (Invitrogen). Subsequently, the cells were incubated with 2 M HCl for 30 min at 37 °C to denature DNA and treated with 0.1 M borate buffer (Na2B4O7, pH 8.5) for 5 min to neutralize the acid. After 3% BSA blocking for 1 h at room temperature (RT), the cells were incubated with anti-Lon antibody (1 : 200) or anti-NDUFS8 (1 : 200) 1 h at 37 °C, then with goat anti-mouse DyLight fluorescence-conjugated secondary antibody for 1 h at RT. The above experimental results were analyzed by an Leica TCS SP5 II confocal fluorescence microscopy.
Cell migration assay
The cell migration was detected with the Oris cell migration assay kit (Platypus Technologies, Fitchburg, WI, USA). About 5 × 104 cells were seeded in the 96-well plate and incubated 16 h to let cell attachment. The seeding stoppers were removed to permit cells migration into the detection zone and change the medium to 200 μl DMEM containing 0.1% FBS. The cell motility was observed on microscope after 0, 24, and 48 h. Finally, the diameter of the cell-free area was measured to quantify the migration ability.
IHC staining
IHC analysis was performed on an automatic staining machine (BenchMark XT, Ventana Medical Systems, Tucson, AZ, USA) using the iVIEW 3, 3-diaminobenzidine (DAB) detection kit (Ventana Medical Systems). Paraffin sections (4 μm) containing human of the 104 OSCC tissues were routinely deparaffinized, hydrated, and heated to 95–100 oC for 4 min to induce antigen retrieval. After inactive the endogenous peroxidase activity, IHC staining was performed with anti-Lon (dilution 1 : 600).23 All sections were finally incubated with iVIEW copper for 4 min to enhance the signal intensity, then counterstained with hematoxylin, dehydrated, mounted, and observed by a Nikon Eclipse E600 light microscope (Tokyo, Japan). Pictures were acquired using Tissue-Faxs software (TissueGnostics GmbH, Vienna, Austria). The staining intensity was estimated in a 4-scored scale (0, negative staining; 1+, weak; 2+, moderate; 3+, strong intensity). The fraction of stained cells was scored according to the following criteria: score 0 (no stained or <10% stained cells), score 1 (11–50% stained cells), score 2 (51–80% stained cells), and score 3 (>80% stained cells).
Reverse transcription-PCR (RT-PCR)
Total RNA was extracted using Trizol reagent (Invitrogen) and reverse-transcribed at 37 °C with SuperScript II reverse transcriptase (Invitrogen). The resulting cDNA was used as the template for PCR reactions. Real-time PCR reactions were performed on a RotorGene 3000 system (Corbett Research, Mortlake, Victoria, Australia) using SYBR Green PCR Master Mix (Cambrex Co., East Rutherford, NJ, USA). The sets of forward and reverse primers, the corresponding PCR conditions, and the lengths of PCR products were described as follows: Lon (5′-GGACGAGGAGCTGAGCAA-3′, 5′-AGGTAGTTGCGGGTGACATT- 3′; 94 °C (30 s), 60 °C (60 s) and 72 °C (30 s) for 40 cycles, 72 bp); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-GAAGGTGAAGGTCGGAGT-3′, 5′-GAAGATGGTGATGGGATTTC-3′; 94 °C (30 s), 52 °C (30 s) and 72 °C (1 min) for 28 cycles, 220 bp). All the PCR reactions were started at 94 °C for 5 min and terminated at 72 °C for 5 min. Finally, the data were analyzed using RotorGene software v5.0 (Corbett Research). Differential RNA expressions between various samples were calculated using GAPDH as an internal control.
Clinical sample
A total of 104 cases were chosen for IHC analysis based on availability of archival human oral tissue blocks from diagnostic resection specimens. Specimens including 99 OSCC patients and 5 benign tissues of epithelium were retrieved from the archives of the Departments of Pathology at Mackay Memorial Hospital, Taipei, Taiwan from January 1999 to December 2006 with approval from the Institutional Review Board (IRB number: 11MMHIS067).
Statistical methods
Parametric Student’s t-test was used to judge the significance of difference between conditions of interest. In all analysis, a P value of <0.05 was considered as statistically significant (Student’s t-test, *P<0.05, **P<0.01, and ***P<0.001).
Abbreviations
- DAPI:
-
4′,6 diamidino-2-phynylindole
- DCFDA:
-
dichlorodihydrofluorescein diacetate
- DMEM:
-
Dulbecco’s modified Eagle’s essential medium
- EMT:
-
epithelial–mesenchymal transition
- ETC:
-
electron transport chain
- HIF-1α:
-
hypoxia inducible factor-1α
- HSP:
-
heat shock protein
- IHC:
-
immunohistochemistry
- MAPK:
-
mitogen-activated protein kinase
- NAC:
-
N-acetylcysteine
- OXPHOS:
-
oxidative phosphorylation
- OSCC:
-
oral squamous cell carcinoma
- PBS:
-
phosphate buffer saline
- ROS:
-
reactive oxygen species
- RT:
-
room temperature
- shRNA:
-
short hairpin RNA
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- UV:
-
ultra-violet
References
Gogvadze V, Orrenius S, Zhivotovsky B . Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 2008; 18: 165–173.
Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Saavedra E, Moreno-Sanchez R . The causes of cancer revisited: "mitochondrial malignancy" and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med 2010; 31: 145–170.
Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E et al. Mitochondrial gateways to cancer. Mol Aspects Med 2010; 31: 1–20.
Green DR, Reed JC . Mitochondria and apoptosis. Science 1998; 281: 1309–1312.
Murphy MP . How mitochondria produce reactive oxygen species. Biochem J 2009; 417: 1–13.
Stowe DF, Camara AK . Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal 2009; 11: 1373–1414.
Pan JS, Hong MZ, Ren JL . Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol 2009; 15: 1702–1707.
Schumacker PT . Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 2006; 10: 175–176.
Lee HC, Wei YH . Mitochondrial DNA instability and metabolic shift in human cancers. Int J Mol Sci 2009; 10: 674–701.
Hamanaka RB, Chandel NS . Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 2010; 35: 505–513.
Wu WS . The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 2006; 25: 695–705.
Runchel C, Matsuzawa A, Ichijo H . Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal 2011; 15: 205–218.
Wada T, Penninger JM . Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004; 23: 2838–2849.
Hong J, Zhou J, Fu J, He T, Qin J, Wang L et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res 2011; 71: 3980–3990.
Wagner EF, Nebreda AR . Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–549.
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010; 107: 8788–8793.
Lee AY, Hsu CH, Wu SH . Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding: role of N-terminal and sensor and substrate discrimination domains. J Biol Chem 2004; 279: 34903–34912.
Venkatesh S, Lee J, Singh K, Lee I, Suzuki CK . Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim Biophys Acta 2012; 1823: 56–66.
Bota DA, Davies KJ . Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol 2002; 4: 674–680.
Matsushima Y, Goto Y, Kaguni LS . Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc Natl Acad Sci USA 2010; 107: 18410–18415.
Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol 2002; 157: 1151–1160.
Bota DA, Ngo JK, Davies KJ . Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic Biol Med 2005; 38: 665–677.
Wang HM, Cheng KC, Lin CJ, Hsu SW, Fang WC, Hsu TF et al. Obtusilactone A and (-)-sesamin induce apoptosis in human lung cancer cells by inhibiting mitochondrial Lon protease and activating DNA damage checkpoints. Cancer Sci 2010; 101: 2612–2620.
Ngo JK, Davies KJ . Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med 2009; 46: 1042–1048.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL . HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007; 129: 111–122.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6: 1–6.
Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 2007; 8: 140.
Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 2008; 3: e1651.
Fulda S, Gorman AM, Hori O, Samali A . Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010: 214074.
Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996; 379: 88–91.
Zhou X, Ferraris JD, Burg MB . Mitochondrial reactive oxygen species contribute to high NaCl-induced activation of the transcription factor TonEBP/OREBP. Am J Physiol Renal Physiol 2006; 290: F1169–F1176.
Brandt U . Energy converting NADH:quinone oxidoreductase (complex I). Ann Rev Biochem 2006; 75: 69–92.
Bayot A, Basse N, Lee I, Gareil M, Pirotte B, Bulteau AL et al. Towards the control of intracellular protein turnover: mitochondrial Lon protease inhibitors versus proteasome inhibitors. Biochimie 2008; 90: 260–269.
Bayot A, Gareil M, Rogowska-Wrzesinska A, Roepstorff P, Friguet B, Bulteau AL . Identification of novel oxidized protein substrates and physiological partners of the mitochondrial ATP-dependent Lon-like protease Pim1. J Biol Chem 2010; 285: 11445–11457.
Downward J . Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22.
Pani G, Galeotti T, Chiarugi P . Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev 2010; 29: 351–378.
Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008; 29: 2267–2278.
Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci 2010; 101: 293–299.
Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005; 8: 393–406.
Ruan K, Song G, Ouyang G . Role of hypoxia in the hallmarks of human cancer. J Cell Biochem 2009; 107: 1053–1062.
Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR . Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci 2006; 31: 164–172.
Jego G, Hazoume A, Seigneuric R, Garrido C . Targeting heat shock proteins in cancer. Cancer Lett 2010; 332: 275–285.
Korde AS, Yadav VR, Zheng YM, Wang YX . Primary role of mitochondrial Rieske iron-sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic Biol Med 2011; 50: 945–952.
Guillon B, Bulteau AL, Wattenhofer-Donze M, Schmucker S, Friguet B, Puccio H et al. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. Febs J 2009; 276: 1036–1047.
Major T, von Janowsky B, Ruppert T, Mogk A, Voos W . Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease pim1. Mol Cell Biol 2006; 26: 762–776.
Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB . Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal 2012; 16: 1295–1322.
Wada T, Stepniak E, Hui L, Leibbrandt A, Katada T, Nishina H et al. Antagonistic control of cell fates by JNK and p38-MAPK signaling. Cell Death Differ 2008; 15: 89–93.
Kim MS, Lee EJ, Kim HR, Moon A . p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res 2003; 63: 5454–5461.
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M . Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160: 1–40.
Tapia PC . Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: "Mitohormesis" for health and vitality. Med Hypotheses 2006; 66: 832–843.
Ristow M, Zarse K . How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 2010; 45: 410–418.
Chang SE, Foster S, Betts D, Marnock WE . DOK, a cell line established from human dysplastic oral mucosa, shows a partially transformed non-malignant phenotype. Int J Cancer 1992; 52: 896–902.
Lu YC, Chen YJ, Wang HM, Tsai CY, Chen WH, Huang YC et al. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer Prev Res 2012; 5: 665–674.
Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB et al. Telomerase maintains telomere structure in normal human cells. Cell 2003; 114: 241–253.
Granot Z, Kobiler O, Melamed-Book N, Eimerl S, Bahat A, Lu B et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: the unexpected effect of proteasome inhibitors. Mol Endocrinol 2007; 21: 2164–2177.
Liu E, Lee AY, Chiba T, Olson E, Sun P, Wu X . The ATR-mediated S phase checkpoint prevents rereplication in mammalian cells when licensing control is disrupted. J Cell Biol 2007; 179: 643–657.
Acknowledgements
The authors thank Dr. C-T Liao (Department of Otorhinolaryngology, Chang Gung Memorial Hospital, Taiwan), Dr. Y-J Chen, Ms. Y-C Lu, and Dr. A-J Cheng (Department of Medical Biotechnology, Chang Gung University, Taiwan) for kindly providing oral normal primary cells (CGHNK2 and CGK6). This work was supported by grants from National Science Council (NSC 98-2311-B-037-001-MY3), National Health Research Institutes (101A1-CA-PP-08 and 102A1-CA-PP-08), and Department of Health (DOH99-TD-C-111-004), Taiwan to A Y-L Lee.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Finazzi-Agró
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cheng, CW., Kuo, CY., Fan, CC. et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis 4, e681 (2013). https://doi.org/10.1038/cddis.2013.204
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.204
Keywords
This article is cited by
-
Mitochondrial Lon-induced mitophagy benefits hypoxic resistance via Ca2+-dependent FUNDC1 phosphorylation at the ER-mitochondria interface
Cell Death & Disease (2023)
-
A Fc-VEGF chimeric fusion enhances PD-L1 immunotherapy via inducing immune reprogramming and infiltration in the immunosuppressive tumor microenvironment
Cancer Immunology, Immunotherapy (2023)
-
Lon upregulation contributes to cisplatin resistance by triggering NCLX-mediated mitochondrial Ca2+ release in cancer cells
Cell Death & Disease (2022)
-
Evidence for mitochondrial Lonp1 expression in the nucleus
Scientific Reports (2022)
-
Structures of the human LONP1 protease reveal regulatory steps involved in protease activation
Nature Communications (2021)
