
Abstract
Inflammation enhances the secretion of sphingomyelinases (SMases). SMases catalyze the hydrolysis of sphingomyelin into phosphocholine and ceramide. In erythrocytes, ceramide formation leads to exposure of the removal signal phosphatidylserine (PS), creating a potential link between SMase activity and anemia of inflammation. Therefore, we studied the effects of SMase on various pathophysiologically relevant parameters of erythrocyte homeostasis. Time-lapse confocal microscopy revealed a SMase-induced transition from the discoid to a spherical shape, followed by PS exposure, and finally loss of cytoplasmic content. Also, SMase treatment resulted in ceramide-associated alterations in membrane–cytoskeleton interactions and membrane organization, including microdomain formation. Furthermore, we observed increases in membrane fragility, vesiculation and invagination, and large protein clusters. These changes were associated with enhanced erythrocyte retention in a spleen-mimicking model. Erythrocyte storage under blood bank conditions and during physiological aging increased the sensitivity to SMase. A low SMase activity already induced morphological and structural changes, demonstrating the potential of SMase to disturb erythrocyte homeostasis. Our analyses provide a comprehensive picture in which ceramide-induced changes in membrane microdomain organization disrupt the membrane–cytoskeleton interaction and membrane integrity, leading to vesiculation, reduced deformability, and finally loss of erythrocyte content. Understanding these processes is highly relevant for understanding anemia during chronic inflammation, especially in critically ill patients receiving blood transfusions.
Similar content being viewed by others


Main
During their stay in the circulation for ∼120 days, erythrocytes undergo age-related changes involving senescent cell antigen formation, cell shrinkage, vesiculation, and loss of deformability. The binding of autologous IgG and exposure of recognition molecules, possibly including phosphatidylserine (PS), ultimately lead to recognition of senescent erythrocytes by macrophages of the reticulo-endothelial system.1 Also, the deformability of the erythrocyte constitutes an important survival parameter, as splenic sequestration of abnormal erythrocytes with reduced deformability leads to a decreased erythrocyte life span and anemia in several erythrocyte membranopathies.2
Chronic inflammation, as occurring in various pathological conditions, is associated with anemia and poor disease outcome.3 This inflammation-driven anemia is especially troublesome in patients with severe sepsis. Furthermore, blood transfusions to counter sepsis-induced anemia only have a limited beneficial effect, and may lead to severe complications.4 Reduced erythrocyte production can only partly explain the low erythrocyte count. Enhanced erythrocyte clearance is likely to contribute to the anemia, but its causes are largely unknown.5 Therefore, understanding the underlying mechanisms of anemia of inflammation is of critical importance.
Inflammation triggers the secretion of acid sphingomyelinase (SMase), which is involved in various pathologies, including diabetes, sepsis, cardiovascular and pulmonary diseases.6 SMases catalyze the hydrolysis of sphingomyelin (SM), a major lipid component of cell membranes, into phosphocholine and ceramide.7 Ceramide functions as a lipid second messenger in many cellular processes including apoptosis, senescence and inflammation, which is partially explained by the tendency of signaling receptors to cluster in ceramide-enriched platforms.8, 9 Also, the formation of these rigid platforms alters membrane curvature and decreases plasma membrane integrity.7, 10
Although erythrocytes do not possess SMase activity of their own,11 and SMases have not been reported as part of the erythrocyte proteome,12 erythrocytes can be exposed to SMase secreted by vascular endothelium, leukocytes, and platelets.6 SMase treatment and direct ceramide incorporation into erythrocytes induces PS exposure.13, 14, 15 Also, erythrocytes expose PS and ceramide after incubation with SMase-containing plasma of patients suffering from sepsis or Wilson’s disease.16, 17 So far, however, only PS exposure and size have been studied. Pathophysiologically relevant molecular processes on the membrane and protein level and their association with functionally relevant parameters have not been addressed as yet.
In the current study, we comprehensively addressed the changes that erythrocytes undergo with respect to morphology, deformability, membrane lipid organization, and protein–protein interactions upon exposure to SMase. Time-lapse microscopy revealed that SMase-driven PS exposure precedes loss of membrane integrity. We also demonstrate a clear erythrocyte age-related increase in sensitivity to SMase, both in erythrocytes from freshly drawn blood and from blood bank units. Next to PS exposure as a critical removal signal, enhanced vesiculation, which is associated with several erythrocyte-associated pathologies,18 was observed. These findings reveal SMase-induced ceramide formation as a critical event in erythrocyte membrane reorganization on both the lipid and protein level, and provide a possible explanation for the reduced effectiveness and harmful side effects of erythrocyte transfusion in patients with sepsis.
Results
SMase alters erythrocyte morphology and increases PS exposure
Secretion of SMase is enhanced during inflammation and involved in the pathophysiology of various diseases.6 As SMase-induced ceramide formation changes membrane lipid organization, enhanced SMase activity could also affect erythrocyte physiology including deformability and exposure of removal signals such as PS, thereby explaining excessive erythrocyte clearance in anemia of inflammation. This hypothesis is supported by the observation that plasma of septic patients induced ceramide formation and PS exposure on the erythrocyte membrane.16
To determine the sensitivity of erythrocytes to SMase, erythrocytes were incubated with ascending activities of SMase and analyzed by flow cytometry. Bacterial SMase was used as a valid surrogate for acid and neutral SMase encountered by erythrocytes in vivo.19 Cytoplasmic proteins were labeled by incubation with membrane-permeable carboxyfluorescein succinimidyl ester diacetate (CFSE diacetate). In this way, a potential loss of membrane integrity was detectable through loss of fluorescence from the cytoplasm.
Erythrocyte size, as reflected by forward scatter, significantly decreased already at 1 mU/ml, with a further reduction in size and a high increase in PS exposure at activities of 10 mU/ml and higher (Figures 1a–c). Also, the asymmetric shape of the forward-versus-sideward scatter plot of the erythrocyte population was lost upon treatment, suggesting that the cell shape was affected (Figure 1a). The sideward scatter increased, which is indicative of the degree of cell granularity. In the absence of extracellular calcium during 10 and 100 mU/ml SMase treatment PS exposure was not induced (0.92% and 0.72% respectively), suggesting the involvement of a calcium-activated scramblase in this process.20

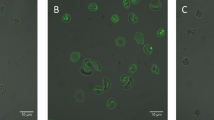
Erythrocyte shape and PS exposure during SMase treatment. (a–c) Flow cytometry of erythrocytes treated with increasing activities of SMase for 15 min at 37 °C. Annexin V-FLUOS staining was performed to determine the percentage of PS-exposing erythrocytes. (a) Forward and sideward scatter (FSC and SSC, respectively) density plots are presented. (b and c) The graphs represent mean values of three healthy volunteers; error bars represent S.D.; *P<0.05. (d) Time-lapse confocal laser scanning microscopy of CFSE-labeled erythrocytes treated with 10 mU/ml SMase at 37 °C in the presence of Annexin V-Alexa 647. Confocal laser scanning microscopy was used to image fluorescence as described in Materials and Methods. Scale bar=5 μM. A typical result from one of three experiments is presented
Although the flow cytometry analyses provided information on the erythrocyte population at a fixed time point, time-lapse confocal laser scanning microscopy revealed a characteristic sequence of events upon SMase treatment: first, erythrocytes lost their discoid shape, which was followed by a gradual increase in the percentage of PS-exposing cells (Figure 1d and Supplementary Movie S1). Subsequently, some erythrocytes lost their cellular content, as indicated by the loss of the CFSE signal. After 45 min of SMase treatment, a considerable part of the cells had an irregular membrane surface, as observed by bright-field microscopy (Supplementary Figure S1). Intriguingly, most of these cells showed high-intensity CFSE spots, which colocalized with the membrane irregularities (Supplementary Figure S1). These clusters are most likely large protein clusters, as CFSE covalently binds to proteins. Furthermore, only a few of the irregularly shaped cells exposed PS, as compared with the spherical cells with a smooth surface and high PS exposure (Supplementary Figure S1 and Supplementary Movie S1).
SMase specifically hydrolyzes SM in erythrocytes
To confirm enzyme specificity and to estimate the extent of SM breakdown and ceramide formation, lipid analyses were performed on the membrane fractions of erythrocytes treated with various SMase activities.
Consistent with the expected activity of SMase, positive-ion matrix-assisted laser desorption and ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) revealed the presence of ceramide and absence of SM in membrane lipid fractions of 10 and 100 mU/ml SMase-treated erythrocytes (Figure 2a). In controls and 1 mU/ml-treated cells, no ceramide was detected. Apparently, at 1 mU/ml concentration the extent of ceramide formation was below the detection limit of this method. Alternatively, the small amount of ceramide might have been suppressed by more abundant lipids, such as phosphatidylcholines (PCs).21 We were not able to detect ceramide subsequent to analysis by combined thin-layer chromatography/MALDI as previously described.22 However, after 1 and 5 mU/ml SMase treatment the percentage of cells with ceramide-enriched platforms was increased, as detected by immunofluorescence (see below). This is a strong indication that the amount of the generated ceramide at 1 mU/ml SMase is beyond the detection limit of MS. 31P nuclear magnetic resonance (NMR) spectroscopy confirmed the nearly complete disappearance of SM after 100 mU/ml SMase treatment of erythrocytes, while the major membrane phospholipids phosphatidylethanolamine (PE), PC and PS were not affected (Figure 2b). Thus, the used enzyme preparation specifically hydrolyzed SM in the erythrocyte plasma membrane.

Erythrocyte membrane lipid analyses after SMase treatment. Erythrocytes were treated with SMase for 1 h at 37 °C. (a) Positive-ion MALDI-TOF MS of isolated membrane lipids. All samples were 1:1 (v/v) diluted with the matrix (0.5 M 2,5-dihydroxybenzoic acid in methanol). All peaks are labeled based on their m/z ratios and matrix cluster ions are marked by an asterisk.39 (b) 31P NMR spectra of membrane lipid extracts. All samples were investigated in the presence of 200 mM sodium cholate to suppress the aggregation of phospholipids.40 Cer, ceramide
SMase-induced erythrocyte membrane reorganization
The changes in erythrocyte morphology, PS externalization, and ceramide formation, suggest that SMase induces major membrane reorganization. This reorganization was studied in more detail using electron microscopy and confocal laser scanning microscopy.
Consistent with our observations by confocal microscopy, electron microscopy also showed an SMase treatment-induced loss of the typical biconcave shape (Figure 3a). A striking finding was the presence of structures resembling intracellular membrane vesicles, or endovesicles, in the SMase-treated erythrocytes. In most cells these structures were confined to one side of the cell in one large cluster.

SMase-induced erythrocyte morphology, membrane lipid rafts, and intracellular invaginations. SMase treatment of erythrocytes was performed for 15 min at 37 °C. (a) Electron microscopy of control and SMase-treated erythrocytes as described in Materials and Methods. (b) Erythrocytes stained with an anti-CD59 antibody. (c) Quantification of CD59 cluster-positive erythrocytes. The graph presents mean values (N=2), error bars represent S.D. Two-hundred cells were scored by eye per experiment. (d) Acrolein-fixed erythrocytes stained with anti-stomatin (green) and anti-band 3 (red) antibodies. (e) Z-stack of an anti-stomatin-stained, tubulated, erythrocyte after treatment with 10 mU/ml SMase. A Z-slice spacing of 0.25 μM was used. (f) Flow cytometry and confocal microscopy of erythrocytes treated with 5 mU/ml SMase, stained with anti-ceramide or isotype control antibody. Confocal laser scanning microscopy was used to image fluorescence as described in Materials and Methods. Bright-field images are shown in combination with fluorescence images. Signal intensity in b and e: low=blue, intermediate=red, and high=yellow/white. Scale bar=5 μM. Representative images from one of two experiments are shown in all cases
We also investigated the presence of microdomains in SMase-treated erythrocytes.23 In untreated erythrocytes, the glycosyl phosphatidylinositol-anchored microdomain marker, CD59,24 was distributed evenly over the erythrocyte membrane (Figure 3b). SMase treatment at activities of 1 mU/ml and higher induced CD59 clusters that colocalized with the membrane irregularities observed in the bright-field images (Figure 3b).
SMase-treated erythrocytes were also stained for stomatin, which is anchored at the cytoplasmic side of the membrane and is associated with lipid rafts in erythrocytes.25 The findings with stomatin were similar to those obtained with CD59, although many additional small clusters were observed upon SMase treatment (Figure 3d). Moreover, a combined staining for stomatin and band 3, which is an abundant erythrocyte membrane protein involved, among others, in linking the plasma membrane to the cytoskeleton, revealed the presence of tubular plasma membrane invaginations in cross-sections of a subpopulation of the SMase-treated erythrocytes (Figure 3d, right-most images and Figure 3e).26 The tubular invaginations also colocalized with membrane irregularities seen in bright field. A three-dimensional reconstruction and animation showing these invaginations is provided in the Supplementary Data (Supplementary Movies S2 and S3).
Finally, anti-ceramide antibody staining revealed the presence of ceramide-enriched platforms on SMase-treated erythrocytes (Figure 3f). It is noteworthy that following acrolein fixation seven to eight times more positive events were detected (after correction for isotype control) than following paraformaldehyde fixation. The recognition of ceramide on only a subpopulation of the cells after 5 mU/ml SMase treatment and the high dependence of antibody binding on the fixative used suggest that the specificity and activity of this antibody is strongly affected by membrane organization.
Next, we investigated whether SMase-induced membrane reorganization also affected the interaction between the plasma membrane and the cytoskeleton. Differential membrane protein extraction showed that the insoluble fraction of band 3 increased upon SMase treatment (Supplementary Figure S2). In contrast, the extractability of β-actin with detergent increased upon SMase treatment. These changes implicate that in addition to the membrane microdomain organization at the protein and lipid levels, the cytoskeletal integrity and the interaction between plasma membrane and cytoskeleton are also altered by SMase.
SMase enhances erythrocyte vesicle formation
During aging, the erythrocyte produces numerous vesicles that expose removal signals including PS, that are enriched in damaged membrane patches, and are rapidly removed from the circulation. Physiologically, erythrocyte vesiculation may constitute a protective mechanism to prevent untimely erythrocyte removal.27 Increased erythrocyte vesicle numbers were observed in several inflammation-associated diseases, including chronic renal failure, sickle cell disease, and β-thalassemia.18 Therefore, we investigated whether erythrocyte vesiculation rate and vesicle composition are affected by SMase treatment.
Erythrocytes were allowed to vesiculate in vitro in the presence or absence of SMase. The vesicles generated in vitro and the plasma vesicles from the corresponding healthy volunteers were assessed for PS exposure and the presence of the erythrocyte-specific marker glycophorin A (CD235a). Erythrocyte-derived plasma vesicles, control vesicles generated in vitro, and vesicles from SMase-treated erythrocytes all exposed PS and expressed CD235a (Figure 4a). Plasma vesicles and control vesicles formed a single population with a comparable, high PS exposure and CD235a expression. An additional population with clearly reduced PS exposure and CD235a expression was observed in the SMase-induced vesicles (Figures 4a and b). In contrast, glycophorin C expression was found to be identical in all conditions (data not shown). On the basis of the forward scatter, all PS+CD235a+ vesicles had similar dimensions (Figure 4b). In addition to these qualitative differences between control and SMase-induced vesicles erythrocyte, vesiculation was enhanced more than 20-fold when the cells were exposed to SMase (Figure 4c). This heterogeneity in PS exposure and CD235a expression indicates that SMase-induced vesiculation differs qualitatively from vesiculation in the absence of SMase. Finally, CFSE-loaded erythrocytes generated CFSE-positive vesicles during SMase treatment (Figure 4d), showing that these vesicles contain cytoplasmic content of the parent cells.

SMase-induced vesiculation of erythrocytes. Erythrocytes were allowed to vesiculate in the presence or absence of 10 mU/ml SMase for 1 h at 37 °C. The vesicles were analyzed by flow cytometry. (a) Density plots of forward/sideward scatter (FSC and SSC, respectively), and Annexin V-FLUOS (PS)/anti-CD235a-PE staining of plasma-derived vesicles and vesicles generated in vitro. Vesicles were gated according to FSC/SSC for fluorescence analysis. (b) PS and CD235a mean fluorescence intensity (MFI), and forward scatter of plasma-derived and in vitro CD235a+ vesicles. (c) Quantification of CD235a+ vesicles after erythrocyte vesiculation in the absence or presence of SMase. Fluorescent 10 μM counting beads were used as an internal standard. (d) Bottom left and center: density plots of CFSE-loaded erythrocytes treated with SMase and stained with anti-CD235a-PE. The vesicles generated during the treatment of these cells were stained with anti-CD235a-PE and Annexin V-Alexa 647. Bottom right: vesicles that were within the standard FSC/SSC vesicle range and that were positive for Annexin V are shown in the CFSE/CD235a density plot. For the assessment of dimensions, only PS+CD235a+ vesicles were considered. Data from five healthy volunteers are presented. The graphs present mean values, error bars represent S.D., and *P<0.05
Loss of osmotic responsiveness in erythrocytes treated with SMase
Following the analysis of the impact of SMase treatment on the structural organization of erythrocytes, we were interested in potential functional implications of SMase exposure. In the circulation, erythrocytes are constantly exposed to changing osmolalities, in particular when passing through the kidneys, and osmotic stress is known to induce PS exposure.28 To study the effect of SMase on the osmotic responsiveness and fragility of erythrocytes, SMase-treated erythrocytes were exposed to different osmolalities and analyzed by flow cytometry.
When erythrocytes were treated with SMase activities of 1 mU/ml and higher, a population could be discerned with a low sideward scatter that was not seen in control erythrocytes (Figure 5). These populations had a nearly identical CFSE content and forward scatter, indicating that they had intact membranes and were of comparable size. When exposed to a hypertonic or hypotonic buffer, the SMase-treated erythrocytes with a normal sideward scatter and the control erythrocytes shrank and swelled, respectively (Figure 5). In contrast, the size of the additional population in the SMase-treated erythrocytes did not change upon incubation with these buffers, indicating a loss of osmotic responsiveness. When erythrocytes were treated with higher (≥10 mU/ml) SMase activities and subsequently incubated in hypotonic buffer, nearly all cells, including this additional population, were lysed, showing an SMase-induced increase in membrane fragility (Figure 5).

Osmotic responsiveness and fragility of SMase-treated erythrocytes. CFSE-labeled erythrocytes from three healthy volunteers were treated with increasing activities of SMase for 15 min at 37 °C, and subsequently incubated with hypotonic, isotonic-, and hypertonic buffer for 15 min at room temperature. The cells were analyzed by flow cytometry. A forward/sideward scatter (FSC and SSC, respectively) density plot of SMase-treated erythrocytes is shown in the upper right. A FSC/CFSE density plot of erythrocyte subpopulation A is shown in the upper left. Forward scatter of subpopulations A and B are depicted in the bottom graphs. Populations of<2500 cells are not shown in the graphs. The graphs present mean values, error bars represent S.D., and *P<0.05
SMase enhances erythrocyte retention in a spleen-mimicking model
Sequestration of poorly deformable erythrocytes by the spleen is known to be critically involved in the decreased erythrocyte life span and anemia in several erythrocyte disorders.2 The observed changes in erythrocyte morphology, ceramide formation, and membrane organization upon SMase treatment, likely also affect erythrocyte deformability. To determine whether SMase treatment affected deformability and splenic retention, we used a bead-sorting device that mimics the mechanical deformation that erythrocytes experience in the spleen.
Erythrocytes were treated with SMase for 15 min at 37 °C, labeled with CFSE and perfused through the spleen-mimicking model. Treatment with SMase induced a decrease in the permeation of the model spleen, concomitant with an increase in the retention (Figure 6). As before, SMase treatment reduced erythrocyte size, as reflected by forward scatter (Figure 6).

Perfusion of SMase-treated erythrocytes through a spleen-mimicking device. A suspension consisting of ∼5% CFSE-labeled erythrocytes treated with SMase for 15 min at 37 °C and 95% untreated/unlabeled erythrocytes were passed through a bead-sorting device at a flow rate of 60 ml/h (see Materials and Methods). Flow cytometry was used to determine the percentage of labeled cells in the initial upstream compartment, the bead compartment, and the downstream fractions. Forward scatter (FSC) is presented as a measure for erythrocyte diameter. Representative data from one of two experiments are shown
Age and storage of erythrocytes increase their sensitivity to SMase
Erythrocytes become more sensitive to stressors such as energy depletion and hyperosmotic conditions with age and storage time.28, 29, 30 As SMase plasma levels increase during inflammation, and critically ill patients often receive erythrocyte transfusions, the sensitivity of stored erythrocytes for SMase-induced changes in cell size and PS exposure were studied by flow cytometry.
Erythrocytes from six healthy donors were obtained immediately after donation, and after 14 and 35 days of storage under blood bank conditions. After incubation with 1 mU/ml SMase for 15 min at 37 °C, we observed a much more pronounced size reduction and increase in PS exposure for 35 day-stored erythrocytes, compared with fresh and 14 day-stored cells (Figure 7a).

SMase-induced size change and PS exposure in stored and aged erythrocytes. Flow cytometry of erythrocytes treated with SMase. Annexin V-FLUOS staining was performed to determine the percentage of PS-exposing erythrocytes. Forward scatter is presented as a measure of erythrocyte diameter. (a) Fresh, 14, and 35 days stored erythrocytes from six healthy donors treated with 1 mU/ml SMase for 15 min at 37 °C. (b) Erythrocytes of various mean age obtained from a 27-year-old male donor by combined density and volumetric separation (see Materials and Methods), treated with 1 and 10 mU/ml SMase for 15 min at 37 °C. t, total erythrocyte product. Erythrocyte fractions 1 and 5 have the lowest and highest mean age, respectively. The graphs present mean values, error bars represent S.D., and *P<0.05
As erythrocyte age might also affect the response to SMase-induced ceramide formation in the plasma membrane, a combination of volume and density separation was used to obtain well-defined erythrocyte populations of different mean age. These were treated with 1 and 10 mU/ml SMase, and showed an age-dependent increase in the sensitivity for SMase-induced PS exposure (Figure 7b).
Discussion
The chronic inflammation that arises in patients with various diseases, including sepsis, can induce anemia, which is associated with poor disease outcome.3 Inflammation enhances SMase secretion, and ceramide formation and PS exposure were observed when erythrocytes were incubated with plasma from septic patients ex vivo.16 PS exposure is a marker for cellular stress and contributes to erythrocyte removal.31 So far, studies concerning SMase activity on erythrocytes focused on membrane lipid rearrangement and shape changes.13, 32 In this study we show that ceramide-induced membrane lipid rearrangement affects multiple processes that are critical for erythrocyte function and removal, including protein–protein interactions, membrane stability, and vesiculation. Furthermore, we directly correlated these molecular changes to parameters relating to erythrocyte function in vivo by addressing sensitivity to osmotic stress, permeation through a spleen-mimicking device, and dependence on erythrocyte age.
In agreement with earlier findings,13 we observed a reduction in cell size and increase in the percentage of PS-exposing erythrocytes upon SMase treatment of erythrocytes. Our time-lapse confocal microscopy experiments revealed that the transition of their typical discoid to a spherical shape preceded PS exposure, which was followed by disintegration and loss of cytoplasmic content. The combination of shape change and enhanced vesiculation explain the reduction in size observed by flow cytometry and confocal microscopy after SMase treatment.
Anchorage of the plasma membrane to the cytoskeleton maintains the discoid shape and membrane stability of the erythrocyte, as several erythrocyte disorders that affect membrane anchorage to the cytoskeleton induce abnormal cell shape and enhanced hemolysis.2 Similar to the observations in pathological erythrocytes, our analyses using differential membrane protein extraction demonstrated that this anchorage is also affected by SMase. This may be mediated by protein phosphorylation, which is known to regulate protein–protein interactions in the erythrocyte.33 Furthermore, membrane lipid interaction with cytoskeletal proteins might be affected by SMase-induced membrane reorganization.34
An intriguing finding was the presence of large CFSE-positive protein clusters that appeared in conjunction with the detected membrane irregularities. As CFSE aspecifically reacts with the intracellular proteins – of which >95% is hemoglobin – the observed high-intensity CFSE signals are most likely hemoglobin clusters. This hemoglobin clustering is probably the indirect result of the effect of SMase on the conformation of band 3, which has a high affinity binding site for hemoglobin in its cytoplasmic domain.26
As shown by high-resolution, immunofluorescence microscopy analysis of CD59 and stomatin localization, SMase treatment not only led to morphological changes, but also to a reorganization of membrane microdomains. The resulting larger domains were comparable in size to the endovesicle-like structures observed with electron microscopy. Owing to their small head group in comparison with the original phospholipids, ceramides induce negative membrane curvature, which promotes both lipid membrane invagination and blebbing.10 This provides a plausible explanation for the SMase-induced tubular plasma membrane invaginations. Endovesicles could result from membrane fusion in such tubular structures.35 Membrane blebbing may also further contribute to the large, SMase-induced increase in vesiculation.27
Also, we observed ceramide-enriched membrane platforms, which are known to originate in plasma membranes with high ceramide content.8 The increase in membrane fragility can be attributed to the formation of these large ceramide-enriched membrane platforms, as earlier studies revealed them to be very rigid, resulting in a weak interface between the platform and the surrounding plasma membrane.10 Small pores are known to form at this interface, which may readily explain the loss of osmotic responsiveness of the affected erythrocytes.10 Splenic sequestration of abnormal erythrocytes with reduced deformability has been observed in several erythrocyte disorders, and is associated with a decreased life span and resulting hemolytic anemia in afflicted patients.2 The presence of rigid ceramide platforms in SMase-treated erythrocytes may lead to enhanced splenic retention. Indeed, we could link SMase-induced ceramide formation to reduced deformability by showing that SMase treatment enhanced erythrocyte retention in a bead-sorting device that mimics the mechanical deformation that erythrocytes experience in the spleen.
Vesiculation is an integral part of erythrocyte aging and appears to constitute a protective mechanism to enhance erythrocyte survival.27 Vesicles from erythrocyte concentrates were found to modulate platelet function in vitro.36 Furthermore, increased erythrocyte vesicle numbers were observed in several disease states (e.g., chronic renal failure, sickle cell disease, and β-thalassemia).18 SMase-induced increase in erythrocyte vesiculation, probably resulting from a weakening of membrane anchorage, may contribute to disease pathology because of the pro-coagulant nature of the vesicles.37 The presence of two distinct vesicle populations, as observed by annexin V and anti-CD235a double staining, indicates that vesiculation occurs in distinct types of microdomains, which are present on the erythrocyte membrane during SMase treatment. Furthermore, the high CFSE content of the microparticles generated by treatment of CFSE-loaded erythrocytes with SMase indicates that the microparticles contain a high concentration of cytoplasmic proteins. This suggests that they are sealed vesicles containing hemoglobin, similar to those generated in the circulation.27 This is supported by the reddish color of the vesicle pellet.
Positive-ion MALDI-TOF MS after chromatographic separation of the total lipid extract is a very sensitive analytical method for the detection of cell membrane ceramide content. The observation that 1 mU/ml SMase affected erythrocyte shape, membrane organization, and osmotic responsiveness, even though membrane ceramide content was below the detection limit of the positive-ion MALDI-TOF MS, suggests that erythrocytes are very sensitive to ceramide-induced changes in membrane organization. The absence of other non-specific breakdown products at higher enzyme activities excludes alternative degradation processes as the basis for these observations.
Critically ill patients, in particular those suffering from severe inflammation and sepsis, frequently receive blood transfusions. We showed that erythrocyte storage under blood bank conditions increased the sensitivity to SMase. This may reduce erythrocyte survival after transfusion, especially in patients with enhanced SMase activity. Also, in healthy individuals, physiological erythrocyte age is associated with enhanced sensitivity. Therefore, cellular age likely underlies the observed heterogeneity in susceptibility to SMase per individual cell, as is particularly apparent in Figure S1 and Supplementary Movie S1. These findings imply that the use of older erythrocyte concentrates in patients suffering from systemic inflammation might result in reduced transfusion efficacy and a higher risk of harmful side effects, such as extravascular hemolysis and increased plasma iron concentrations, which are associated with a SMase-induced rapid clearance.38
In summary, we have shown that multiple, pathophysiologically relevant erythrocyte parameters are affected by SMase. These parameters are associated with changes that exceeded mere membrane lipid rearrangement and included altered protein–protein interaction, membrane stability, deformability, and enhanced erythrocyte vesiculation. Furthermore, our findings suggest that enhanced SMase secretion during chronic inflammation contributes to the enhanced erythrocyte clearance that is often associated with inflammation. In particular, these data suggest that transfusion in patients with increased SMase activity should be restricted to fresh erythrocyte concentrates.
Materials and Methods
Collection, isolation, and storage of erythrocytes
Fresh erythrocytes were isolated from whole blood (EDTA) donated by healthy human volunteers, using Ficoll (GE Healthcare, Waukesha, WI, USA) density centrifugation. To obtain erythrocyte populations of different age, a combination of volume and density separation was used.27 Erythrocyte concentrates were obtained using standard blood bank procedures. In short, whole blood (500 ml) was collected in a Composelect quadruple CPD-SAGM top-and-bottom bag system (Fresenius Kabi, Bad Homburg, Germany), containing 70 ml CPD as an anticoagulant. After cooling and centrifugation, erythrocytes were isolated using a Compomat G4 (Fresenius Kabi), after which 110 ml SAGM was added to the erythrocytes. The erythrocyte suspension was leukocyte-depleted by in-line filtration and subsequently stored at 2–6 °C. The study was performed following the guidelines of the local medical ethical committee and in accordance with the declaration of Helsinki. Written informed consent was obtained from all blood donors participating in this study.
Solutions
Erythrocytes were kept in Ringer solution (125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 32 mM HEPES, 5 mM glucose, 2.5 mM CaCl2, pH 7.4). NaCl concentrations of 162.5 mM and 87.5 mM NaCl were used for hyper- and hypotonic Ringer, respectively. Calcium-free Ringer was used during whole-blood erythrocyte and vesicle isolation to prevent clotting.
Erythrocyte lysis was performed using lysis buffer (10 mM HEPES, 1 mM EDTA, 1 mM EGTA, 1 mM benzamidine, and 0.005 mM leupeptin, pH 8.0). Protein extractions were performed using extraction buffer (25 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM benzamidine, 0.005 mM leupeptin, 1% TritonX-100, pH 7.4). When indicated, TritonX-100 was replaced by 10 mM n-Dodecyl-β-D-Maltoside (DDM).
SMase treatment
Unless mentioned otherwise, 2 × 106 erythrocytes were incubated in 100 μl Ringer containing various concentrations of SMase (Sigma-Aldrich, St. Louis, MO, USA; 159 U/mg; one unit of SMase hydrolyzes 1 μM of SM per minute at pH 7.4 at 37 °C) for 15 min at 37 °C. After treatment, cells were washed twice with Ringer and centrifuged at 1000 × g for 3 min. When treated in the absence of extracellular calcium, cells were washed five times with calcium-free Ringer before treatment in this buffer. Subsequent washing and staining of cells in Ringer was performed at 4 °C.
Flow cytometry
To probe PS exposure, cells were washed with Ringer containing 0.2% bovine serum albumin (BSA, Sigma-Aldrich) and stained with Annexin V-FLUOS (Roche, Basel, Switzerland; 1/25) for 1 h at room temperature. Alternatively, erythrocytes were stained with 1 μM CFSE diacetate (Invitrogen, Carlsbad, CA, USA) in Ringer for 15 min at 37 °C before SMase treatment. Excess CFSE diacetate was removed by washing cells three times with Ringer containing 0.2% BSA. Flow cytometry was performed on a FACSCalibur system using Cellquest Pro software version 6.0 (BD Biosciences, Franklin Lakes, NJ, USA). Per sample, 25 000 cells were measured and data analysis was performed using Cyan Summit software version 4.3 (Dako, Glostrup, Denmark).
Confocal microscopy
For time-lapse confocal laser scanning microscopy experiments, 200 μl Ringer containing 1 × 106 CFSE diacetate-labeled erythrocytes, Annexin V-Alexa 647 (Roche; 1/20), and 10 mU SMase were added to a Lab-Tek chambered coverglass (Thermo Fisher Scientific, Rochester, NY, USA). One minute after addition of SMase to the erythrocytes, recording of images was started with 30-sec intervals for a total of 60 min.
Mouse anti-CD59-Alexa 647 monoclonal antibody (clone MEM-43, AbD Serotec, Düsseldorf, Germany; 1/200) staining was performed on erythrocytes in Ringer buffer. Acrolein-fixated erythrocytes were used for combined antibody staining with mouse anti-stomatin (GARP-50, kindly provided by Rainer Prohaska, University of Vienna, Austria; 1/200) and rabbit polyclonal antiserum against the membrane domain of human band 3 (K2N6B/PMB3;39 1/100). Acrolein fixation and subsequent staining were performed as described by Matte et al.40 BSA (1%) was used instead of fish skin gelatin for blocking purposes. Secondary antibodies were goat anti-mouse-Alexa 488 and goat anti-rabbit-Alexa 633 (Invitrogen; 1/1000). Ceramide staining on acrolein-fixated erythrocytes was performed using anti-ceramide IgM (clone MID 15B4, Enzo life sciences, Farmingdale, NY, USA, 1/50) and isotype control IgM (BD Biosciences, 1/50), followed by goat anti-mouse-Alexa 647 staining (Invitrogen; 1/200).
Confocal laser scanning microscopy was performed on a TCS SP5 confocal laser scanning microscope (Leica Microsystems, Mannheim, Germany) equipped with a HCX Plan-Apochromat 63X/N.A. 1.2 water immersion lens. Cells were imaged at a temperature-controlled stage set at 37 °C. The Leica TCS SP5 LAS AF software was used for image acquisition. Image J version 1.45J (National Institutes of Health, Bethesda, MD, USA) was used for further image analysis.
Electron microscopy
Erythrocytes were fixated with 1% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) for 1 h at room temperature. The cells were then washed twice in cacodylate buffer and post-fixated for 1 h in 1% osmium tetraoxide. After two more washes with cacodylate buffer, samples were pelleted in 2% agarose (ultra-low gelling temperature, Sigma-Aldrich) and cells were dehydrated by incubation in an ascending series of aqueous ethanol for 15 min per incubation step. Cells were then incubated overnight in ethanol and Epon at a 1/1 ratio, subsequently incubated for 4 h in pure Epon and embedded at 37 °C overnight in fresh Epon. The samples were then stored at 60 °C overnight, after which 80-nm sections were made using a Reichert Ultracut 6 Microtome (Reichert, Depew, NY, USA). After drying, sections were stained in uranyl acetate for 20 min and subsequently in lead citrate for 10 min. Sections were examined with a JEOL TEM 1010 (JEOL Ltd, Tokyo, Japan).
Erythrocyte vesiculation
All buffers used for vesicle generation, isolation, and analysis were complemented with 0.2% BSA and filtered (0.22 μM) before use. Erythrocytes (1 × 109) were incubated with 5 ml Ringer containing 10 mU/ml SMase for 1 h. The cell suspension was centrifuged for 10 min at 1500 × g, and the supernatant was centrifuged again for 20 min at 1500 × g. Vesicles were then pelleted by centrifugation at 21 000 × g and 4 °C for 20 min. The vesicles were washed once with Ringer and resuspended in 25 μl Ringer. Vesicles from whole blood were isolated from 200 μl plasma as described above, with an additional calcium-free Ringer wash of the vesicle pellet after high-speed centrifugation.
Vesicles were stained with Annexin V-FLUOS and anti-CD235a-PE monoclonal antibody (clone KC16, Beckman Coulter, Brea, CA, USA; 1/50) in a total volume of 50 μl Ringer for 45 min at room temperature, and washed once with Ringer. Vesicles were resuspended in 150 μl Ringer, and washed Flow-Count Fluorospheres (Beckman Coulter) were added (1 × 104) for quantification. Samples were analyzed by flow cytometry as described for erythrocytes at high speed for 1 min, using a protocol optimized for vesicle analysis. Sulfate latex microspheres (Invitrogen; 0.9 μM) were used to determine the maximum upper boundary allowed for forward and sideward scatter gating. All staining solutions were centrifuged at 21 000 × g and 4 °C for 20 min before use to remove fluorescent aggregates. PE-conjugated IgG1 isotype control (Dako) did not show any aspecific binding.
Membrane protein extraction, SDS-PAGE, and immunoblot
Erythrocytes (1 × 109) were incubated with 5 ml Ringer containing 10 mU/ml SMase for 1 h and subsequently lysed in 1 ml lysis buffer. The membrane fraction was washed repeatedly by centrifugation at 21 000 × g for 10 min to remove free hemoglobin. Membrane proteins were extracted by adding 200 μl extraction buffer and samples were vortexed for 30 s. The samples were incubated for 30 min with regular vortexing and were subsequently centrifuged at 21 000 × g for 15 min. Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) containing 5% 2-mercaptoethanol was added to the supernatant at a 1/1 ratio (v/v). The remaining pellet was washed once with phosphate-buffered saline (PBS, pH 7.4) and resuspended in 50 μl sample buffer.
SDS-PAGE was performed using 12.5% running gels within the Mini Protean 3 system (both Bio-Rad), according to the method of Laemmli Either band 3 protein or actin were used as loading controls. Apparent molecular masses were calculated based on the Precision Plus Protein Standard (Bio-Rad). After SDS-PAGE, the proteins were transferred to PVDF membranes using the iBlot system (Invitrogen). The membranes were then blocked with Odyssey Blocking Buffer (OBB, LI-COR, Lincoln, NE, USA), and incubated for 16 h at 4 °C in OBB containing 0.1% Tween-20, rabbit anti-band 3 serum (K2N6B/PMB3;39 1/5000), and mouse anti-β actin monoclonal antibody (clone AC-15, Sigma-Aldrich; 1/5000). After washing with PBS containing 0.1% Tween-20, the blots were incubated for 1 h at room temperature in OBB, 0.1% Tween-20, 0.01% SDS, goat anti-rabbit IgG-Alexa Fluor 680 (Invitrogen; 1/10 000), and goat anti-mouse IgG-IRDye 800 (LI-COR; 1/10 000). This final incubation was followed by a single washing step with PBS containing 0.1% Tween-20, and three subsequent washes with PBS. Immunoblots were scanned using the Odyssey Infrared Imaging System (LI-COR), and analyzed using Odyssey Software version 2.1.
Positive-ion MALDI-TOF MS and 31P NMR analyses of membrane lipids
Packed erythrocytes (300 μl) were incubated with 5 ml Ringer containing SMase for 1 h and subsequently lysed and washed as described for membrane protein extraction. Membrane pellets (200 μl) were transferred to glass tubes and 2 ml methanol and 1 ml dichloromethane were added. After vortexing, another 1 ml dichloromethane and 1.6 ml 0.5% acetic acid were added and vortexed once more. After 10-min centrifugation at 800 × g, the bottom dichloromethane layer was isolated. Residual lipids were extracted from the aqueous layer by extraction with 1 ml dichloromethane. The two dichloromethane fractions were pooled and the solvent evaporated. The lipid fraction was analyzed by positive-ion MALDI-TOF MS and 31P NMR spectroscopy as described by Dannenberger et al.41 Shortly, the lipid extract was mixed 1:1 (v/v) with the matrix solution (0.5 M 2,5-dihdyroxybenzoic acid in methanol), and 1 μl of this mixture was directly applied onto the MALDI target. All MALDI-TOF mass spectra were acquired on an Autoflex I mass spectrometer (Bruker Daltonics, Bremen, Germany) with ion reflector. The system utilizes a pulsed 50-Hz nitrogen laser, emitting at 337 nm. The extraction voltage was 20 kV and gated matrix suppression was applied to prevent the saturation of the detector by matrix ions. All spectra were acquired in the reflector mode using delayed extraction conditions. Peak identities were additionally confirmed by recording post source decay spectra as previously described.42 As the focus of this study is on SM, no negative-ion spectra were acquired because SM is much more sensitively detectable in the positive-ion mode.43
Erythrocyte bead-sorting device
Erythrocyte retention/deformability was assessed using a bead-sorting device that mimics the mechanical deformation that erythrocytes experience in the spleen.44 After SMase treatment, erythrocytes (1 × 108) were labeled with CFSE diacetate. A 2% hematocrit suspension (600 μl) consisting of 5% treated/labeled and 95% untreated/unlabeled erythrocytes in Ringer with 1% BSA was passed through the bead-sorting device at a flow rate of 60 ml/h. The untreated/unlabeled erythrocytes acted as both facilitators of steady perfusion of the cells of interest, and as an internal control for retention/deformability. Flow cytometry measurement (1 00 000cells) of the initial upstream compartment, the bead compartment, and downstream fractions was performed to determine the ratio of labeled versus unlabeled erythrocytes in each separate fraction.
Statistical analysis
Differences between two groups of data were determined using a paired ttest. Differences between multiple groups were assessed with a repeated measures one-way ANOVA in combination with Tukey’s post-test. Reported P-values are two sided, and a P-value of<0.05 was used to assess statistical significance.
Abbreviations
- CFSE:
-
carboxyfluorescein succinimidyl ester
- MALDI-TOF:
-
matrix-assisted laser desorption and ionization time-of-flight
- MS:
-
mass spectrometry
- NMR:
-
nuclear magnetic resonance
- PC:
-
phosphatidylcholine
- PE:
-
phosphatidylethanolamine
- PS:
-
phosphatidylserine
- SM:
-
sphingomyelin
- SMase:
-
sphingomyelinase
References
Bosman GJ, Werre JM, Willekens FL, Novotny VM . Erythrocyte ageing in vivo and in vitro: structural aspects and implications for transfusion. Transfus Med 2008; 18: 335–347.
Mohandas N, Gallagher PG . Red cell membrane: past, present, and future. Blood 2008; 112: 3939–3948.
Roy CN . Anemia of inflammation. Hematology. Am Soc Hematol Educ Program 2010; 2010: 276–280.
Gould S, Cimino MJ, Gerber DR . Packed red blood cell transfusion in the intensive care unit: limitations and consequences. Am J Crit Care 2007; 16: 39–48.
Weiss G, Goodnough LT . Anemia of chronic disease. N Engl J Med 2005; 352: 1011–1023.
Jenkins RW, Canals D, Hannun YA . Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal 2009; 21: 836–846.
Goni FM, Alonso A . Sphingomyelinases: enzymology and membrane activity. FEBS Lett 2002; 531: 38–46.
Grassme H, Riethmuller J, Gulbins E . Biological aspects of ceramide-enriched membrane domains. Prog Lipid Res 2007; 46: 161–170.
Hannun YA, Obeid LM . Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9: 139–150.
Lopez-Montero I, Monroy F, Velez M, Devaux PF . Ceramide: from lateral segregation to mechanical stress. Biochim Biophys Acta 2010; 1798: 1348–1356.
Hanada K, Mitamura T, Fukasawa M, Magistrado PA, Horii T, Nishijima M . Neutral sphingomyelinase activity dependent on Mg2+ and anionic phospholipids in the intraerythrocytic malaria parasite Plasmodium falciparum. Biochem J 2000; 346 (Pt 3): 671–677.
D'Alessandro A, Righetti PG, Zolla L . The red blood cell proteome and interactome: an update. J Proteome Res 2010; 9: 144–163.
Lang KS, Myssina S, Brand V, Sandu C, Lang PA, Berchtold S et al. Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ 2004; 11: 231–243.
Lang F, Gulbins E, Lang PA, Zappulla D, Foller M . Ceramide in suicidal death of erythrocytes. Cell Physiol Biochem 2010; 26: 21–28.
Lang F, Lang KS, Lang PA, Huber SM, Wieder T . Osmotic shock-induced suicidal death of erythrocytes. Acta Physiol (Oxf) 2006; 187: 191–198.
Kempe DS, Akel A, Lang PA, Hermle T, Biswas R, Muresanu J et al. Suicidal erythrocyte death in sepsis. J Mol Med 2007; 85: 273–281.
Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med 2007; 13: 164–170.
Hind E, Heugh S, Ansa-Addo EA, Antwi-Baffour S, Lange S, Inal J . Red cell PMVs, plasma membrane-derived vesicles calling out for standards. Biochem Biophys Res Commun 2010; 399: 465–469.
Ago H, Oda M, Takahashi M, Tsuge H, Ochi S, Katunuma N et al. Structural basis of the sphingomyelin phosphodiesterase activity in neutral sphingomyelinase from Bacillus cereus. J Biol Chem 2006; 281: 16157–16167.
Daleke DL . Phospholipid flippases. J Biol Chem 2007; 282: 821–825.
Schiller J, Suss R, Fuchs B, Muller M, Zschornig O, Arnold K . MALDI-TOF MS in lipidomics. Front Biosci 2007; 12: 2568–2579.
Fuchs B, Schiller J, Suss R, Schurenberg M, Suckau D . A direct and simple method of coupling matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS) to thin-layer chromatography (TLC) for the analysis of phospholipids from egg yolk. Anal Bioanal Chem 2007; 389: 827–834.
Simons K, Gerl MJ . Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 2010; 11: 688–699.
Civenni G, Test ST, Brodbeck U, Butikofer P . In vitro incorporation of GPI-anchored proteins into human erythrocytes and their fate in the membrane. Blood 1998; 91: 1784–1792.
Salzer U, R Prohaska . Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood 2001; 97: 1141–1143.
van den Akker E, Satchwell TJ, Williamson RC, Toye AM . Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol Dis 2010; 45: 1–8.
Willekens FL, Werre JM, Groenen-Dopp YA, Roerdinkholder-Stoelwinder B, de Pauw B, Bosman GJ . Erythrocyte vesiculation: a self-protective mechanism? Br. J Haematol 2008; 141: 549–556.
Bosman GJ, Cluitmans JC, Groenen YA, Werre JM, Willekens FL, Novotny VM . Susceptibility to hyperosmotic stress-induced phosphatidylserine exposure increases during red blood cell storage. Transfusion 2010; 51: 1072–1078.
Gevi F, D'Alessandro A, Rinalducci S, Zolla L . Alterations of red blood cell metabolome during cold liquid storage of erythrocyte concentrates in CPD-SAGM. J Proteomics 2012 doi:10.1016/j.jprot.2012.03.012.
Bennett-Guerrero E, Veldman TH, Doctor A, Telen MJ, TL Ortel, Reid TS et al. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci USA 2007; 104: 17063–17068.
Kuypers FA, de Jong K . The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol Biol (Noisy-le-grand) 2004; 50: 147–158.
Montes LR, Lopez DJ, Sot J, Bagatolli LA, Stonehouse MJ, Vasil ML et al. Ceramide-enriched membrane domains in red blood cells and the mechanism of sphingomyelinase-induced hot-cold hemolysis. Biochemistry 2008; 47: 11222–11230.
Gauthier E, Guo X, Mohandas N, An X . Phosphorylation-dependent perturbations of the 4.1R-associated multiprotein complex of the erythrocyte membrane. Biochemistry 2011; 50: 4561–4567.
An X, Guo X, Sum H, Morrow J, Gratzer W, Mohandas N . Phosphatidylserine binding sites in erythroid spectrin: location and implications for membrane stability. Biochemistry 2004; 43: 310–315.
Hagerstrand H, Kralj-Iglic V, Fosnaric M, Bobrowska-Hagerstrand M, Wrobel A, Mrowczynska L et al. Endovesicle formation and membrane perturbation induced by polyoxyethyleneglycolalkylethers in human erythrocytes. Biochim Biophys Acta 2004; 1665: 191–200.
Xiong Z, Cavaretta J, Qu L, Stolz DB, Triulzi D, Lee JS . Red blood cell microparticles show altered inflammatory chemokine binding and release ligand upon interaction with platelets. Transfusion 2011; 51: 610–621.
Kozuma Y, Sawahata Y, Takei Y, Chiba S, Ninomiya H . Procoagulant properties of microparticles released from red blood cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol 2011; 152: 631–639.
Hod EA, Zhang N, Sokol SA, Wojczyk BS, Francis RO, Ansaldi D et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood 2010; 115: 4284–4292.
Bosman GJ, Visser FE, de Man AJ, Bartholomeus IG, de Grip WJ . Erythrocyte membrane changes of individuals with Down's syndrome in various stages of Alzheimer-type dementia. Neurobiol Aging 1993; 14: 223–228.
Matte A, Low PS, Turrini F, Bertoldi M, Campanella ME, Spano D et al. Peroxiredoxin-2 expression is increased in beta-thalassemic mouse red cells but is displaced from the membrane as a marker of oxidative stress. Free Radic Biol Med 2010; 49: 457–466.
Dannenberger D, Suss R, Teuber K, Fuchs B, Nuernberg K, Schiller J . The intact muscle lipid composition of bulls: an investigation by MALDI-TOF MS and 31P NMR. Chem Phys Lipids 2010; 163: 157–164.
Fuchs B, Schober C, Richter G, Suss R, Schiller J . MALDI-TOF MS of phosphatidylethanolamines: different adducts cause different post source decay (PSD) fragment ion spectra. J Biochem Biophys Methods 2007; 70: 689–692.
Fuchs B, Suss R, Schiller J . An update of MALDI-TOF mass spectrometry in lipid research. Prog Lipid Res 2010; 49: 450–475.
Deplaine G, Safeukui I, Jeddi F, Lacoste F, Brousse V, Perrot S et al. The sensing of poorly deformable red blood cells by the human spleen can be mimicked in vitro. Blood 2011; 117: e88–e95.
Acknowledgements
We thank Mietske Wijers-Rouw from the Department of Cell Biology, RUNMC, NCMLS, for assisting with the electron microscopy. We are grateful to Lucas van Eijk from the Department of Intensive Care Medicine, RUNMC, for blood sampling of healthy volunteers. We also thank the people who have volunteered to donate blood for this study. This work was supported by the German Research Council (DFG Schi 476/12-1 and Fu 771/1-2).
Author contributions
SD, KW, WV, IJ, RB, and GB designed the research; SD, KW, WV, CT, JC, BF, and JS performed the experiments and analyzed the data; SD, KW, WV, JF, IJ, RB, and GB wrote and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Ciliberto
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Dinkla, S., Wessels, K., Verdurmen, W. et al. Functional consequences of sphingomyelinase-induced changes in erythrocyte membrane structure. Cell Death Dis 3, e410 (2012). https://doi.org/10.1038/cddis.2012.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.143
Keywords
This article is cited by
-
Increased acid sphingomyelinase levels in pediatric patients with obesity
Scientific Reports (2022)
-
Evaluation of eryptosis in patients with chronic kidney disease
International Urology and Nephrology (2022)
-
Validation and application of a protocol for the extraction and quantitative analysis of sphingomyelin in erythrocyte membranes of patients with non-alcoholic fatty liver disease
JPC – Journal of Planar Chromatography – Modern TLC (2021)
-
Blunted apoptosis of erythrocytes in mice deficient in the heterotrimeric G-protein subunit Gαi2
Scientific Reports (2016)
-
Eryptosis Indices as a Novel Predictive Parameter for Biocompatibility of Fe3O4 Magnetic Nanoparticles on Erythrocytes
Scientific Reports (2015)

{kind=link}
{kind=link}