
Abstract
We reported a relevant activity of the combination between sorafenib and octreotide long-acting release (LAR) in advanced hepatocellular carcinoma (HCC) patients. In this work, we have studied if oxidative stress in both serum and peripheral blood mononuclear cells (PBMC) and pERK activation status in PBMC could be predictive of response. In the 20 responsive patients, the decrease of reactive oxygen species levels was already detectable after 10 days (T10) from the beginning of sorafenib administration, and this effect was enhanced by the combined treatment with sorafenib+octreotide LAR (T21). This effect correlated with the modulation of superoxide dismutase (SOD) activity (physiological scavenger of O2−) and of serum nitric oxide (NO) levels. Sorafenib alone induced an increase of about 40% of NO levels and of about two-fold of SOD activity in responsive patients, and both effects were significantly potentiated by the combined treatment. We found a gradual reduction of Erk1/2 activity, as evaluated by cytofluorimetric analysis, in 15 responsive patients reaching about 50% maximal decrease at T21. On the other hand, in 17 resistant patients, Erk1/2 activity was about 80% increased at T21. The determination of both the oxidative stress status and pERK activity in PBMC has high value in the prediction of response to sorafenib+octreotide therapy in HCC patients.
Similar content being viewed by others


Main
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world, and the third most common cause of cancer-related death. The number of new cases is estimated to be 564 000 per year, including 398 000 in men and 166 000 in women. It accounts for up to 75–85% of primary liver cancer in the United States and for over 90% in high-risk areas. It has predominantly affected those in developing countries, such as sub-Saharan Africa, China, Taiwan, Korea, or Vietnam, where the hepatitis B virus (HBV) is endemic.1, 2 On the other hand, HCC is relatively rare in Europe.3 In Italy, the incidence and mortality rates are at a median frequency, compared with other populations, and it represents the seventh cause of death from tumor, with about 5000 deaths per year.4, 5 In the Unites States, the incidence is increased.6 In the majority of the cases, there is underlying cirrhosis, mainly caused by HBV and hepatitis C viruses (HCV).1, 7
Ablative therapies, surgical resection or liver transplantation are the first-line treatment for patients affected with HCC.8 Nonetheless, advanced tumor stage and poor liver function preclude the majority of patients from these surgical interventions. In addition to this obstacle, transplantation is indicated only for early small HCC, and its application is limited by the availability of liver grafts.9 Therefore, there is an urgent need to develop an effective systemic therapy for patients with advanced HCC.10
In the last years, many advances in the knowledge of the molecular mechanisms that govern tumor development and progression have been made.11 More recently, single agent sorafenib (Bayer 43-9006; Nexavar, Bayer AG Inc., Leverkusen, Germany), a putative multi-targeted kinase inhibitor, has shown to prolong the overall survival (OS) of patients with advanced HCC in the pivotal phase III Sorafenib HCC Assessment Randomized Protocol (SHARP) and Oriental study.9 Currently, sorafenib is the only approved targeted therapy for patients with advanced HCC.
Sorafenib is an oral multikinase inhibitor able to block both tyrosine kinase and serin-threonin-kinase activities. Mutation or over-activation of related components in the Raf/MAPK cascade would lead to acceleration of cell proliferation and extension of survival, thus contributing to human oncogenesis.12 This pathway has been implicated in the molecular pathogenesis of HCC for three principal reasons: (i) the Ras gene is mutationally activated in 30% of HCCs;13 (ii) the over-expression of its substrate Raf kinase occurs in most HCCs;14 (iii) a variety of upstream growth factors, such as epidermal growth factor, vascular endothelial growth factor (VEGF), platelet-derived growth factor-β (PDGF) and transforming growth factor-α, which are generally overexpressed in HCC, can activate this pathway through binding their receptor tyrosine kinases.15, 16 Sorafenib blocks tumor cell proliferation and tumor angiogenesis, and increases the rate of apoptosis in a wide range of tumor models by targeting the Raf/MAPK- and VEGF-mediated pathways.16, 17
The combination of sorafenib with agents active in the control of the HCC-derived symptoms could be useful in the clinical strategy of HCC.
Differential somatostatin receptor subtypes (SSTR 1, 2, 3 and 5) are expressed in HCC.18 Analogs of somatostatin, such as octreotide, which display high binding affinity to SSTR2 and lower affinity to SSTR5 and SSTR3 (affinity rank order: SSTR2>SSTR5>SSTR3) are efficacious in the treatment of neuroendocrine tumors and exhibit only mild toxicity.19
Octreotide long-acting release (LAR) is a formulation of octreotide encapsulated into microspheres of the biodegradable glucose star polymer.20 This synthetic version of somatostatin differs from the latter for the prolonged half-life that allows to administer the drug every 28 days to obtain active plasmatic concentrations.
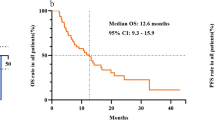
As somatostatin, octreotide reduces the release of growth factors and inhibits neo-angiogenesis. Octreotide was previously used in HCC patients with conflicting results.20, 21 However, approximately 40% of patients respond to octreotide with improved survival and an impressive quality of life.22 We showed, in a previous study, that combination of octreotide and radiofrequency ablation produced about 80% of disease control and interesting mean OS (31.4 months) in a series of advanced HCC patients.23 Investigations on octreotide in HCC are still ongoing also as National Cancer Institute sponsored trials.
On the basis of these premises, our group started a phase II multicenter study, based on the combination between sorafenib and octreotide LAR (So.LAR protocol) in order to assess its safety and activity in advanced HCC patients.24 It was recorded five partial responses (PR, 10%), 33 stationary diseases (SD, 66%) and 12 progressions of disease (PD, 24%). Overall disease control rate (CR+PR+SD) was 76%. In conclusion, the combination between So.LAR was active and well tolerated in advanced HCC.24
Continuous oxidative stress, which results from the generation of reactive oxygen species (ROS) by environmental factors or cellular mitochondrial dysfunction, has recently been associated with the progression of chronic liver diseases and hepatocarcinogenesis. On the other hand, a distinctive pathological hallmark of HCC is a dramatic downregulation of oxidoreductase enzymes that constitute the most important free radical scavenger systems represented by catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase.25, 26
The aim of this study was to define predictor parameter of outcome to treatment in advanced HCC patients enrolled in the phase II multicenter So.LAR study. Moreover, we have investigated on the pharmaco-dynamic interference between the two agents and the level of Erk activation that serves as a surrogate of the activity of sorafenib.
In details, we have evaluated possible variations in oxidative stress induced by treatment: we have assessed the O2− levels in peripheral blood mononuclear cells (PBMC) and the nitric oxide (NO), SOD and CAT serum levels. Moroever, we have evaluated the effects of treatment on tyrosine phosphorylation levels of Erk-1/2 in PBMC.
Results
Modulation of intracellular levels of ROS
To evaluate the intracellular levels of ROS, PBMC of 23 resistant and 20 responsive patients enrolled in the So.LAR study were incubated with dihydroethidine followed by FACS analysis of the oxidative product, ethidium, which emits red fluorescence. The mean fluorescence intensity (MFI) corresponds to ROS levels and to intracellular oxidative stress due to superoxide anion (O2−) generation induced by their presence.
In the patients responsive to treatment (SD), there was a gradual time-dependent reduction of fluorescence, indicating a decrease of oxidative phenomena (Figure 1a). In detail, the decrease of ROS levels was already detectable after 10 days (T10) from the beginning of sorafenib administration (12% mean reduction) and further decreased after 3 weeks (T21) from the beginning of combined treatment with sorafenib+octreotide LAR (about 50%; P<0.005).

Modulation of intracellular levels of ROS. PBMC of 23 patients resistant to treatment (PD) and 20 patients responsive to treatment (SD) enrolled in the So.LAR study were incubated with dihydroethidine and analyzed by FACS as described in ‘Materials and Methods’. (a) Representation of the ROS levels expressed as the percentage of MFI derived by dihydroethidine oxidation of 43 patients. T0, T3, T10 and T21 indicate the time of sample collection before the treatment and after 3 days, 10 days and 21 days from the beginning of the schedule, respectively. Bars, S.D.s. (b and c) Flow cytometric analysis of PBMC of a resistant and a responsive patient, respectively, exposed to dihydroethidine used as a probe for measurement of O2− at different times of treatment. CTR−, PBMC not labeled; T0, T3, T10, T21 indicate the measurement performed before the treatment, and after 3 days, 10 days and 21 days from the beginning of the schedule, respectively. Inset, table representing the P-values and the statistical significance of the changes of the different values
In patients resistant to treatment (PD), there was an increase of about 60% of MFI after 10 (T10) and 21 (T21) days from the beginning of treatment (P<0.004). In Figures 1b and c, two example of ROS determination in PBMC of a resistant (B) and responsive (C) patient are reported, at different times of treatment. We observed an opposite trend of intracellular ROS levels with a gradual increase of fluorescence in the resistant patient (Figure 1b) and a gradual decrease of fluorescence in the responding patient (Figure 1c) reaching a decrease of about 50% of MFI at T21 time. In conclusion, in responding patients, we observed a reduction of intracellular ROS levels that was evident after 21 days from the beginning of the treatment (Cut-off: 19.3%). Moreover, the addition of octreotide did not interfere with the reduction of ROS induced by sorafenib, even potentiating its effect.
Modulation of SOD activation levels
On the basis of the effect of treatment on intracellular oxidative stress regulation, we have evaluated the levels of enzymatic activity of SOD (physiological scavenger of ROS) in serum. We have demonstrated that, in resistant patients, the treatment with sorafenib for 10 days (T10) induced an about two-fold increase of SOD activity, and this effect was significantly potentiated after the addition of octreotide LAR (P<0.002) (Figure 2). Also in resistant patients, the treatment with sorafenib for 10 days (T10) induced an about two-fold increase of SOD activity, but it resumed to basal levels after addition of octreotide LAR (T21) (Figure 2). These effects were correlated to the modulation of intracellular ROS levels described above.

Modulation of SOD activation levels. Variations of SOD serum levels in 23 patients resistant to treatment (PD) and 20 patients responsive to the treatment (SD) enrolled in the So.LAR study, using an enzyme spectrophotometric assay as described in ‘Materials and Methods’. T0, T3, T10 and T21 indicate the measurement performed before the treatment, and after 3 days, 10 days and 21 days from the beginning of the schedule, respectively. Bars, S.D.s. Inset, table representing the P-values and the statistical significance of the changes of the different values
In the same patients, we have evaluated the levels of CAT activity, recording no significant changes in both resistant and responding patients (data not shown).
These data suggest that the determination of serum SOD at T21 is useful to predict the sensitivity of the patients to the combination therapy (cut off: 7.7 U/ml).
Modulation of serum levels of NO
We have investigated if the reduction of superoxide anions following the increase of SOD activity in responding patients could be caused by the increase in the serum levels of NO. We observed an increase in NO levels of 75 and 50%, in resistant and responder patients, respectively, after 3 days (T3) from the beginning of treatment with sorafenib alone (P<0.005). Moreover, in resistant patients, the NO levels resumed to basal levels from T10 up to T21, whereas in responder patients NO was about 40% higher than basal levels in all the time points assessed during the present study (Figure 3). These data suggest again that the increase of NO levels recorded after 21 days from the beginning of the treatment can be predictive of response to therapy. Again, the addition of octreotide did not modify the trend of NO levels in the responsive patients.

Modulation of serum levels of NO. Variations of NO serum levels in 23 patients resistant to treatment (PD) and 20 patients responsive to the treatment (SD) enrolled in the So.LAR study, using an enzyme spectrophotometric assay as described in ‘Materials and Methods’. T0, T3, T10 and T21 indicate the measurement performed before the treatment, and after 3 days, 10 days and 21 days from the beginning of the schedule, respectively. Bars, S.D.s. Inset, table representing the P-values and the statistical significance of the changes of the different values
Modulation of Erk1/2 activity
Sorafenib inhibits the phosphorylation of both Erk and Mek (downstream targets of raf) and Erk1/2 activity can be considered a surrogate target of sorafenib and then a marker of biological activity of the drug.
Therefore, we have evaluated the activation status of Erk1/2 in the PBMC of 17 resistant and 15 sensitive patients, with both western blotting and FACS analysis for pERK-1/2 in PBMCs. The results obtained with the two techniques were absolutely overlapping.
We have found a gradual reduction of Erk1/2 activity in SD patients reaching a maximal decrease of 50% after 21 days (T21) from the beginning of therapy (Figure 4a). On the other hand, Erk1/2 activity was only about 10% reduced after 10 days (T10) of treatment with sorafenib alone (P<0.05). On the other hand, in resistant patients, the Erk1/2 activity was about 80% increased after 21 days (T21) from the beginning of therapy. This increase was detected also after 10 days from the beginning of therapy with sorafenib alone, but the change was again not statistically significant (P>0.05). The trend of Erk1/2 activity was confirmed by the analysis of 20 patients through western blotting analysis. In Figure 4b, western blotting for pERK1/2 in a responder and a resistant patient, respectively, are shown. The results clearly suggest the inverse trend of pERK1/2 in the two patients.

Modulation of Erk1/2 activity. PBMC of 17 patients resistant to treatment (PD) and 15 patients responsive to the treatment (SD) enrolled in the So.LAR study were processed for the evaluation of the intracytoplasmic expression of Erk 1/2 activity. (a) Representation of the percentage of pERK1/2 levels compared with PBMC labeled with a specific antibody obtained by cytofluorimetric analysis of indirectly-labeled PBMC of 32 patients. (b) Detection of phosphorylation levels of Erk1/2 evaluated by western blot analysis as described in ‘Materials and Methods’. T0, T3, T10 and T21 indicate the measurement performed before the treatment, and after 3 days, 10 days and 21 days from the beginning of the schedule, respectively. Bars, S.D.s. Inset, table representing the P-values and the statistical significance of the changes of the different values
In conclusion, the addition of octreotide LAR did not interfere with the biological effects induced by sorafenib and even increased the effects of sorafenib in responsive patients. The phosphorylation of Erk can be a surrogate marker of sorafenib+octreotide activity if evaluated after 21 days from the beginning of the treatment (cut-off: 21 MFI). Moreover, the detection of pERK at FACS was a valid assay for the evaluation of pERK intracellular levels.
Discussion
The activation of Ras → MAPK-dependent pathway has a key role in the genesis and progression of human HCC.27, 28, 29 This signaling pathway, found in all proliferative cells, determines the activation of Erk protein by its phosphorylation that favours the HCC progression and promotes the tumor growth and angiogenesis. In fact, it was demonstrated a progressive and aberrant Erk1/2 activation in the tumor liver tissue compared with the normal counterpart, with a more pronounced increase in tumors with poor prognosis, without mutations of Ras protein. Mutations of Ras, Raf and EGFR genes, that both activate the Ras → MAPK-dependent pathway, are very rare in human HCC.27, 28, 29 However, studies on mutations of Raf family genes in significantly wide series are not available in the literature.30 The development of HCC is closely related to angiogenesis and to disruption of liver vascular architecture, which contributes both to increased hepatic vascular resistance and portal hypertension. On the other hand, it was recently reported that angiogenesis modulates the formation of portal-systemic collaterals and the increased splanchnic blood flow, which are involved in the life threatening complications of cirrhosis. Finally, angiogenesis has a key role in the growth of tumors, suggesting that interference with angiogenesis may prevent or antagonize the development of HCC.31 Moreover, VEGF levels in HCC tissues correlated to the recurrence of HCC after hepatectomy.32
Sorafenib is a multikinase inhibitor able to antagonize different molecular mechanisms involved in the progression of HCC inhibiting both Raf and VEGF-R. Octreotide is also able to inhibit angiogenesis and is an effective agent in improving the QoL of patients affected by HCC.24 Important topics in the field of target-based therapy of human tumors including HCC is represented by the need of predicting the response of the patients to the treatment in order to both avoid unuseful side effects and to limit the cost of the therapy. At least, at our knowledge, up-to-date reports on definitive predictive markers of response to octreotide are not still available. On the other hand, it has been recently demonstrated that the phosphorylation levels of Erk in the tumor tissues correlated with the time to progression of patients affected by HCC.32 Zhang and colleagues17 confirmed that Erk phosphorylation is a potential surrogate marker of sorafenib activity in human HCC cell lines. It is also possible to evaluate the effects of sorafenib on PBMC, where the pERK activity levels were correlated with oxidative stress induced by Vibrio vulnificus.33 However, non-invasive methods for the determination of Erk activity during the treatment are strongly warranted. In this view, quick, easy and quantitative methods for the ex vivo determination of pERK in PBMC have been developed on the basis of cytofluorimetric techniques.34 On these basis, we have evaluated the effects of So.LAR treatment on Erk activity in PBMC of patients affected by HCC with cytofluorimetric technique.
Our results have confirmed the absolute overlapping of data obtained by FACS and those determined with western blotting, suggesting the feasibility of molecular screening studies on biological agents through the determination of pERK in the PBMC. Our data suggested that the treatment with octreotide LAR did not change the effects of sorafenib on Erk phosphorylation. In fact, in resistant patients, pERK levels increased already before the administration of octreotide LAR. This effect can be likely induced by the triggering of a feed-back pathway activated by the sorafenib-mediated inhibition of Raf kinase that can elicit alternative modes of Mek activation that, in turn, causes Erk phosphorylation. In fact, protein kinase C alpha can trigger Ras and Raf-independent Mek/Erk activation in human hepatoma cell HepG235 and cannot be excluded, that also in PBMNC alternative Erk-activation pathway exist in specific patients. On the other hand, in sensitive patients, the decrease of pERK levels detected at day 10 (T10) was potentiated by the addition of octreotide LAR (T21). These results demonstrated, at least in our series of patients, the absence of a pharmaco-dynamic interaction between the two agents, suggesting the chance to use them in combination in the HCC treatment.
It is known that anti-oxidative enzymes, such as CAT and SOD are significantly downregulated in HCC, because of a gene re-organization to support the proliferating ability of tumor cells. In fact, cancer cells carry out different survival mechanisms to increase their proliferative ability and the presence of ROS has a pivotal role in these processes. Moreover, cancer cells escape from apoptosis induced by excessive levels of ROS activating both Erk e PI3 K signal transduction pathways.
On these bases, we have evaluated the levels of ROS in the PBMC of patients treated with So.LAR schedule by cytofluorimetric analysis based on incubation of PBMC with dihydroethidine, which is oxidized in cells to fluorescent ethidium bromide in the presence of superoxide anions.
We have found a significant increase of ROS levels after 10 days (T10) from the beginning of the treatment that continues to go up also after 21 days (T21) of treatment in the resistant patients. On the other hand, in the sensitive patients, there was a gradual decrease of ROS levels that reached the maximal effect after 21 days from the beginning of the treatment. These effects were paralleled by the regulation of serum activity of SOD that was unchanged in resistant patients, whereas gradually increased in sensitive patients after 21 days from the beginning of the treatment. These data suggest once again the absence of pharmaco-dynamic interaction between the two agents and the possible use of oxidative stress levels as predictor markers of clinical outcome to this treatment.
In cancer biology, NO can be involved either in promotion or in prevention of tumor occurrence dependently from tumor microenvironment, NO concentration and time of exposure. NO is a product of endothelial cells that binds and activates the guanylate cyclase, which catalyzes the conversion of GTP to the second messenger molecule cyclic GMP. Concentrations of NO ranging between 1 and 30 nM produce high levels of cyclic GMP, promoting angiogenesis and proliferation of endothelial cells. In these conditions, Erk phosphorylation stimulates the proliferation of endothelial cells. Concentrations of NO ranging between 30 and 100 nM correspond to an increase of proliferative and anti-apoptotic Akt and Erk-dependent pathways in tumor cells.36, 37 This range of concentrations seems to protect tumor cells from apoptosis and enhance angiogenic effects. In these conditions, the molecules activated by NO can be considered as factors correlated to poor prognosis events. On the other hand, higher NO levels (>300 nM) promote apoptosis and are responsible for anti-tumor activity. NO levels are influenced also by ROS, in particular by superoxide anions that can attenuate the NO-mediated pathway. In fact, superoxide anion and ROS, through the scavenging of NO, can lower NO levels favouring its tumor-promoting activity.38 Usually, tumors have high levels of ROS and low levels of SOD.
In this study, we have evaluated, using a spectrophotometric assay, serum NO levels of patients treated with So.LAR schedule. After 3 days (T3) from the beginning of the treatment, resistant patients presented a strong increase of NO levels that resumed to basal levels at day 10 and thereafter. On the other hand, in sensitive patients NO levels increased at T3 and remained elevated during all the pharmacological treatment.
These data indicate that the trend of NO activity is correlated with the prognosis of HCC patients treated with So.LAR schedule. In fact, in sensitive patients, the increased levels of NO probably had an anti-tumor and pro-apoptotic role, as demonstrated by the decrease of Erk activation. On the other hand, resistant patients presented low levels of NO correlated to the induction of Erk activation. Also in this case, the addition of octreotide LAR to sorafenib did not interfere with the biological and biochemical effects of the latter, and in some cases octreotide LAR even potentiated the effects of sorefenib.
In conclusion, the combination used in So.LAR represents a valid approach in the treatment of patients affected by advanced HCC. However, the treatment of the patients with the combination induces also important side effects and has a relevant cost. Therefore, the possibility to select the sensitive patients on the basis of easy-to-detect clinical and molecular markers has a high clinical impact. The determination of both pERK expression in PBMC with FACS analysis and the oxidative stress status have high value in the prediction of response to sorafenib+octreotide therapy in HCC patients.
Materials and Methods
Patient characteristics, treatment schedule and isolation of peripheral blood mononuclear cells (PBMC)
A total of 50 patients (43 M/7 F) were enrolled between July 2007 and July 2008. Patients were required to have HCC confirmed by biopsy or diagnosed by radiological and clinical criteria, as previously described.27 Patients received sorafenib 400 mg bid for 28 days, with a following week of rest and long-acting octreotide at a dose of 40 mg every 28 days. Treatment was continued until disease progression or unacceptable toxicity. The first octreotide injection was administered 10 days after sorafenib, starting in order to evaluate eventual pharmcodynamic interference between the two agents. Dose reduction of sorafenib (200 mg bid) and octreotide (20 mg) were allowed for drug-related toxicities (National Cancer Institute Common Toxicity Criteria version 2.0). Response to treatment was assessed by at least two independent radiologists, using RECIST criteria every 2 months. Patients were considered ‘responsive to the treatment’ if a PR or a SD lasting equal or more than 6 months was recorded and ‘resistant to treatment’ if a PR or SD lasting less than 6 months or a PD was recorded. The biological samples (both sera and PBMC) were collected before the treatment (T0), and before 3 days (T3), 10 days (T10) and 21 days (T21) from the beginning of the schedule.
PBMC were isolated from freshly obtained blood by FICOLL density gradient centrifugation, as previously described.39 PBMC were stored at −80°C for all the following experiments. Sera were collected from the patients at the selected times and stored at −20°C for all the following experiments.
Cytofluorimetric analysis of oxidative stress
PBMC of patients were incubated with dihydroethidine (20 ng/ml for 1 h) and analyzed by FACScan flow cytometer (FacSCAN, Becton Dickinson, San Josè, CA USA). For each sample, about 20 000 events were acquired and the fluorescence was measured on FL2-H channel by the FACScalibur (Applied Biosystems, Carlsbad, CA, USA) software. The MFI indicates ROS (O2−) levels.
SOD assay
SOD are metalloenzymes that catalyze the dismutation of superoxide radical into hydrogen peroxide (H2O2)+molecular oxygen (O2) and consequently provide an important protection mechanism against superoxide radical toxicity.
To determine SOD activity, we have applied the xanthine/xanthine oxidase (XOD) system to generate superoxide anions and a chromogen to produce a water-soluble formazan dye upon reduction by superoxide anions. The rate of the reduction with O2− is linearly related to the xanthine oxidase (XO) activity, and is inhibited by SOD. Therefore, the IC50 (50% inhibition activity of SOD) can be determined by this colorimetric method. Absorbance can be measured at 440 nm.
Spectrophotometric determination of NO serum levels
The determination of serum NO concentration, expressed in μM, has been performed with an indirect method through the measure of serum concentrations of nitrates and nitrites. For NO determination, the spectrophotometric reaction of Griess was used.40
Cytofluorimetric analysis of Erk activity
The PBMCs were fixed with 3% paraformaldehyde in PBS for 30 min at room temperature and permeabilized with 0.5% Tween 20 in PBS for 5 min. The PBMCs were exposed to a specific antibody, directly against pMAPK p42/44 (10 μg/ml per sample) for 30 min at 4°C, and then incubated with FITC-conjugated anti-mouse goat antiserum (25 μl per sample) for 30 min at 4°C. After final washes, samples were analyzed by FACScan flow cytometer (FACScan, Becton Dickinson, Mountain View, CA, USA). For each sample, about 20 000 events were acquired and the fluorescence was measured on FL2-H channel by the FACScalibur (Applied Biosystems) software.
Determination of Erk activity by immunoblotting
The PBMC were lysed for 30 min at 4°C in lysis buffer (1% Triton, 0.5% sodium deoxycholate, 0.1 NaCl, 1 mM EDTA, pH 7.5, 10 mM Na2HPO4, pH 7.4, 10 mM PMSF, 25 mM benzamidine, 1 mM leupeptin, 0.025 units per ml aprotinin). About 100 μg of proteins were incubated with a specific antibody directed against pMAPK p42/44 and immunoprecipitated with Protein A sepharose.41 Proteins were run on SDS-PAGE, transferred on nitrocellulose film and incubated with the same primary antibody. Films were developed using the chemoluminescence assay (SuperSignal West Pico, Pierce, Rockford, IL, USA).
Determination of cut-off value
This method for choosing a cut-off is based on the application of 95% CI of mean. Mean and standard deviation (S.D.) of trial values are calculated. The interval obtained by subtracting 2 × S.D. from mean and by adding 2 × S.D. to mean (that is, μ±2σ) shows that the chance of a trial value coming outside this interval will be less than 5%. The lower limit of this interval (i.e. mean−2 S.D.) may be considered as cut-off point. If a subject's trial value comes less than this cut-off, then it may be considered negative (normal), and if value comes greater than or equal to cut-off value, then it is considered positive (diseased).
Statistical analysis
All data are expressed as mean±S.D. Statistical analysis was performed by analysis of variance (ANOVA) with Neumann–Keul's multiple comparison test or Kolmogorov–Smirnov, where appropriate. The asterisks in the figure indicate significant difference between the basal patient with respect to the patients treated with SO.LAR therapy (**P<0.003; *P<0.05; n.s., not significant). The differences between patients with PD and those with SD were performed using the Mann–Whitney U-test for non-parametric independent and continuous variables.
Abbreviations
- HCC:
-
hepatocellular carcinoma
- PBMC:
-
peripheral blood mononuclear cells
- SOD:
-
superoxide dismutase
- NO:
-
nitric oxide
- HBV:
-
hepatitis B virus
- HCV:
-
hepatitis C virus
- VEGF:
-
vascular endothelial growth factor
- PDGF:
-
platelet-derived growth factor-β
- SSTR:
-
somatostatin receptor
- OS:
-
overall survival
- LAR:
-
long-acting release
- CR:
-
complete remission
- PR:
-
partial remission
- SD:
-
stationary disease
- CAT:
-
catalase
- ROS:
-
reactive oxygen species
- PMSF:
-
phenyl-methyl-sulfonyl-fluoride
- SDS-PAGE:
-
sodium-dodecyl-sulphate-polyacrilammide-gel-eletrophoresis
- MFI:
-
mean fluorescence intensity
- S.D.:
-
standard deviation
- XOD:
-
xanthine/xanthine oxidase
- H2O2:
-
hydrogen peroxide
References
Bosch FX, Ribes J, Díaz M, Cléries R . Primary liver cancer: worldwide incidence and trends. Gastroenterology 2004; 127: S5–S16.
Larson AM . The epidemiology of hepatocellular carcinoma in HCV. Curr Hepat Reps 2005; 4: 145–152.
Levi F, Lucchini F, Negri E, La Vecchia C . Continuing declines in cancer mortality in the European Union. Ann Oncol 2007; 18: 593–595.
Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA . Epidemiology; risk factors; and natural history of hepatocellular carcinoma. Ann N Y Acad Sci 2002; 963: 13–20.
La Vecchia C, Negri E, Pilucchi C . The rise and fall in primary liver cancer mortality in Italy. Dig Liver Dis 2002; 34: 169–171.
McGlynn KA, Tsao L, Hsing AW, Devesa SS, Fraumeni Jr JF . International trends and patterns of primary liver cancer. Int J Cancer 2001; 94: 290–296.
Yang JC, Teng CF, Wu HC, Tsai HW, Chuang HC, Tsai TF et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009; 49: 1962–1971.
Forner A, Hessheimer AJ, Real IM, Bruix J . Treatment of hepatocellular carcinoma. Crit Rev Oncol Hematol 2006; 60: 89–98.
Yau T, Chan P, Epstein R, Poon RTP . Management of advanced hepatocellular carcinoma in the era of targeted therapy. Liver Int 2009; 29: 10–17.
Llovet JM, Bruix J . Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003; 37: 429–442.
Thorgeirsson SS, Grisham JW . Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002; 31: 339–346.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–279.
Downward J . Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22.
Hwang YH, Choi JY, Kim S, Chung ES, Kim T, Koh SS et al. Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma. Hepatol Res 2004; 29: 113–121.
Roberts PJ, Der CJ . Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26: 3291–3310.
Caraglia M, Tassone P, Marra M, Budillon A, Venuta S, Tagliaferri P . Targeting Raf-kinase: molecular rationales and translational issues. Ann Oncol 2006; 17(Suppl 7): 124–127.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D et al. Sorafenib blocks the RAF/MEK/ERK pathway; inhibits tumor angiogenesis; and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006; 66: 11851–11858.
Bläker M, Schmitz M, Gocht A, Burghardt S, Schulz M, Bröring DC et al. Differential expression of somatostatin receptor subtypes in hepatocellular carcinomas. J Hepatol 2004; 41: 112–118.
Lamberts SW, van der Lely AJ, de Herder WW, Hofland LJ . Octreotide. N Engl J Med 1996; 334: 246–254.
Dimitroulopoulos D, Xinopoulos D, Tsamakidis K, Zisimopoulos A, Andriotis E, Panagiotakos D et al. Long acting octreotide in the treatment of advanced hepatocellular cancer and overexpression of somatostatin receptors: randomized placebo-controlled trial. World J Gastroenterol 2007; 13: 3164–3170.
Attia S, Holen KD, Thomas JP, Richie K, Dzelak T, Teeter K et al. Biologic study of the effects of octreotide-LAR on growth hormone in unresectable and metastatic hepatocellular carcinoma. Clin Adv Hematol Oncol 2008; 6: 44–54.
Samonakis DN, Notas G, Christodoulakis N, Kouroumalis EA . Mechanisms of action and resistance of somatostatin analogues for the treatment of hepatocellular carcinoma: a message not well taken. Dig Dis Sci 2008; 53: 2359–2365.
Montella L, Addeo R, Caraglia M, Faiola V, Guarrasi R, Vincenzi B et al. Vascular endothelial growth factor monitoring in advanced hepatocellular carcinoma patients treated with radiofrequency ablation plus octreotide: a single center experience. Oncol Rep 2008; 20: 385–390.
Del Prete S, Montella L, Caraglia M, Maiorino L, Cennamo G, Montesarchio V et al. Sorafenib plus octreotide is an effective and safe treatment in advanced hepatocellular carcinoma: multicenter phase II So.LAR. study. Cancer Chemother Pharmacol 2010; 66: 837–844.
Clemente C, Elba S, Buongiorno G, Guerra V, D’Attoma B, Orlando A et al. Manganese superoxide dismutase activity and incidence of hepatocellular carcinoma in patients with Child-Pugh class A liver cirrhosis: a 7-year follow-up study. Liver Int 2007; 27: 791–797.
Liaw KY, Lee PH, Wu FC, Tsai JS, Lin-Shiau SY . Zinc; copper; and superoxide dismutase in hepatocellular carcinoma. Am J Gastroenterol 1997; 92: 2260–2263.
Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest 2007; 117: 2713–2722.
Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998; 27: 951–958.
Stowers SJ, Wiseman RW, Ward JM, Miller JA, Miller EC, Anderson MW et al. Detection of activated proto-oncogenes in N-nitrosodiethylamine-induced liver tumors: a comparison between B6C3F1 mice and Fisher 344 rats. Carcinogenesis 1988; 9: 271–276.
Calvisi DF, Pinna F, Meloni F, Ladu S, Pellegrino R, Sini M et al. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res 2008; 68: 4192–4200.
Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J . Angiogenesis in liver disease. J Hepatol 2009; 50: 604–620.
Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24: 4293–4300.
Kim WH, Goo SY, Lee KH, Park SJ . Vibrio vulnificus-induced cell death of human mononuclear cells requires ROS-dependent activation of p38 and ERK 1/2 MAPKs. Immunol Invest 2009; 38: 31–48.
Chow S, Hedley D, Grom P, Magari R, Jacobberger JW, Shankey TV . Whole blood fixation and permeabilization protocol with red blood cell lysis for flow cytometry of intracellular phosphorylated epitopes in leukocyte subpopulations. Cytometry 2005; 67: 4–17.
Wen-Sheng W . Protein kinase C alpha trigger Ras and Raf-independent MEK/ERK activation for TPA-induced growth inhibition of human hepatoma cell HepG2. Cancer Lett 2006; 28: 27–35.
Ridnour LA, Thomas DD, Switzer C, Flores-Santana W, Isenberg JS, Ambs S et al. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 2008; 19: 73–76.
Prueitt RL, Boersma BJ, Howe TM, Goodman JE, Thomas DD, Ying L et al. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer 2007; 120: 796–805.
Pervin S, Singh R, Freije WA, Chaudhuri G . MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: implications in breast cancer. Cancer Res 2003; 63: 8853–8860.
Correale P, Campoccia G, Tsang KY, Micheli L, Cusi MG, Sabatino M et al. Recruitment of dendritic cells and enhanced antigen specific immune-reactivity in cancer patients treated with hrGM-CSF (Molgramostim) and hr IL-2: results from a phase Ib clinical trial. Eur J Cancer 2001; 37: 892–902.
Titheradge A . The enzymatic measurement of nitrate and nitrite. Methods Mol Biol 1998; 100: 83–91.
Lamberti A, Longo O, Marra M, Tagliaferri P, Bismuto E, Fiengo A et al. C-Raf antagonizes apoptosis induced by IFN-alpha in human lung cancer cells by phosphorylation and increase of the intracellular content of elongation factor 1A. Cell Death Differ 2007; 14: 952–962
Acknowledgements
MC received a grant from Italian Association for Cancer Research. AA received a grant from Italian Ministry for Education (PRIN 2008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Caraglia, M., Giuberti, G., Marra, M. et al. Oxidative stress and ERK1/2 phosphorylation as predictors of outcome in hepatocellular carcinoma patients treated with sorafenib plus octreotide LAR. Cell Death Dis 2, e150 (2011). https://doi.org/10.1038/cddis.2011.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.34
Keywords
This article is cited by
-
Prognostic stratification based on HIF-1α signaling for evaluating hypoxia status and immune landscape in hepatocellular carcinoma
Journal of Big Data (2023)
-
Identification of DNA repair-related genes predicting pathogenesis and prognosis for liver cancer
Cancer Cell International (2021)
-
A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo
Cell Death & Disease (2018)
-
Molecular mechanisms governing microRNA-125a expression in human hepatocellular carcinoma cells
Scientific Reports (2017)
-
Curcumin ameliorate DENA-induced HCC via modulating TGF-β, AKT, and caspase-3 expression in experimental rat model
Tumor Biology (2015)
