
Abstract
The molecular determinants of Alzheimer's (AD) disease are still not completely known; however, in the past two decades, a large body of evidence has indicated that an important contributing factor for the disease is the development of an unbalanced homeostasis of two signaling cations: calcium (Ca2+) and zinc (Zn2+). Both ions serve a critical role in the physiological functioning of the central nervous system, but their brain deregulation promotes amyloid-β dysmetabolism as well as tau phosphorylation. AD is also characterized by an altered glutamatergic activation, and glutamate can promote both Ca2+ and Zn2+ dyshomeostasis. The two cations can operate synergistically to promote the generation of free radicals that further intracellular Ca2+ and Zn2+ rises and set the stage for a self-perpetuating harmful loop. These phenomena can be the initial steps in the pathogenic cascade leading to AD, therefore, therapeutic interventions aiming at preventing Ca2+ and Zn2+ dyshomeostasis may offer a great opportunity for disease-modifying strategies.
Similar content being viewed by others


Main
Although the majority of Alzheimer's disease (AD) cases, referred as sporadic AD (sAD), are idiopathic with no obvious implication of genetic factors, 5% of all the AD patients show a Mendelian pattern of inheritance (familial AD, fAD). In both sAD and fAD, the accumulation of the amyloid-β peptide (Aβ) and neurofibrillary tangles (NFTs) is a key determinant in AD pathogenesis, the most common form of dementia. Aβ, the main constituent of the amyloid plaques, is a 40–42 amino-acid peptide formed by the proteolytic cleavage of the membrane-bound amyloid precursor protein (APP) through the enzymatic activity of the β- and γ-secretase complexes. NFTs are intraneuronal aggregates composed of hyperphosphorylated tau (h-tau) protein, a peptide that is involved in stabilizing the microtubular structures of the cytoskeleton. In addition to Aβ dysmetabolism and the appearance of h-tau deposits, a growing body of evidence suggests that the deregulation of brain calcium (Ca2+) and zinc (Zn2+) homeostasis facilitates the development and progression of AD.1, 2, 3
Cytosolic Ca2+ levels ([Ca2+]i) are kept in a low range (∼100 nM) compared with the levels present in the extracellular space (2 mM) or what is found inside of intracellular stores (100–500 μM). These low [Ca2+]i are maintained by the activity of Ca2+-binding buffering proteins (e.g., calbindin and parvalbumin), whereas Ca2+ extrusion is operated by the Ca2+-ATPase pump, and the Na+/Ca2+ exchanger as well as by the sequestration of the cation in intracellular stores like the endoplasmic reticulum (ER) and mitochondria. Conversely, [Ca2+]i rises are the result of an influx across the plasma membrane via store-operated Ca2+ channels (SOCCs), voltage-gated Ca2+ channels (VGCCs), ionotropic glutamate receptors (N-Methyl-D-Aspartic acid receptors, NMDARs; α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate, AMPARs) or the release from intracellular stores. The ER is the largest intracellular store in neurons and loaded with Ca2+ at high concentrations (100–500 μM) through the unidirectional pumping of cytosolic Ca2+ into the ER lumen, a process operated by the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump. Ca2+ release from the ER occurs through the activation of two types of Ca2+ receptors: the inositol 1,4,5-trisphosphate (IP3R) and the ryanodine (RyR) receptors. Moreover, capacitative calcium entry (CCE), a process by which the depletion of intracellular Ca2+ stores causes the opening of plasma membrane Ca2+-permeable channels, has been described in neuronal subpopulations.
Zn2+ is the second most abundant metal in the body after iron (∼ 3 g). Much like [Ca2+]i, cytosolic Zn2+ ([Zn2+]i) is kept at low concentrations by the combined activity of Zn2+ transporters (ZnTs), Zn2+-importing proteins (ZIPs), and by the buffering action of proteins like the metallothioneins (MTs; reviewed in ref. 1). In the central nervous system (CNS), Zn2+ is selectively stored at high levels (∼1 mM) in presynaptic vesicles of some, but not all, glutamatergic neurons and co-released with the neurotransmitter.4 Thus, during intense synaptic activity, Zn2+ can accumulate in the synaptic cleft and enter postsynaptic spines sharing some entry routes that are also employed by Ca2+ (VGCCs, Ca2+-permeable AMPARs (Ca-ARs) and, with less efficacy, NMDARs1).
Although Ca2+ and Zn2+ are involved in a variety of CNS functions (neurotransmission, mitochondrial functioning, axonal transport, and a plethora of enzymatic activities), their dysfunctional homeostasis can be a potent trigger for Aβ oligomerization and NFT formation, as well as overproduction of reactive oxygen species (ROS),1, 3 all of which are factors critically involved in AD-related neurodegeneration. Furthermore, as both Ca2+ and Zn2+ modulate the physiology of synaptic transmission and plasticity, several lines of evidence now reveal that the deregulation of the two ions can help to explain the synaptic dysfunction and cognitive decline associated with AD.
Thus, given the importance of Ca2+ and Zn2+ homeostasis for brain functioning, strategies aimed at restoring the brain balance of these ions are emerging as promising avenues for the treatment of AD with several therapeutic approaches based on the so-called ‘metals hypothesis’ already showing encouraging results.5, 6
However, an important question that still needs to be addressed concerns the time frame in which such ionic dyshomeostasis occurs in AD. Does the ionic deregulation precede or follow the onset of the pathology? Is this a cause or a consequence of the disease?
In this review, we analyze the most recent findings indicating the genetic and environmental factors that promote the early establishment of Ca2+ and Zn2+ dyshomeostasis and show how the process can contribute to AD pathogenesis.
Ca2+ Dyshomeostasis, Synaptic Dysfunction, and AD
The idea that the perturbation of Ca2+ homeostasis can have a pivotal role in the cascading events leading to AD was introduced more than 20 years ago,7 and an impressive amount of evidence obtained from experiments on dissociated cells, brain slices, and, more recently and importantly, on live AD animal models is strongly supporting this hypothesis.8, 9
The most important evidence for the ‘Ca2+/AD hypothesis’ came from a large number of studies indicating that fAD cases are associated with mutations of three genes: APP, presenilin-1 (PS1), and presenilin-2 (PS2). Presenilins are transmembrane proteins synthesized in the ER that have been shown to greatly affect ER-Ca2+ dynamics through the activation of the SERCA pump,10 via IP3R and RyR opening11, 12 as well as by allowing passive Ca2+ leakage from the ER. Notably, all these homeostatic Ca2+ mechanisms are found to be profoundly deregulated by mutant presenilins both in vitro and in vivo9, 11, 13, 14 substantiating the idea that intraneuronal Ca2+ deregulation can be part of the pathogenic cascade leading to AD. Interestingly, a recent study suggests that PS2 can critically regulate the mitochondrial uptake of Ca2+ that is released from the ER, thereby promoting a potentially harmful cationic overload in the organelles.15 Presenilins have also been shown to diminish CCE in AD neurons and increase Ca2+ release from intracellular stores, thereby leading to diminished Ca2+ storage.16 However, another study has reported that AD neurons show impaired CCE along with augmented store-dependent Ca2+ release, as well as increased Ca2+ storage.17 Regardless of the mechanisms involved, it seems reasonable that a diminished CCE could, in some way, contribute to AD-related neurodegeneration. Besides their functions in Ca2+ homeostasis, PS1 and PS2 may have an additional role in AD, as they are part of the γ-secretase catalytic domain and presenilin mutations accelerate (although with different degrees of activity) the production of ‘aggregation-prone’ forms of Aβ1−42.18
Contrary to what has been demonstrated for presenilins, the role of APP in Ca2+ deregulation is still controversial. Whether or not, independently of Aβ production, APP can directly modulate Ca2+ homeostasis, is still a matter of debate. Evidence for a direct effects of APP on [Ca2+ ]i homeostasis is supported by studies performed on transgenic mice overexpressing wild-type APP19 or carrying fAD-linked APP mutations.20 These models show neurons undergoing increased [Ca2+]i levels at rest.20 However, it should not be overlooked that conflicting results also indicate the absence of a direct effect of fAD-linked APP mutations on [Ca2+]i levels and/or Ca2+ release from intracellular stores.21, 22
Factors linked to sAD are less clear; however, there is evidence to back the idea that some of them are associated with [Ca2+]i perturbation. For instance, the expression of the Apoɛ4 allele that is associated with sAD promotes a strong increase in [Ca2+]i levels by triggering Ca2+ influx in neurons.23 Another sAD risk factor is the presence of a polymorphism for the Ca2+ homeostasis modulator 1 (CALHM1) protein, a multipass transmembrane glycoprotein with a large Ca2+ conductance across the plasma membrane.24 The P86 L mutation of the peptide decreases CALHM1-mediated Ca2+ currents, thereby favoring an increase in Aβ production, which supports the idea that alterations of specific Ca2+-pools, rather than a global increase of [Ca2+]i, may be critical contributing factors for AD development and progression.
But how do [Ca2+]i alterations influence the propagation of the AD-related pathology? The introduction of transgenic animal models as experimental tools has helped to elucidate, at least in part, the complex interactions between AD-linked mutations, ionic deregulation, and the development of Aβ and tau pathology. For example, transgenic mice expressing human mutant APP, generate when aging Aβ plaques in the hippocampus and cortex, ultimately mimicking the AD-like pathology found in patients.
APP processing involves the activation of several Ca2+-dependent proteins and therefore it is likely that increased or decreased Ca2+ levels may have aberrant effects on APP cleavage. For instance, many studies have shown that the mobilization of different Ca2+ pools leads to Aβ production.25 To date, the global effect of [Ca2+]i rises on Aβ levels is still unclear as some evidence shows that increased [Ca2+]i rises25, 26, 27 promote the production of the non-amyloidogenic αAPP fragment, but other studies have indicated that [Ca2+]i increases, acting on either the extracellular milieu or intracellular stores, induce a shift toward the amyloidogenic pathway.28 A critical revision of the most recent studies seems to support the idea that the homeostasis of the ER-Ca2+ pool is crucial in controlling the fate of APP processing.9, 10, 11 In fact, ER overfilling appears to be the first clinical sign of the pathogenic effect of mutant presenilins, a phenomenon caused by either SERCA overactivation or by the loss of Ca2+ leakage, a process ultimately resulting in higher Ca2+ efflux through RyRs and IP3Rs. All of these mechanisms have been shown to lead to increased Aβ secretion.9, 10, 11
It should be noted that there is consensus by which AD can start as a de-afferentation process, thereby isolating neurons and promoting synaptic dysfunction. Thus, given the importance of Ca2+ signaling in the modulation of synaptic efficacy and integrity, many investigators have started to explore the consequences of disrupted intracellular Ca2+ signaling in the spines and dendrites of neurons of transgenic AD models. In that respect, ER-Ca2+ signaling appears, again, to have a key role in modulating synaptic transmission by promoting presynaptic transmitter release and paired pulse facilitation. Lending support to this idea, it has been shown that CA1 hippocampal 3 × Tg-AD neurons in slices present an aberrant, yet seemingly functional, interplay between ER-Ca2+ signaling and synaptic transmission, an event that appears long before the onset of AD-like pathology.29
A large body of evidence also demonstrates the deleterious effects of Aβ in driving further Ca2+ deregulation. Many studies have shown that Aβ directly interferes with Ca2+ homeostasis and disrupts the activity of ionotropic glutamate receptors, the P/Q or L-type VGCCs,30, 31 ER-Ca2+ stores,32 and CCE,33 thereby producing increased [Ca2+]i levels.20 Recent in vivo experiments employing multiphoton imaging indicate that Aβ is promoting Ca2+ deregulation in transgenic AD neurons, and neurites positioned in the proximity of Aβ plaques,8 a phenomenon that leads to the downstream activation of the Ca2+-dependent protein phosphatase, calcineurin.34 This phenomenon is a key component of the pathogenic cascade as inhibiting calcineurin with K506 ameliorates dendritic spine density in these animals.35 Furthermore, several studies indicate that Aβ oligomers can directly induce Ca2+ entry through the plasma membrane by promoting the formation of Ca2+ permeable channels.36, 37
Ionic Deregulation Implication for Synaptic Plasticity
A crucial issue that deserves particular attention is the link between Aβ-driven Ca2+ deregulation and synaptic efficacy (long-term potentiation (LTP) and long-term depression (LTD)). Synaptic loss and dysfunction are the most prominent pathological features of AD and these processes best correlate with the development of AD-related cognitive decline.38 In vitro and in vivo studies have shown that high levels of oligomeric Aβ can negatively interfere with glutamatergic synaptic transmission and plasticity.39, 40, 41 Several studies have investigated the pathogenic interactions between altered synaptic Ca2+ signaling and the aggregation state of Aβ, supporting the idea that Ca2+ and Aβ may interact in several different ways to promote synaptic dysfunction.2
In the past years, the complex interplay between Ca2+, APP, and Aβ in the modulation of synaptic transmission and plasticity has been the subject of a great number of studies. Through these investigations, it has become clear that Aβ can be secreted in the extracellular space during neuronal activity40, 42 and that pathologically elevated synaptic Aβ concentrations can alter transmission. Physiological levels of endogenous Aβ may act presynaptically as a positive regulator to enhance the release probability of synaptic vesicles of glutamatergic terminals,43 thereby increasing the glutamatergic transmission. Conversely, pathologically elevated Aβ concentrations may negatively affect plasticity by inducing a deficit of NMDAR-mediated excitatory post-synaptic currents as well as a shift toward NMDAR-dependent induction of LTD and synaptic loss.44, 45, 46, 47, 48 The process is set by Aβ promoting NMDAR endocytosis and, in this view, Ca2+ (through calcineurin activation by Aβ) may be considered the intracellular trigger for Aβ–driven synaptic dysfunction. Supporting this hypothesis, a recent study has proposed an intriguing mechanism linking Ca2+ deregulation, Aβ dysmetabolism, and synaptic dysfunction.45
According to this study, Ca2+ influx through synaptic NMDAR activates at least two pathways that regulate spine density and postsynaptic responses.45 On one hand, high [Ca2+]i levels obtained upon tetanic or suprathreshold synaptic stimulation induce LTP by activating the Ca2+/calmodulin-dependent protein kinase II (CAMKII)-dependent pathway. On the other hand, low levels of [Ca2+]i, obtained during low-frequency subthreshold stimulation can induce LTD by activating a calcineurin-dependent pathway (reviewed in ref. 49). Further evidence indicates effects of Ca2+ and Aβ on synaptic efficacy and shows that NMDAR-mediated Ca2+ influx into active spines is reduced by soluble Aβ oligomers, thereby causing a decrease in spine density via a NMDAR/calcineurin/cofilin-dependent process that leads to LTD.50 The overall scenario is however more complex as other studies indicate that Aβ oligomers bind to NMDARs and promote excitotoxic Ca2+ influx by enhancing NMDAR activation.51, 52
Further studies have dissected the link between Ca2+, AMPAR activation, and Aβ-dependent synaptic dysfunction.44 Acute application of Aβ in the dentate gyrus of rat hippocampal slices has been shown to block the activity-dependent autophosphorylation of CaMKII, thereby leading to reduced phosphorylation and functioning of the GluA1 subunit of AMPARs.53 Another study has shown that soluble Aβ oligomers selectively cause the loss of surface AMPARs.44 The study suggests that this detrimental process is driven by Aβ binding to Ca-ARs at the dendritic surface and by the subsequent activation of calcineurin, an event that leads to the internalization of Aβ and AMPARs.44
A growing body of evidence also supports the idea that AD-related neuronal damage may result not only from the actions of synaptically released Aβ, but also derives from the intraneuronal accumulation of the peptide. It is known that elevated intraneuronal Aβ immunoreactivity precedes the appearance of plaques and better correlates with the onset of the synaptic and behavioral abnormalities shown in transgenic AD models.54 Moreover, in neuronal cultures, the depolarization and subsequent Ca2+-mediated transient phosphorylation of both tau and APP induce the accumulation of intraneuronal Aβ.55 However, decreased synaptic activity has also been shown to be detrimental by promoting an increase in intracellular Aβ that is followed by synaptic damage.56, 57
As far as tau-dependent pathology, in recent years evidence strongly substantiates the idea that Ca2+ deregulation is also involved in NFTs formation. This phenomenon is particularly relevant given the renewed interest in the pathogenic action of tau pathology in promoting neurodegeneration and cognitive decline in AD.58
It should be remembered that the anatomical distribution of NFTs follows a typical spreading pathway that, compared with the distribution of Aβ pathology, correlates in a better way with the progression of AD symptoms. Recent evidence is in fact starting to question the exact sequence of events previously described in the ‘Aβ amyloid cascade hypothesis’ and suggests that, in fact, Aβ-mediated toxicity is largely modulated (and not in a secondary way) by tau-dependent processes.59, 60, 61 For instance, a recent study has shown that some hippocampal cultured neurons exposed to Aβ oligomers relocate tau from axons to cell bodies and dendrites.61 Interestingly, this Aβ-driven dendritic missorting of tau appears to be the key to promote downstream pathogenic events like dendritic Ca2+ rises, changes in tau phosphorylation, loss of spines and mitochondria, and relocation of other cytoskeletal elements, as well as changes in the activity of crucial kinases.61 A similar change in tau distribution and phosphorylation has been shown in neurons exposed to oxidative or excitotoxic stress, suggesting a common signaling cascade that culminates in regional tau pathology and eventually in the breakdown of neuronal architecture.61 Furthermore, recent evidence has linked tau dysmetabolism to Aβ-dependent excitotoxic damage.60, 62 According to this model, tau is moving to dendrites, promoting the activation of a Src kinase (Fyn), and leading to the phosphorylation of the GluN2B NMDAR subunit. This process facilitates the interaction of NMDARs with the scaffolding protein PSD-95, increasing the stability of NMDARs within the PSD, thereby coupling the receptors to Aβ-dependent excitotoxic signaling.
The full extent by which tau dyshomeostasis mediates synaptic transmission, neuronal degeneration, or both, is actually matter of an exciting current debate58, 63 and new intriguing evidence is emerging. For instance, genetic deletion of tau completely prevents the deleterious effects on LTP elicited by the exposure to either human or rodent Aβ1–42, thereby suggesting that the protein is instrumental for the development of the synaptotoxic effects of Aβ.64 Moreover, tau-induced defects in synaptic plasticity are reversible in transgenic conditional mice after switching off the toxic tau mutant.63 Data linking Ca2+ dyshomeostasis with the neurotoxic effects of a tau fragment of 17 kD are associated with calpain activation.65 Interestingly, another study has shown that [Ca2+]i rises evoked by glutamate or thapsigargin (a SERCA inhibitor) are also able to promote the same cleavage of this ‘17 kD’ fragment, thereby confirming the Ca2+ dependency of the phenomenon.66
Further studies are lending support to the idea that AD-related alterations have an important role in altering glutamate neurotransmission (and indeed, AD patients are now treated with memantine, a low-affinity NMDAR blocker). As described above, a major source for [Ca2+]i rises results from the overactivation of glutamatergic receptors, a phenomenon that occurs in experimental models of AD and triggers robust oxidative stress.20 It is worth noticing that Aβ itself can increase glutamatergic neurotransmission and render neurons more vulnerable to excitotoxicity by interfering with glutamate re-uptake.67
As both Aβ and h-tau further facilitate Ca2+ dyshomeostasis, the process appears to take the shape of an injurious cycle that leads to AD-related synaptic dysfunction and neuronal death.
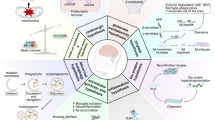
Thus, glutamate and Aβ can operate synergistically to promote oxidative stress and set the stage for a self-perpetuating harmful loop. Oxidative stress can also enhance tau phosphorylation and tau-dependent pathology that further exacerbates Aβ-driven pathology68 (Figure 1). It should also be noted that, suggesting a feed forward process, recent data indicate that injection of oligomers, obtained from the cortex of AD patients, triggers tau hyperphosphorylation, disrupts the microtubule architecture, and sets in motion neuritic dystrophy.69 Finally, NMDAR activation can also promote the intraneuronal release of Zn2+, thereby suggesting a pathogenic link between the dyshomeostasis of the two cations70 (Figure 2).

Aβ, oxidative stress, tau, glutamate receptor overactivation, Ca2+, and Zn2+ dyshomeostasis may synergistically promote AD-related synaptic and neuronal loss. The sequestration of Zn2+ by the amyloid contained in extracellular plaques (1) may remove the cation from the synaptic cleft and release the Zn2+-dependent NMDAR blockade (1). Aβ also directly activates Ca-ARs (2). Both phenomena promote Ca2+ overload, increased superoxide generation from mitochondria (3), as well as nitric oxide (NO) production from Ca2+-dependent activation of NO synthase (NOS) (4). Reactive oxygen and nitrosative species (ROS and RNS) then mobilize Zn2+ from MTs leading to toxic Zn2+ concentrations (5) that further impair mitochondrial function, promote more ROS generation (6), and release pro-apoptotic factors (7). ROS-driven Zn2+ mobilization may also facilitate intra-neuronal Aβ aggregation (8). Aβ may amplify this vicious loop by increasing the generation of ROS that when extracellularly released reduce glutamate reuptake (9) and overactivate NMDARs and Ca-ARs. Mutant PS1 and APP enhance Ca2+ dyshomeostasis by altering the ER-mitochondrial network (10). NMDAR-mediated [Ca2+]i rises and oxidative stress may also accelerate tau hyperphosphorylation (11) and recent evidence indicates that oligomers from AD patients are directly promoting tau hyperphosphorylation as well (12). Zn2+ sequestration also impairs BDNF signaling (13) (by blocking TrkB and or MMPs), and also reduces the modulating effects exerted by the activation of its own ZnR (14). All these processes, set in motion at the level of synaptic spines, may be the primum movens of synaptic dysfunction, neuronal deafferentation, and death

Glutamate receptor activation: a link between Ca2+ and Zn2+ dyshomeostasis. NMDAR activation promotes a Ca2+ dependent release of intraneuronal Zn2+. (a) Neuronal cultures, loaded with the Zn2+-sensitive (but Ca2+-insensitive) fluorescent probe, FluoZin-3, are shown, before, during, and after a 10 min exposure to NMDA (50 μM), in a Ca2+ containing (black) or nominally Ca2+ free (red) buffer. (b) Pseudocolor images of FluoZin-3 fluorescent changes upon the experimental condition described in A. Note that the addition of the Zn2+ chelator compound, TPEN, completely quenches the FluoZin-3 fluorescence changes, demonstrating the Zn2+ dependency of the signal (modified from ref 70)
Zn2+ Deregulation, Synaptic Functioning, and AD
Like Ca2+, Zn2+ is involved in a wide array of physiological functions1 and, as with Ca2+, the deregulation of intracellular Zn2+ ([Zn2+]i) has an important role in AD.
The mammalian brain contains a pool of loosely bound Zn2+ also defined as ‘chelatable Zn2+’.1 Zn2+-containing neurons form a complex and elaborate associational network that interconnects most of the cerebral cortices and limbic structures and Zn2+-containing axon terminals are particularly abundant in the neocortex and hippocampus.
Zn2+ is often localized within synaptic vesicles of a subset of glutamatergic axon terminals, although there is also evidence indicating enrichment of Zn2+ at selected inhibitory γ-Aminobutyric Acid (GABA) terminals in the cerebellum and spinal cord.71, 72 In the subpopulation of glutamatergic neurons, Zn2+ is transported into small, clear, and round presynaptic vesicles by the neuronal-specific Zn2+ transporter, ZnT3, throughout a process that stores the cation inside vesicles together with glutamate.1 Thus, Zn2+ and glutamate are co-released in the synaptic cleft during neuronal activity. Previous studies showed that sustained stimulation of hippocampal mossy fibers (MFs), a brain structure that shows the strongest Zn2+ amount in the brain, produces a great increase in synaptic Zn2+ levels and more recent evidence has demonstrated that such synaptic Zn2+ is a principal factor in modulating neurotransmission.1
Both excitatory ionotropic glutamate receptors (iGluRs) and inhibitory ionotropic receptors (GABAA) are affected by Zn2+.73 The best characterized synaptic Zn2+ target is represented by the NMDAR. Zn2+ modulates NMDAR properties through a dual mechanism: low micromolar concentrations selectively inhibit NMDAR-mediated currents through a voltage-independent, non-competitive (allosteric) inhibition whereas high Zn2+ concentrations (more than 20 μM) are responsible of a voltage-dependent inhibition of the receptor-mediated currents.74, 75, 76 The N-Terminal-Domain (NTD) of the NMDAR GluN2A subunit binds Zn2+ with high (nanomolar) affinity and this high sensitivity to Zn2+ has important consequences. The most relevant is that synaptic Zn2+ can exert its modulatory effects not only during fast phasic synaptic release but also in a tonic mode. In other words, under normal resting conditions, the GluN2A-specific Zn2+ binding site can be partially occupied by the cation, thereby inhibiting the receptor.75 Interestingly, Zn2+ inhibition can be also maximized by the tyrosine kinase src-mediated phosphorylation of the receptor.77 AMPARs are also sensitive to extracellular Zn2+,78 however, the phenomenon appears to require higher (micromolar) Zn2+ levels.
In addition to ionotropic receptors, synaptically released Zn2+ modulates the activity of key synaptic proteins. For instance, Zn2+ can interfere with the biochemical activity of proteins like glutamate transporters and matrix metalloproteinases (MMPs) in physiological and pathological conditions (Figure 3).79 Furthermore, a recent study indicates a novel mechanism by which Zn2+ can modulate synaptic functions as the cation is able to activate mitogen-activated protein kinase (MAPK) and CaMKII by acting on a putative Zn2+-sensing receptor (ZnR).80

Zn2+ supplementation is beneficial in animal model of AD. (A) Zn2+ supplementation prevents the development of hippocampus-dependent memory deficits in 3 × Tg-AD mice. 3 × Tg-AD mice were treated with either water containing 30 p.p.m. of ZnSO4 or tap water for 11–13 months and tested for the spatial memory version of the MWM. Mice were tested when the platform was removed 1.5 h (to investigate short-term memory) and 24 h (to investigate long-term memory) after the last training trial. Compared with untreated 3 × Tg-AD mice, Zn2+-fed 3 × Tg-AD mice did not show any significant difference in their short-term memory performance, but exhibited a marked recovery in their long-term memory as indicated by the decreased time (latency) they employed to reach the point where the platform used to be (Error bars indicate mean values±S.E.M.; *P<0.05). (B) Zn2+ supplementation reduces both Aβ and tau pathology in the hippocampus of 3 × Tg-AD mice. Immunohistochemistry shows deposits of intraneuronal Aβ (a–d) and h-tau (f–j) in brain slices from treated and untreated 3 × Tg-AD mice (left column: 20 × magnification; right column: 40 × magnification). Compared with untreated mice (a, b), treated 3 × Tg-AD mice showed a significant decrease of intraneuronal Aβ deposits in the hippocampus of (c, d). (b, d) × 40 magnification of the hippocampal CA1 area as shown in the rectangle. (e) Quantification of Aβ load as shown in a and c. Compared with untreated mice (f, g), Zn2+-fed 3 × Tg-AD mice showed a strong decrease of intraneuronal h-tau immunoreactivity in the hippocampus of (h, i). (g, i) × 40 magnification of the CA1 area as shown in the rectangle. (j) Quantification of h-tau levels from f and h. Error bars indicate mean values±S.E.M.; *P<0.05. (C) Zn2+ supplementation promotes MMPs activation in 3 × Tg-AD mice. (a) Gelatine zimography indicates that Zn2+ feeding induced a significant increase of MMP-2 and MMP-9 activation in 3 × Tg-AD mice brains. (D). Zn2+ supplementation increased brain BDNF levels. BNDF immunoblotting reveals that Zn2+-treated 3 × Tg-AD mice showed a fourfold increase in BDNF levels when compared with untreated animals. *P<0.05; **P<0.01. Error bars indicate mean values±S.E.M. (modified from ref. 79)
A recent study has also shown that Zn2+ transactivates the tropomyosin-related kinase B (TrkB) receptor (the receptor responsive to the brain-derived neurotrophic factor (BDNF)) and activates BDNF signaling in a neurotrophin-independent manner.81 Furthermore, Zn2+ also affects BDNF signaling by promoting the maturation of pro-BDNF to BDNF throughout the activation of MMPs82 and this phenomenon has been recently validated in animal AD model (Figure 3).79 It is also important to underline that the non-amyloidogenic APP processing is mediated by membrane-anchored Zn2+-dependent metalloproteinases like the ones belonging to the ADAM family [a disintegrin and metalloproteinases,83] a phenomenon that suggests that [Zn2+]i might also have a role in regulating the balance between amyloidogenic and non-amyloidogenic pathways.
Finally, intracellular Zn2+ can interfere with neuronal excitability by modulating the activity of the neuronal K+ transporter, KCCN, a key controller of the neuronal resting membrane voltage potential.84 Thus, the specific localization of Zn2+ at certain glutamatergic synapses, together with its multiple synaptic targets, makes the cation a very attractive candidate as an endogenous modulator of synaptic transmission and plasticity; however, deregulated Zn2+ homeostasis also appears to be a strong promoter of AD development (Figure 4).

Alterations of the ‘zinc set point’, implications for AD. The maintenance of brain Zn2+ homeostasis is crucial for neuronal functioning. Perturbations of this equilibrium may be a contributing factor in AD development and progression. Excessive Zn2+ facilitates Aβ oligomerization as well as overproduction of reactive oxygen species (ROS), thereby promoting synaptic dysfunction and neuronal death. However, Zn2+ deficiency may also be deleterious as decreased bioavailability of the cation leads to reduced activation of neuroprotective BDNF signaling. Zn2+ deficiency in fact decreases the maturation of pro-BDNF to BDNF and reduces the transactivation of the BNDF receptor, TrkB. Moreover, decreased levels of brain Zn2+ can further downregulate the expression and/or the activity of neuroprotective Zn2+-dependent proteins that are involved in Aβ clearance (e.g., MMPs) or free radical scavenging (e.g., MTs). Finally, synaptic Zn2+deficiency also leads to neurotoxic activation of NMDARs
Indeed, there are no doubts that a complex alteration of transportation and distribution occurs in the AD brain and that alterations of synaptic Zn2+ are key factors in promoting AD-like pathology. Lending support to this idea, a study has shown that aging is associated with the development of decreased brain levels of ZnT3, a phenomenon that is enhanced in AD brains.85 ZnT3 downregulation leads to a deficit in synaptic Zn2+ that is associated with a strong downregulation of synaptic proteins that are important for memory and learning processes.85 Interestingly, another study has indicated that ZnT3-KO mice show a reduced activity-dependent induction of Erk1/2 signaling at the MF presynaptic terminals. Such diminished Erk1/2 activity derives from the lack of inhibition of the Zn2+-sensitive MAPK tyrosine phosphatase, thereby suggesting that an increased synaptic release of the cation from MFs on learning might enhance the inhibition of tyrosine phosphatases leading to further presynaptic Erk activation. Consistent with this prediction, ZnT3-KO animals show cognitive deficits.86
A deficit of synaptic Zn2+ has also been associated with decreased brain levels of BDNF and other neurotrophins as well as with the development of severe cognitive decline.85 Moreover, data obtained from acute murine hippocampal slices indicate that synaptic Zn2+ facilitates the binding of Aβ oligomers to the GluN2B subunit of the NMDAR in MF terminals and interferes with glutamatergic transmission.87 Furthermore, notable studies by Lovell and colleagues have shown alterations in the expression of the Zn2+ transporters, ZnT4, ZnT1, and ZnT6 in the brain of preclinical and early-stage AD patients, suggesting a concurrent causative role of early Zn2+-dyshomeostasis in triggering AD-related pathogenic events.88, 89
A strong link between Zn2+ and Aβ in AD is also confirmed by several observations: (1) amyloid plaques are highly enriched in Zn2+ (ref 90); (2) genetic ablation of synaptic Zn2+ prevents Aβ deposition;91 (3) trace concentrations of Zn2+, as well as Cu2+ or Fe3+ are sufficient to induce Aβ nucleation and eventually the fibrilization of the peptide;92, 93 (4) Zn2+ enhances Aβ aggregation and precipitation during AD progression whereas Zn2+ chelators induce a rapid resolubilization of Aβ deposits in post-mortem AD brain samples.94
Zn2+ also critically modulates amyloid metabolism.1 Indeed, both APP and Aβ specifically bind Zn2+, with APP having a binding site for zinc (KD=750 nM) in the cysteine-rich region of the APP ectodomain,95 and Aβ exhibiting both a high-affinity binding site (KD=107 nM; with a 1 : 1 (Zn2+/Aβ) stoichiometry) as well as a lower-affinity binding site (KD=5.2 μM; with a 2 : 1 stoichiometry92). Subtle changes in Zn2+ and Aβ brain levels appear therefore crucial in determining a shift from a physiology to pathology (Figure 4).
As mentioned, the brain has one of the highest content of Zn2+ with an overall concentration of ∼150 μM. (ref. 1) Some forebrain neurons store up to 200–600 μM of Zn2+ in synaptic vesicles and release ∼1–10% of this amount as freely exchangeable Zn2+. However, in a healthy brain, the exchangeable amount of Zn2+ in the extracellular fluid is approximately 1–10 nM whereas the free cytosolic [Zn2+]i levels are in the sub-nanomolar range. Aβ is a physiological component of the CSF and normally produced and secreted during cell metabolism at nanomolar concentrations and then quickly degraded (reviewed in ref. 96).
Very little is known on the exact dynamics of the Aβ intracellular pool. Indeed, although an increasing body of evidence indicates that Aβ accumulates intracellularly, it is still not clear whether this accumulation is the result of a failure of Aβ secretion, or whether secreted Aβ is taken back by the cell to build up intracellular pools.96 While under physiological conditions (especially if one considers the Aβ KD for the higher affinity binding site), [Zn2+]i levels are likely too low to promote significant complex formation with Aβ. However, [Zn2+]i release occurring under oxidative stress might promote sufficient cation levels to initiate Aβ oligomerization. Such ROS-dependent intraneuronal [Zn2+]i rises have been found to be particularly high in AD neurons expressing mutant APP, PS1 and h-tau.97
To date, a precise understanding of how Aβ forms complexes with Zn2+ and/or other metal ions is still largely unknown. Even though monomeric Aβ has been shown to form a complex with Zn2+, large oligomeric assemblies seem to be preferentially precipitated upon coordination with the cation.98 The N-terminal region of different Aβ oligomeric populations can access a range of metal-coordination structures,98 thereby leading to an extensive polymorphism.99 It is important to underline that these polymorphic species include antiparallel β-sheet conformations100 increasingly recognized as ‘signature’ structures of toxic Aβ oligomers.101 The neurotoxic fate of these aggregates varies. Depending on their relative stabilities and energy landscapes, some intermediates can be selectively stabilized or precipitated into insoluble, inert fibrils by Zn2+ (ref. 100,102). The scenario changes when they get conjugated with metals. Indeed, high [Zn2+]i levels can promote Aβ-induced toxicity both in vitro (at 50 μM)103 and in vivo (at 1 mM),104 when considering equimolar Aβ/Zn2+ concentrations. These findings suggest that high [Zn2+] rises may preferentially induce a stabilization of small, soluble, toxic intermediates that remain for a longer time in solution without necessarily evolving into fibrils.100 However, a neuroprotective effect can also be seen at lower Zn2+concentrations ((5–0.5 μM; in the case of Aβ/Zn2+ molar ratios of 1 : 0.1 and 1 : 0.01102, 103). In contrast to high Zn2+ levels, low Zn2+ concentrations can selectively destabilize larger soluble intermediates by lowering their kinetic barriers toward the formation of precipitates.102 This concentration-dependent activity of Zn2+ in the modulation of Aβ-associated neurotoxicity has been considered a paradox.105
Besides its activity on Aβ, Zn2+ also promotes tau aggregation106 and it should be noted that NFT-bearing neurons contain abnormally high (millimolar) levels of Zn2+.107 Similarly to what is seen with the Aβ-Zn2+ complex, Zn2+ also binds to single tau monomers with a moderate (micromolar) affinity (KD=3.8 μM). Interestingly, under reducing conditions, tau fibrilization depends on [Zn2+] levels as low Zn2+ (10 μM) accelerates the process (with a tau/Zn2+ molar ratio of 1 : 1) whereas higher levels (100 μM) have opposite effects (with a tau/Zn2+ molar ratio of 0.1 : 1106). Finally, Zn2+ also exerts a bimodal effect on tau phosphorylation, with high Zn2+ levels inducing tau hyperphosphorylation through the activation of glycogen synthase kinase 3β and low Zn2+ concentrations promoting tau dephosphorylation.79, 108
Ca2+ and Zn2+ Dyshomeostasis: a Synergy that Produces a Potentially Self-perpetuating Pathogenic Loop
In the complex scenario of AD, all the factors previously mentioned, Aβ, oxidative stress, tau, glutamate receptor overactivation, Ca2+, and Zn2+ dyshomeostasis may act in concert to promote synaptic loss and, ultimately, neuronal loss (Figure 1). Zn2+ sequestration by Aβ in extracellular amyloid plaques promotes an over-activation of NMDARs, whereas Aβ directly activates Ca-ARs and the two phenomena lead to Ca2+ overload, increased superoxide generation from NADPH oxidase and mitochondria as well as nitric oxide (NO) production from Ca2+-dependent activation of NO synthase (NOS). Ca2+-dependent production of reactive oxygen and nitrosative species (ROS and RNS) then mobilize Zn2+ from MTs leading to toxic Zn2+ concentrations. These Zn2+ rises impair mitochondrial function, promote further ROS generation,109 and release pro-apoptotic factors. ROS-driven Zn2+ mobilization may then promote intraneuronal Aβ aggregation. Aβ may amplify the damaging results of this vicious cycle by increasing oxidative stress, reducing glutamate reuptake and inducing the activation of NMDARs and Ca-ARs. Furthermore, glutamate-driven mitochondrial Ca2+ overload mobilizes Zn2+ from these organelles,70 thereby increasing toxic cytosolic Zn2+ levels. PS1 and APP mutations may enhance these pathways by contributing to Ca2+ dyshomeostasis via increased Ca2+ release from the ER-mitochondrial network.
[Ca2+ ]i rises and oxidative stress also enhance neuronal damage by interfering with tau hyperphosphorylation.61, 110 In that respect, NMDAR overactivation, Ca2+ overload, and the resultant increased superoxide generation may all favor and increase tau hyperphosphorylation.
All these processes, set in motion at the level of synaptic spines, may be the primum movens of synaptic dysfunction, neuronal deafferentation, and death.
Ionic Dyshomeostasis as Potential Therapeutic Target
Consistent with the idea that AD may involve a chronic deregulation in the homeostasis of Ca2+and Zn2+, several therapeutic strategies have aimed at restoring physiological cationic signaling in an effort to counteract the development and progression of the disease. For example, recent evidence points to a possible therapeutic role for Ca2+ chelation, as the cation may have an important role in stabilizing the γ-secretase complex, as confirmed by recent observations showing that EGTA and EDTA reduce its activity.111
Furthermore, targeting NMDAR-mediated Ca2+ influx has produced a promising anti-AD drug. Memantine, an uncompetitive and moderate-affinity NMDAR antagonist, protects neurons against Aβ-related toxicity,112 reduces AD-like pathology in transgenic mice,113 and appears to be an effective, safe, and well-tolerated treatment for moderate-to-severe AD.114 Great attention has also been paid toward the activity of antagonists of a specific NMDAR subunit. Ifenprodil and Ro 25–6981, two compounds that selectively target NMDAR containing the GluN2B subunit protect neurons from Aβ-induced synaptic plasticity impairment.115 Furthermore, EVT-101 (Evotec AG, Hamburg, Germany; http://www.evotec.com/), a newly developed GluN2B antagonist, has been shown to improve the cognitive performance of AD patients and has less side effects when compared with memantine.
In the past few years, efforts have also been made to address AD-related Zn2+ dyshomeostasis. To date, the most promising class of drugs developed for counteracting Zn2+ deregulation are the so called metal-protein attenuation compounds (MPACs). MPACs aim at removing Zn2+ from Aβ and relocate the cation to sites where it can be beneficial, like for instance the TrkB receptor or the MMPs, thereby increasing BDNF signaling. Among the MPACs, the lipophilic quinoline derivative, PBT2, has shown the most promising results by serving as metal exchanger and ionophore. In AD murine models, PBT2 restores cognition, strongly reduces interstitial Aβ, restores spine density, and preserves the maintenance of crucial synaptic proteins.116, 117 In AD patients, PBT2 reduces CSF Aβ levels and promotes cognitive improvement.5, 6 Interestingly, a dual, Ca2+ and Zn2+, modulator compound, DP-109, is also effective in reducing Aβ load and cognitive deficits in AD mice.118 The effect of DP-109 is most likely due to removal of Zn2+ from amyloid plaques as well as its capability to restore intraneuronal Ca2+ homeostasis; however, the full spectrum of its actions is still largely unknown.
Finally, the issue of handling AD-dependent Zn2+ deregulation also poses the question of when and how to intervene. While it is now clear that, in a symptomatic phase of the disease, therapeutic strategies aimed at restoring Zn2+ homeostasis should favor the de-sequestration of Zn2+ from plaques and its relocation from injurious to beneficial sites, early stage intervention may actually take advantage by the daily supplementation of small quantities of Zn2+. In that respect, a recent study in a transgenic AD model, indicates that restoring brain Zn2+ levels with dietary supplementation in a pre-symptomatic stage of the disease can effectively counteract the development of hippocampus-dependent cognitive deficits.79 The beneficial effects of Zn2+ supplementation appear to be linked to the action of the cation in increasing BDNF levels and by its activity in counteracting age-dependent mitochondrial dysfunction79 (Figure 3).
In summary, a better understanding of the complex network linking the AD-dependent deregulation of these two cations poses great challenges but also offers the potential for the development of effective disease-modifying drugs.
Abbreviations
- AD:
-
Alzheimer’s Disease
- Ca2+:
-
calcium
- Zn2+:
-
zinc
- sAD:
-
sporadic AD
- fAD:
-
familial AD
- Aβ:
-
amyloid-β peptide
- NFTs:
-
neurofibrillary tangles
- APP:
-
amyloid precursor protein
- h-tau:
-
hyperphosphorylated tau
- [Ca2+]i:
-
cytosolic Ca2+ concentration
- [Zn2+]i:
-
cytosolic Zn2+ concentration
- ER:
-
endoplasmic reticulum
- VGCCs:
-
voltage-gated Ca2+ channels
- NMDAR:
-
N-Methyl-D-aspartic acid receptor
- AMPAR:
-
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor
- Ca2+:
-
permeable AMPARs (Ca-ARs)
- SERCA:
-
sarco/endoplasmic reticulum Ca2+-ATPase
- IP3R:
-
inositol 1,4,5-trisphosphate receptor
- RyR:
-
ryanodine receptor
- ZnTs:
-
Zn2+ transporters
- ZIPs:
-
Zn2+-importing proteins
- MTs:
-
metallothioneins
- CNS:
-
central nervous system
- iGluRs:
-
Ionotropic glutamate receptors
- ROS:
-
reactive oxygen species
- PS1:
-
presenilin-1
- PS2:
-
presenilin-2
- CCE:
-
capacitative Ca2+ entry
- CALHM1:
-
Ca2+ homeostasis modulator 1
- LTP:
-
long-term potentiation
- LTD:
-
long-term depression
- CAMKII:
-
Ca2+/calmodulin-dependent protein kinase II
- GluR:
-
glutamate receptor
- MFs:
-
mossy fibers
- GABA:
-
γ-Aminobutyric acid
- MMPs:
-
matrix metalloproteinases
- MAPK:
-
mitogen-activated protein kinase
- ZnR:
-
Zn2+-sensing receptor
- TrkB:
-
tropomyosin-related kinase B
- BDNF:
-
brain-derived neurotrophic factor
- ∣MPAC:
-
metal-protein attenuation compounds
References
Sensi SL, Paoletti P, Bush AI, Sekler I . Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 2009; 10: 780–791.
Green KN, LaFerla FM . Linking calcium to Abeta and Alzheimer's disease. Neuron 2008; 59: 190–194.
Mattson MP . ER calcium and Alzheimer's disease: in a state of flux. Sci Signal 2010; 3: pe10.
Qian J, Noebels JL . Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol 2005; 566 (Pt 3): 747–758.
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 2008; 7: 779–786.
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J et al. PBT2 rapidly improves cognition in Alzheimer's Disease: additional phase II analyses. J Alzheimers Dis 2010; 20: 509–516.
Khachaturian ZS . Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann NY Acad Sci 1989; 568: 1–4.
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ . Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008; 59: 214–225.
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 2006; 126: 981–993.
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Gen Physiol 2008; 132: i1.
Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J et al. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008; 58: 871–883.
Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P . The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol 2008; 40: 84–97.
Stutzmann GE, Caccamo A, LaFerla FM, Parker I . Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 2004; 24: 508–513.
Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F . Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann NY Acad Sci 2007; 1097: 265–277.
Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P . Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci USA 2011; 108: 2777–2782.
Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron 2000; 27: 561–572.
Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM . Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 2000; 149: 793–798.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 1996; 2: 864–870.
Rojas G, Cardenas AM, Fernandez-Olivares P, Shimahara T, Segura-Aguilar J, Caviedes R et al. Effect of the knockdown of amyloid precursor protein on intracellular calcium increases in a neuronal cell line derived from the cerebral cortex of a trisomy 16 mouse. Exp Neurol 2008; 209: 234–242.
Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A . Increased intraneuronal resting [Ca2+] in adult Alzheimer's disease mice. J Neurochem 2008; 105: 262–271.
Herms J, Schneider I, Dewachter I, Caluwaerts N, Kretzschmar H, Van Leuven F . Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J Biol Chem 2003; 278: 2484–2489.
Stieren E, Werchan WP, El Ayadi A, Li F, Boehning D . FAD mutations in amyloid precursor protein do not directly perturb intracellular calcium homeostasis. PLoS One 2010; 5: e11992.
Veinbergs I, Everson A, Sagara Y, Masliah E . Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res 2002; 67: 379–387.
Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell 2008; 133: 1149–1161.
Buxbaum JD, Ruefli AA, Parker CA, Cypess AM, Greengard P . Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc Natl Acad Sci USA 1994; 91: 4489–4493.
Hoey SE, Williams RJ, Perkinton MS . Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci 2009; 29: 4442–4460.
Petryniak MA, Wurtman RJ, Slack BE . Elevated intracellular calcium concentration increases secretory processing of the amyloid precursor protein by a tyrosine phosphorylation-dependent mechanism. Biochem J 1996; 320 (Pt 3): 957–963.
Querfurth HW, Selkoe DJ . Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 1994; 33: 4550–4561.
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE . Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3 × Tg-AD mice. J Neurosci 2009; 29: 9458–9470.
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H et al. Amyloid beta oligomers (A beta(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 2008; 28: 788–797.
Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K . Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 1997; 68: 265–271.
Supnet C, Grant J, Kong H, Westaway D, Mayne M . Amyloid-beta-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem 2006; 281: 38440–38447.
Niu Y, Su Z, Zhao C, Song B, Zhang X, Zhao N et al. Effect of amyloid beta on capacitive calcium entry in neural 2a cells. Brain Res Bull 2009; 78: 152–157.
Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci 2010; 30: 2636–2649.
Rozkalne A, Hyman BT, Spires-Jones TL . Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis 2011; 41: 650–654.
Arispe N, Rojas E, Pollard HB . Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA 1993; 90: 567–571.
Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB . Small molecule blockers of the Alzheimer Abeta calcium channel potently protect neurons from Abeta cytotoxicity. Proc Natl Acad Sci USA 2009; 106: 3348–3353.
DeKosky ST, Scheff SW . Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 1990; 27: 457–464.
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 1999; 96: 3228–3233.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T et al. APP processing and synaptic function. Neuron 2003; 37: 925–937.
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000; 20: 4050–4058.
Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci 2003; 23: 8844–8853.
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I . Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 2009; 12: 1567–1576.
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006; 52: 831–843.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL . Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27: 2866–2875.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 2005; 8: 1051–1058.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416: 535–539.
Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R . Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 2010; 13: 190–196.
Collingridge GL, Peineau S, Howland JG, Wang YT . Long-term depression in the CNS. Nat Rev Neurosci 2010; 11: 459–473.
Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T . Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 2004; 44: 759–767.
De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 2007; 282: 11590–11601.
Molnar Z, Soos K, Lengyel I, Penke B, Szegedi V, Budai D . Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport 2004; 15: 1649–1652.
Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ et al. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem 2010; 285: 7619–7632.
Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM . Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 2005; 45: 675–688.
Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN . Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem 2006; 281: 39907–39914.
Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT et al. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer's disease transgenic mice. J Neurosci 2010; 30: 14299–14304.
Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E et al. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci 2009; 29: 9704–9713.
Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH . Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci 2010; 30: 14946–14954.
Gotz J, Nitsch RM . Compartmentalized tau hyperphosphorylation and increased levels of kinases in transgenic mice. Neuroreport 2001; 12: 2007–2016.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 2010; 142: 387–397.
Zempel H, Thies E, Mandelkow E, Mandelkow EM . Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci 2010; 30: 11938–11950.
Gotz J, Chen F, van Dorpe J, Nitsch RM . Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science 2001; 293: 1491–1495.
Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci 2011; 31: 2511–2525.
Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci 2011; 31: 1688–1692.
Park SY, Ferreira A . The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 2005; 25: 5365–5375.
Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y . Cleavage of Tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment. Neurobiol Aging 2011; 32: 1–14.
Harris ME, Wang Y, Pedigo Jr NW, Hensley K, Butterfield DA, Carney JM . Amyloid beta peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 1996; 67: 277–286.
Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2007; 2: e536.
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ . Soluble amyloid {beta}-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 2011; 108: 5819–5824.
Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci USA 2003; 100: 6157–6162.
Velazquez RA, Cai Y, Shi Q, Larson AA . The distribution of zinc selenite and expression of metallothionein-III mRNA in the spinal cord and dorsal root ganglia of the rat suggest a role for zinc in sensory transmission. J Neurosci 1999; 19: 2288–2300.
Wang Z, Li JY, Dahlstrom A, Danscher G . Zinc-enriched GABAergic terminals in mouse spinal cord. Brain Res 2001; 921: 165–172.
Hirzel K, Muller U, Latal AT, Hulsmann S, Grudzinska J, Seeliger MW et al. Hyperekplexia phenotype of glycine receptor alpha1 subunit mutant mice identifies Zn(2+) as an essential endogenous modulator of glycinergic neurotransmission. Neuron 2006; 52: 679–690.
Mayer ML, Vyklicky Jr L . The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J Physiol 1989; 415: 351–365.
Christine CW, Choi DW . Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. J Neurosci 1990; 10: 108–116.
Legendre P, Westbrook GL . The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. J Physiol 1990; 429: 429–449.
Zheng F, Gingrich MB, Traynelis SF, Conn PJ . Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1998; 1: 185–191.
Rassendren FA, Lory P, Pin JP, Nargeot J . Zinc has opposite effects on NMDA and non-NMDA receptors expressed in Xenopus oocytes. Neuron 1990; 4: 733–740.
Corona C, Masciopinto F, Silvestri E, Viscovo AD, Lattanzio R, Sorda RL et al. Dietary zinc supplementation of 3 × Tg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis 2010; 1: e91.
Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT et al. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci 2009; 29: 2890–2901.
Huang YZ, Pan E, Xiong ZQ, McNamara JO . Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008; 57: 546–558.
Hwang JJ, Park MH, Choi SY, Koh JY . Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J Biol Chem 2005; 280: 11995–12001.
Lichtenthaler SF . Alpha-secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J Neurochem 2011; 116: 10–21.
Hershfinkel M, Kandler K, Knoch ME, Dagan-Rabin M, Aras MA, Abramovitch-Dahan C et al. Intracellular zinc inhibits KCC2 transporter activity. Nat Neurosci 2009; 12: 725–727.
Adlard PA, Parncutt JM, Finkelstein DI, Bush AI . Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease? J Neurosci 2010; 30: 1631–1636.
Sindreu C, Palmiter RD, Storm DR . Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci USA 2011; 108: 3366–3370.
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J . A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci 2009; 29: 4004–4015.
Smith JL, Xiong S, Markesbery WR, Lovell MA . Altered expression of zinc transporters-4 and -6 in mild cognitive impairment, early and late Alzheimer's disease brain. Neuroscience 2006; 140: 879–888.
Lyubartseva G, Smith JL, Markesbery WR, Lovell MA . Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer's disease brain. Brain Pathol 2010; 20: 343–350.
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR . Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci 1998; 158: 47–52.
Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY . Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA 2002; 99: 7705–7710.
Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF et al. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994; 265: 1464–1467.
Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI . Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer's Abeta peptides. J Biol Inorg Chem 2004; 9: 954–960.
Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS et al. Aqueous dissolution of Alzheimer's disease Abeta amyloid deposits by biometal depletion. J Biol Chem 1999; 274: 23223–23228.
Bush AI, Pettingell Jr WH, Paradis MD, Tanzi RE . Modulation of A beta adhesiveness and secretase site cleavage by zinc. J Biol Chem 1994; 269: 12152–12158.
LaFerla FM, Green KN, Oddo S . Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci 2007; 8: 499–509.
Sensi SL, Rapposelli IG, Frazzini V, Mascetra N . Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse model of Alzheimer's disease. Exp Gerontol 2008; 43: 488–492.
Miller Y, Ma B, Tsai CJ, Nussinov R . Hollow core of Alzheimer's Abeta42 amyloid observed by cryoEM is relevant at physiological pH. Proc Natl Acad Sci USA 2010; 107: 14128–14133.
Dong J, Canfield JM, Mehta AK, Shokes JE, Tian B, Childers WS et al. Engineering metal ion coordination to regulate amyloid fibril assembly and toxicity. Proc Natl Acad Sci USA 2007; 104: 13313–13318.
Miller Y, Ma B, Nussinov R . Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc Natl Acad Sci USA 2010; 107: 9490–9495.
Sandberg A, Luheshi LM, Sollvander S, Pereira de Barros T, Macao B, Knowles TP et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc Natl Acad Sci USA 2010; 107: 15595–15600.
Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S . Zinc lowers amyloid-beta toxicity by selectively precipitating aggregation intermediates. Biochemistry 2007; 46: 10655–10663.
Lovell MA, Xie C, Markesbery WR . Protection against amyloid beta peptide toxicity by zinc. Brain Res 1999; 823: 88–95.
Bishop GM, Robinson SR . The amyloid paradox: amyloid-beta-metal complexes can be neurotoxic and neuroprotective. Brain Pathol 2004; 14: 448–452.
Cuajungco MP, Faget KY . Zinc takes the center stage: its paradoxical role in Alzheimer's disease. Brain Res Brain Res Rev 2003; 41: 44–56.
Mo ZY, Zhu YZ, Zhu HL, Fan JB, Chen J, Liang Y . Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J Biol Chem 2009; 284: 34648–34657.
Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G et al. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains. Brain Res 2000; 852: 274–278.
Boom A, Authelet M, Dedecker R, Frederick C, Van Heurck R, Daubie V et al. Bimodal modulation of tau protein phosphorylation and conformation by extracellular Zn2+ in human-tau transfected cells. Biochim Biophys Acta 2009; 1793: 1058–1067.
Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH . Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA 1999; 96: 2414–2419.
Lovell MA, Xiong S, Xie C, Davies P, Markesbery WR . Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J Alzheimers Dis 2004; 6: 659–671; discussion 673–681.
Ho M, Hoke DE, Chua YJ, Li QX, Culvenor JG, Masters C et al. Effect of metal chelators on gamma-secretase indicates that calcium and magnesium ions facilitate cleavage of alzheimer amyloid precursor substrate. Int J Alzheimers Dis 2010; 2011: 950932.
Song MS, Rauw G, Baker GB, Kar S . Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 2008; 28: 1989–2002.
Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G et al. Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol 2010; 176: 870–880.
Bakchine S, Loft H . Memantine treatment in patients with mild to moderate Alzheimer's disease: results of a randomised, double-blind, placebo-controlled 6-month study. J Alzheimers Dis 2008; 13: 97–107.
Hu NW, Klyubin I, Anwyl R, Rowan MJ . GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci USA 2009; 106: 20504–20509.
Adlard PA, Bica L, White AR, Nurjono M, Filiz G, Crouch PJ et al. Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer's disease. PLoS One 2011; 6: e17669.
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008; 59: 43–55.
Lee JY, Friedman JE, Angel I, Kozak A, Koh JY . The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol Aging 2004; 25: 1315–1321.
Acknowledgements
We thank Mary Evangeline Oberschlake for the critical revision and editing of the manuscript and to Stefano Lattanzio for figure preparation. SLS is supported by funds from the Italian Dept. of Education (FIRB 2003; PRIN 2006; PRIN 2008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Corona, C., Pensalfini, A., Frazzini, V. et al. New therapeutic targets in Alzheimer's disease: brain deregulation of calcium and zinc. Cell Death Dis 2, e176 (2011). https://doi.org/10.1038/cddis.2011.57
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.57
Keywords
This article is cited by
-
Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer's Disease
Neurotherapeutics (2020)
-
Zinc transporters in Alzheimer’s disease
Molecular Brain (2019)
-
The pharmacological perturbation of brain zinc impairs BDNF-related signaling and the cognitive performances of young mice
Scientific Reports (2018)
-
The SERCA2: A Gatekeeper of Neuronal Calcium Homeostasis in the Brain
Cellular and Molecular Neurobiology (2018)
-
Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death
Molecular Neurodegeneration (2014)
