
Abstract
The IPC-81 cell line is derived from the transplantable BNML model of acute myelogenic leukemia (AML), known to be a reliable predictor of the clinical efficiency of antileukemic agents, like the first-line AML anthracycline drug daunorubicin (DNR). We show here that cAMP acted synergistically with DNR to induce IPC cell death. The DNR-induced death differed from that induced by cAMP by (1) not involving Bim induction, (2) being abrogated by GSK3β inhibitors, (3) by being promoted by the HSP90/p23 antagonist geldanamycin and truncated p23 and (4) by being insensitive to the CRE binding protein (CREB) antagonist ICER and to cyclin-dependent protein kinase (CDK) inhibitors. In contrast, the apoptosis induced by cAMP correlated tightly with Bim protein expression. It was abrogated by Bim (BCL2L11) downregulation, whether achieved by the CREB antagonist ICER, by CDK inhibitors, by Bim-directed RNAi, or by protein synthesis inhibitor. The forced expression of BimL killed IPC-81WT cells rapidly, Bcl2-overexpressing cells being partially resistant. The pivotal role of CREB and CDK activity for Bim transcription is unprecedented. It is also noteworthy that newly developed cAMP analogs specifically activating PKA isozyme I (PKA-I) were able to induce IPC cell apoptosis. Our findings support the notion that AML cells may possess targetable death pathways not exploited by common anti-cancer agents.
Similar content being viewed by others


Main
The second messenger cAMP regulates fundamental cell processes, including cell survival, differentiation and proliferation, mainly by activating cAMP-dependent protein kinase I,II (PKA-I, PKA-II) or cAMP-stimulated exchange factors Epac1,2 (see1 and references therein). The IPC-81 acute myelogenic leukemia (AML) cells2 undergo apoptosis within 4–6 h after cAMP stimulation.3 The cells are also sensitive to first-line anti-leukemic drugs like daunorubicin (DNR) both in vitro and in vivo.4 This may reflect their origin from the BNML model of leukemia, considered reliable to judge the clinical efficacy of anti-leukemic drugs.5 The IPC cell provides therefore a unique opportunity to compare the cell death induced by a physiologically relevant endogenous second messenger and a chemotherapeutically relevant agent.
Intriguingly, the cAMP-induced IPC cell death is blocked by the cyclin-dependent protein kinase (CDK) inhibitor roscovitine (RCV),6 which (under trade name Seliciclib) is used against cancer.7, 8 Equally intriguing, the cAMP-induced death is blocked by overexpression of ICER,9 which competitively inhibits the access of the cAMP-responsive gene promoter elements binding protein (CREB) transcription factor family members to CRE.10 ICER is considered an AML tumor suppressor and CREB an AML oncogene.11, 12, 13
The present study was undertaken to know how the normally proapoptotic RCV and ICER could block the cAMP-induced IPC cell death. We wanted also to know the relative roles of PKA-I, PKA-II and Epac in the death induction. For this purpose, new PKA-I selective cAMP analogs were developed and used, together with previously developed analogs discriminating between PKA and Epac.14, 15 Finally, we wanted to know the relation, if any, between cAMP- and anthracycline-induced IPC cell death.
We found that activation of PKA-I was necessary and sufficient for cAMP-induced IPC cell death. The death was dependent on Bim induction, whose transcription was dependent both on CREB and RCV-inhibited CDK activity. In contrast, neither Bim induction, CREB activity or RCV-inhibitable CDK activity affected anthracycline-induced IPC cell death, which depended on glycogen synthase kinase 3β (GSK3β) activity and was promoted by disruption of the HSP90/cochaperone p23 complex. In spite of having dissimilar apoptosis induction mechanism, cAMP synergized strongly with anthracycline to induce IPC cell death. We conclude that IPC cells have a potent cAMP-induced cell death pathway unexploited by first-line chemotherapeutics.
Results
Strong PKA-I activation induces IPC cell apoptosis, whereas the basal PKA activity is necessary to maintain growth and survival
The apoptosis induced (Figure 1a) by cAMP stimulation of IPCWT allow the probing of both cAMP signaling and apoptosis pathways. We studied first whether the cAMP receptor Epac could be co-responsible with PKA for the death induction. The Epac-specific activator 8-CPT-2′-O-Me-cAMP failed, however, to induce any apoptosis alone (Figure 1a) or to enhance the apoptosis induced by the PKA-specific activator N6-MB-cAMP (not shown; see also Supplementary Section IV). To probe the role of PKA more directly, the cells were stably transduced with short hairpin-based RNAi directed against the catalytic Cα subunit of PKA, and selected for survival after 48 h exposure to an apoptogenic concentration of cAMP analog. The surviving clones expressed from 25–60% of the normal amount of catalytic kinase activity (Figure 1b). This suggests that at least 60% of the normal PKA content is required for cAMP-induced apoptosis to occur. The RI subunit of PKA-I comprises about 75% of the total R subunit expressed in IPCWT cells, the remaining 25% being RII associated with PKA-II.16 An isolated activation of PKA-I might therefore be sufficient to induce apoptosis.

Activation of PKA-I, but not Epac or PKA-II, induces CRE-dependent IPC cell apoptosis counteracted by Bcl2. (a) The upper row show that IPCWT AML cells underwent apoptosis with cell fragmentation and chromatin condensation after 5 h incubation with either 200 μM of the cAMP analog 8-CPT-cAMP, which activates both PKA and Epac or 700 μM of the PKA-specific analog N6-MB-cAMP. The specific Epac activator 8-CPT-2′-O-Me-cAMP (700 μM) did not induce any apoptosis. Rows 2,3 show that the Bcl2 overexpressing IPC cells (IPCBCL2) or IPC cells with enforced expression of the CREB antagonist ICER (IPCICER) were protected against apoptosis by the same cAMP analogs. (b) The PKA activity was determined (at saturating cAMP concentration) in extracts of IPCWT and four cAMP-resistant IPC clones stably transduced with shRNAi against the catalytic (Cα) subunit of PKA. (c) The panel shows the new cAMP analog 2-Cl-8-AHA-cAMP, highly selective for site BI of PKA isozyme I (Supplementary Table S2), to be an inefficient inducer of IPCWT apoptosis alone (○). When combined with the new site AI-specific analog N6-Bnz-8-Pip-cAMP (300 μM), its efficiency was enhanced >20-fold (•). This analog pair is expected to synergize strongly for PKA-I activation (inset), but not for PKA-II (Supplementary Table S3). (d and e) The analogs Sp-5,6-DCl-cBIMPS and N6-MB-cAMP (○) were, respectively, moderately (d) and only marginally (e) more efficient when combined with N6-Bnz-8-Pip-cAMP (•). The insets show their expected PKA-I synergy (from Supplementary Table S3). Note that N6-Bnz-8-Pip-cAMP (300 μM) had only little effect (5% increase of the IPC cell death) when present alone (c–e). Error bars indicate S.E.M., n=3–8
To enable specific activation of PKA-I, cAMP analog combinations with improved isozyme selectivity were synthesized. The new PKA-I selective analogs N6-Bnz-8-Pip-cAMP and 2-Cl-8-AHA-cAMP discriminate better than any previous cAMP analogs between the cAMP binding sites (A,B) of PKA-I and PKA-II (Supplementary Tables S1–S3). They acted in strong synergy to induce cAMP-dependent apoptosis (Figure 1c). When N6-Bnz-8-Pip-cAMP was combined with other cAMP analogs, a moderate (Figure 1d) or barely any (Figure 1e) synergy was achieved. The observed synergies are in accordance with predictions based on the analog cAMP binding site preferences (Supplementary Table S3), and therefore unlikely to be due to nonspecific side effects. Note also that the concentration of paired analogs yielding synergistic death induction was far lower than that required for each analog alone (Figure 1). We conclude that IPC cell apoptosis can be induced by the isolated activation of PKA-I.
Although a strong activation of PKA leads to IPC cell death, the basal activity seemed necessary for growth. In fact, some of the surviving shRNAi-transfected clones showed slow growth, sometimes associated with mitotic arrest. That the slow growth was due to low PKA activity was supported by the demonstration of improved growth of clone Lo45-II in the presence of a low concentration (20 μM) of cAMP analog (8-CPT-cAMP). This clone had about 25% of the normal PKA content, against about 40–60% for three surviving clones that did not depend on added cAMP for optimal growth (Figure 1b). In conclusion, PKA appears to have a dual role. It induces death when strongly activated, but its basal activity is necessary to avoid growth arrest.
Cyclic AMP operates mainly through CRE-dependent factors to regulate the IPC cell transcriptome
The cAMP-induced death is prevented in IPC cells overexpressing the truncated cAMP responsive element modulator (CREM) variant ICER9 (Figure 1a). ICER blocks the access to the cAMP-responsive elements (CRE) of gene promoters.10 A functional CRE is required for the action of the CREB, CREM, ATF-1 family of transcription factors.17, 18 The IPCICER cells might be protected because ICER alters the basal gene expression or because it prevents the acute regulation by cAMP of one or more genes required for apoptosis to occur. The basic transcriptome was very similar in IPCWT and IPCICER cells (not shown). This suggested that few CRE-dependent gene regulation events took place at basal cAMP concentration in the cells, and that ICER acted to modulate the acute effect of cAMP on gene transcription.
We found nearly 40 named genes to be >2-fold upregulated and 15 to be >2-fold downregulated after incubation for 2 h with an apoptogenic concentration of the PKA-directed cAMP analog N6-MB-cAMP (Figures 2a–d). As cAMP can affect mRNA expression via other transcription factors than CREB/CREM19 and affect mRNA stability independently of transcription,20 we expected ICER to block only a subset of the cAMP-induced effects on mRNA expression. This subset would be particularly relevant to search for candidate genes to mediate the cAMP-induced death as IPCICER cells are quite insensitive to cAMP-induced death (see9 and Figure 1a). ICER overexpression blunted most of the effects of cAMP on the IPC cell transcriptome, including the upregulation of Bim (BCL2L11) mRNA (Figure 2a). We conclude that most cAMP-induced regulations of IPC cell gene mRNA expression are mediated via the CRE/CREB system.

Gene expression analysis of IPC cells exposed to the PKA-activating cAMP analog N6-MB-cAMP. IPCWT or IPC cells stably over-expressing ICER (IPCICER) or Bcl2 (IPCBCL2), were exposed for 2 h to the PKA activator N6-MB-cAMP (700 μM). The transcriptome was analyzed by Affymetrix gene array (U230A, 15 866 probe sets). (a) The 82 transcripts increased (red) or decreased (blue) >2-fold in either IPCWT or IPCICER or IPCBCL2 are shown in a hierarchical clustering diagram. Genes in green have known cAMP responsive elements (CRE) in the promoter region from −3000 to +300 bases from the transcriptional start site. (b and c) The Venn diagram circles show the number and overlap of transcripts upregulated (b) or downregulated (c) >2 fold by N6-MB-cAMP in IPCWT (grey), IPCICER (cyan), and IPCBCL2 (pink) cells. (d) The panel shows that the fold regulation by N6-MB-cAMP in IPCWT cells correlates well (r2=0.866) with that in IPCBCL2 cells (Δ), but poorly (r2=0.287) with that in the IPCICER cells (O). The dotted line represents perfect correlation. Note that only transcripts regulated more than 2-fold are included. (e) Biological processes over-represented among the N6-MB-cAMP regulated genes according to the http://www.pantherdb.org tool are shown. Two processes had significantly fewer and seven had more regulated genes than the average. The P-values ranged from 0.003* (intracellular protein transport) to 0.047 (protein metabolic process). The number of regulated genes for each process is shown above each bar (see Supplementary Table S5 for details)
That Bim is regulated by cAMP via the CRE/CREB system was unexpected. The rat Bim promoter contains CRE-sites able to recruit CREB,21 but as they are TATA-less,22 the bound CREB is not supposed to recruit the cofactors necessary for cAMP/CREB-induced transcription17 (see also Supplementary Section V). In general, the IPC cells obey the rule that a TATA-box is required in the proximity of a CRE element to allow CREB-dependent regulation via cAMP. Thus, the TATA-less DNA-damage inducible transcript 4 (Ddit4) was induced equally by cAMP in wild-type and ICER-overexpressing cells (Figure 2a; Supplementary Table S4).
In IPCBCL2 cells, cAMP induces maturation/differentiation rather than death (see23 and Figure 1a). We used them therefore to exclude that any of the cAMP-induced effects on transcript expression in IPCWT cells were secondary to events linked to the apoptotic process. We found similar cAMP regulation of the transcriptome of IPCWT and of IPCBCL2 cells, also for genes considered to be potentially pro-apoptotic or involved in development and cell differentiation (Figures 2a–d). In conclusion, cAMP regulates IPC cell gene expression mainly through CRE-dependent transcription, unaffected by Bcl2 overexpression.
Five genes (JAK2, CEBPB, MADD, cMyc, Bim/BCL2L11) with cAMP-upregulated transcripts were classified (Figure 2e) as potential apoptosis inducers (Supplementary Table S5; panther database). Of these, Bim showed the strongest upregulation. Closer study revealed seven Bim transcript variants in the cAMP-stimulated IPC cells (Figure 3). In addition to the previously described isoforms BimEL, BimL and BimS and Bimβ4,24 we identified novel (η) variants of BimEL, BimL, and BimS, characterized by an extra exon (Figure 3b). The novel transcript variants were expressed at 0.1–10% the level of their previously described counterparts. The induction by cAMP based on qRT-PCR was robust (about 30-fold) and similar for both the novel and known Bim transcripts (Figure 3d). That cAMP induced all gene variants suggested that cAMP had little effect on the Bim splicing pattern.

Several Bim isoforms are expressed in cAMP-stimulated IPC cells. (a) Overview of the Bim gene exons (size in base pairs) with localization of the BH3-domain, start and stop translational codons, sites targeted by the Affymetrix probe sets, and the epitope of the anti-Bim antibody used. (b) Scheme of the Bim mRNA forms detected in IPC cells exposed to 8-CPT-cAMP (100 μM) for 2 h. The known major isoforms (BimEL/L/S), Bim β4, the novel variant η-BimL with an additional exon downstream of the BH3 encoding exon, were sequenced from amplified cDNA. Two additional variants (η-BimEL and η-BimS) were detected by RT-PCR (see Supplementary Table S7a and Materials and Methods for details). The predicted MW (right column) is shown for each variant. (c) The level of the six BH3-containing Bim transcript variants was estimated by qRT-PCR, using specific primer pairs. The fold induction is shown after 3 h incubation with 8-CPT-cAMP (200 μM), when the expression relative to the BimL transcript reached 65% for BimEL and for BimS, and 5, 11, and 0.1% for the novel variants η-BimEL, η-BimL, and η-BimS, respectively. The results are the mean of three independent experiments±S.E.M. (d) The fold change in Bim-transcript after 2 h incubation with N6-MB-cAMP, based on hybridization to the three microarray probe sets (color code as in panel a). Note the blunted response to N6-MB-cAMP in the IPCICER, but not the IPCBCL2 cells
The endogenous Bim expression is tightly coupled to death in cAMP-stimulated IPC cells
The CDK inhibitor RCV nearly abolished the induction of Bim mRNA, indicating that Bim mRNA transcription involves a CDK-dependent step. The Bim mRNA expression appeared to be independent of new protein synthesis as it was unaffected by cycloheximide (CHX) (Figure 4a).

The cAMP-induced apoptosis correlates closely with Bim protein expression. (a) The Bim-mRNA expression in IPCWT cells exposed for 0, 0.5, 1, 2, or 5 h to 200 μM 8-CPT-cAMP was determined by qRT-PCR, using primers covering all detected isoforms (see Figure 3b and Supplementary Table S7a). Co-incubation with the protein synthesis inhibitor CHX (36 μM) had only marginal effect on the transcript expression, whereas the CDK inhibitor RCV (15 μM) prevented the induction of Bim-mRNA. (b) The inset shows an immune-blot probed with anti-pan-Bim antibody of extracts from IPC cells treated for 4 h with 8-CPT-cAMP (200 μM) with no added RCV (−) or 15 μM RCV added simultaneously with cAMP analog (0), or 1,2 or 3 h thereafter. The numbers on the left correspond to MW of standard proteins. Actin was used as loading control. The main part of the panel shows the relative expression of BimEL after 4 h incubation with 8-CPT-cAMP, as a function of time after RCV addition (Δ), and its correlation with the % apoptotic cells, as determined in parallel experiments where the incubations were continued until 6 h after 8-CPT-cAMP was added (•). (c) Panel c is similar to panel b except that CHX (36 μM) rather than RCV was added. The data for all panels show average values±S.E.M. from 3–7 independent experiments. A close correlation is noted between apoptosis and BimEL expression (r2=0.95 for panel b and 0.97 for panel c)
As the cAMP-induced IPC cell death is blocked by both CHX and RCV,3, 6 these findings suggested that Bim induction might be important for the death. If so, a correlation should exist between cell death and Bim protein expression, whether manipulated by interference with Bim-mRNA synthesis (RCV) or Bim-mRNA translation (CHX). We achieved a graded expression of Bim protein (see insets and right hand axes of Figures 4b and c) by adding RCV (Figure 4b) or CHX (Figure 4c) at successive time points after the addition of cAMP. In either case, Bim protein expression correlated tightly with apoptosis, as determined 6 h after the onset of cAMP stimulation.
To provide a more direct link between cAMP-induced apoptosis and Bim transcript induction, we knocked down the Bim transcript. The restriction enzyme-based method used to achieve IPC cells with knocked-down Cα (Figure 1b) was not feasible owing to lack of suitable restriction enzyme sites in the Bim-cDNA. We therefore designed synthetic RNAi hairpins to target the part of the Bim transcript coding for the common N-terminal part of all the detected Bim isoforms. The hairpin DNA was inserted into vectors to produce retrovirus and stably transduce IPCWT cells. Two of the selected clones were compared with a clone expressing non-target (luciferase) hairpin RNAi for apoptosis induction after incubation with 8-CPT-cAMP. The hairpin-transduced cells were compared with non-target RNAi-transduced cells for ability to develop apoptosis in response to the cAMP analog 8-CPT-cAMP. The hairpin1 transduced cells showed very little apoptosis (Figures 5a and c) and low expression of Bim (Figure 5c, inset). Hairpin 2 gave less-efficient knockdown of Bim (Figure 5d) and protection against apoptosis (Figures 5a and d), but still provided significant protection as compared with the vector control (Figure 5b).

Bim-shRNAi protects against cAMP-induced apoptosis, whereas ectopic BimL expression induces apoptosis. (a) IPC cells were retrovirally transfected for stable expression using two different constructs against Bim (Bim-shRNA1 or Bim-shRNA2). The non-target control was shRNAi against luciferase (LUC-shRNA). After selection, the cells were exposed to various concentrations of 8-CPT-cAMP (6 h) and % apoptotic cells determined. (b) IPCLUC−shRNA cells were treated with 80 μM 8-CPT-cAMP for various periods of time and % apoptotic cells determined. The inset shows an immune-blot as for Figures 4b and c. The position of BimEL and BimL is indicated. (c and d) IPCBim−shRNA1 (c) or IPCBim−shRNA2 (d) cells were treated and analyzed for apoptosis as for panel (b). In addition, the level of Bim was assayed by immunoblots (insets) of extracts from cells exposed to 8-CPT-cAMP for 1, 1.5 or 2 h. (e) IPCWT cells were infected with retrovirus for bicistronic expression of BimL and GFP or for expression of GFP alone (pMIG). Only the BimL-transduced cells showed apoptosis 18 h after infection. (f) IPCBCL2 cells similarly infected and scored for apoptosis showed less, but significant apoptosis. The dying IPCBCL2 cells infected with vector for bicistronic expression of BimL and GFP showed GFP expression (left inset)
In conclusion, an excellent correlation is observed between Bim expression and apoptosis in cAMP-stimulated IPC cells, whether the Bim expression is manipulated by RCV, CHX or various Bim-directed shRNAi hairpins.
Enforced BimL expression induces IPC apoptosis independently of cAMP and more strongly in IPCWT than IPCBCL2 cells
In order to test whether Bim expression alone is sufficient to induce apoptosis in the IPCWT cells, they were transfected retrovirally with a bicistronic vector for BimL and GFP (BimL). Cells transfected with vector coding only for GFP served as control (pMIG). GFP-positive IPCWT and IPCBCL2 cells were observed 18 h after virus transduction with control vector. About 50% of the cells were detectably green 36 h after the transfection (not shown). No GFP positive cells were apparent after transduction with vector for with BimL+GFP. Nevertheless, a high proportion of the cells were apoptotic (Figure 5e). A likely explanation is that the retroviral-driven BimL expression induces apoptosis too rapidly for the cells to synthesize detectable GFP before undergoing death. The death morphology was classically apoptotic (Figure 5e, left inset), and strikingly similar to that induced by cAMP (Figure 1a).
The IPCBCL2 cells subjected to BimL+GFP transduction showed much less overall apoptosis than the similarly transduced IPCWT cells (Figure 5f). Some of the apoptotic cells were GFP positive (Figure 5f, left inset). This suggests that some of the BimL-transfected IPCBCL2 cells underwent apoptosis slowly enough to synthesize a detectable amount of GFP before dying. We conclude that ectopically expressed BimL is a highly efficient inducer of IPC cell apoptosis, whose effect is counteracted, but not completely abolished, by Bcl2 overexpression.
Cyclic AMP and DNR induce IPC cell death synergistically via distinct mechanisms
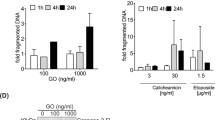
Cyclic AMP induces early mitochondrial damage in IPC cells.25 It resembles in this respect the intrinsic death induced by common chemotherapeutic drugs like DNR.26 In spite of this similarity DNR does not affect Bim protein expression (Figure 6a). This may explain why DNR-induced death is independent on CDK activity (Supplementary Figure S1d) and insensitive to ICER overexpression (Supplementary Figure S3). Another distinction was that the effect of DNR, but not cAMP, was abrogated by inhibitors of the generally pro-apoptotic GSK3β protein kinase (Supplementary Figures S1b and c).

The cAMP-induced IPC cell apoptosis differs from DNR-induced apoptosis with respect to Bim expression and role of HSP 90 modulators. (a) Extracts from IPC cells treated with vehicle for 2 h (left Ctr) or 6 h (right hand Ctr), with 8-CPT-cAMP (200 μM) for 2 h, 4 h, or 6 h, or with 0.4 μM DNR for 6 h were probed for Bim, HSP90 cochaperone p23 and actin by immunoblotting. Note that p23 cleavage is unaccompanied by Bim induction of the DNR-exposed cells. (b) Apoptosis was scored for IPC cells exposed to 0.5 μM GA alone or together with 8-CPT-cAMP (200 μM). (c) IPC cells retrovirally transfected with CRU5-IRES-GFP (empty vector), CRU5-IRES-GFP-p23 D142/145A (p23 D142/145A) or CRU5-IRES-GFP-p23 1–142 aa (Δp23) were treated with 8-CPT-cAMP (200 μM; 6 h) and scored for apoptosis. The 3–5% apoptosis in vehicle-treated cells is subtracted. (d) Cell lysates from cells transfected as for panel c were treated with vehicle (Ctrl) or DNR (0.4 μM; 6 h), and analyzed for p23, HSP90, and actin expression by immunoblotting. Note that cells transduced with Δp231–142 expressed Δp23 also before challenge with DNR. (e) IPC cells were treated with DNR (0.4 μM) in the presence or absence of GA (500 μM) and apoptosis scored. (f) The clones described in (c) were treated with DNR (0.4 μM; 6 h) and scored for apoptosis. Bars represent mean±S.E.M., n=3–8, and P-values are estimated using student t-test
The ansamycin antibiotic geldanamycin (GA) antagonizes HSP90 by disrupting its association with p23.27 The p23 function is also compromised by caspase-mediated p23 truncation.28 We show that DNR and cAMP-stimulation both lead to p23 truncation (Figure 6a). On the other hand, GA only enhances the DNR-induced IPC cell death (Figures 6b and e). This questioned the importance of the observed p23 cleavage for cAMP-induced death. To probe its importance, we prepared retroviral stable expression vectors for truncated p23 (Δp23), mimicking the endogenously formed p23 cleavage product, as well as a caspase cleavage-resistant (p23D142/145A) p23 (Figure 6d). We found no significant difference in cAMP-sensitivity between mock-transfected cells and cells expressing caspase-resistant p23 or Δp23 (Figure 6c). In contrast, the DNR-induced apoptosis was significantly higher in the Δp23-transduced cells than in the p23D142/145A-transduced cells (Figure 6f). That p23 cleavage can sensitize cells to DNR was supported further by experiments on HEK293T cells (Supplementary Figure S2).
We conclude that the cAMP- and DNR-induced cell death differs fundamentally regarding both Bim induction and effect of inhibition of CDK, GSK3ß and HSP90/p23 activity. This suggests that DNR-resistant AML cells may have alternative death pathways that can be exploited therapeutically. In fact, cAMP and DNR synergized for IPC cell death induction (Figure 7). A considerable decrease of the cAMP analog dose required to induce apoptosis was achieved by using PKA-I synergistic cAMP analog pairs instead of single analogs (Figure 1). The use of analog pairs should therefore allow lessening of the cAMP analog load for potential clinical use. A further decrease should be achieved by acetoxymethyl-ester prodrugs of the analogs, which have improved cell permeability.29

Synergism between death induced by anthracycline and cAMP. IPCWT cells were preincubated for 2 h in the absence or presence of N6-MB-cAMP at a sub-apoptogenic concentration (175 μM) (Figure 1e). Thereafter the cells were added vehicle (0) or DNR to 0.1, 0.2, 0.3 or 0.4 μM concentrations. 3.5 h thereafter, the cells were fixed and assessed for apoptosis. Note the strikingly enhanced effect of DNR for cells pre/co-incubated with the cAMP analog. Data represent mean±S.E.M. (n=4)
Discussion
Cyclic AMP signaling has versatile roles in hematopoiesis. They range from stimulation of proliferation to maturation and cell death.30, 31 The presently studied IPC cell line reflects this versatility and places PKA as the pivotal cAMP mediator. PKA appears to have a delicate role in balancing the IPC cell proliferation. Cells expressing <40% of the C subunit of PKA or being treated with PKA inhibitor (Rp-8-Br-cAMPS) are growth arrested. The high basal (resting state) cAMP level in IPC cells32 may therefore be an adaption required to activate the PKA enough to sustain proliferation. On the other hand, strong PKA activators arrest the cells at the G1/S restriction point.3
Although the basal cAMP level is sufficient to sustain proliferation, it appears not to induce net CRE-dependent IPC cell transcription. Thus, the basal state IPC transcriptome is similar in wild-type cells and cells overexpressing the CRE-blocking ICER protein (not shown). In contrast, the gene expression is profoundly altered in ICER-transfected HL-60 cells.11 It is also puzzling in view of the notion of CREB as an AML oncogene and marker for therapy resistance and ICER as a tumor suppressor.11, 13 The lack of CRE-dependent transcription in the basal state is not due to deficient CREB response. A number of transcripts are induced in an ICER-inhibitable manner in cells incubated with a PKA-directed cAMP analog. It may rather be related to the nature of the induced transcripts. They code for proteins involved in apoptosis or differentiation/development (Figure 1e), rather than growth and survival.
A near complete neutrophilic maturation can be achieved by cAMP stimulation of IPC cells stably overexpressing Bcl2.23 Bcl2 can enhance maturation by directly affecting gene expression, as shown for the hematopoietic differentiation of murine embryonic stem cells.33 We find similar cAMP-induced transcriptomes of IPCWT and IPCBCL2 cells. Bcl2 may therefore promote differentiation indirectly, by simply allowing the cAMP-induced maturation program to progress without being overwhelmed by apoptosis, as in the IPCWT cells.
The most striking effect of IPCWT cell PKA activation is apoptosis induction, which occurs much more rapidly than in the S49 thymocyte cell line, which also depends on Bim-induction.34 We found that Bim protein expression correlates closely with apoptosis. This is true whether Bim expression is manipulated by the CDK inhibitor (RCV), Bim-directed shRNAi, or by protein synthesis inhibitor (CHX). Bim is a plausible death inducer. It elicits intrinsic cell death by neutralizing the anti-apoptotic effects of Bcl2, Bcl-XL and Mcl-1, as well as through more direct pathways35, 36 (see also Figure 8). In fact, the forced expression of BimL led to rapid death of IPCWT cells and more attenuated death of IPCBCL2 cells (Figure 5). A high proportion of the cAMP-induced Bim protein was of the BimEL variant, which is a substrate for PKA.37 It is therefore possible that cAMP may contribute to IPC cell death also by activating phosphorylation37 of the already induced BimEL protein. The role of Bim may reflect that the IPC cells have retained death pathway components operating in normal myeloid-derived cells. Bim is a negative regulator of normal hematopoesis35 and accelerates the demise of mature neutrophilic granulocytes.38 It appears therefore that the IPC cells have retained normal bone marrow properties like pleiotropic response to cAMP and Bim-dependent death induction.

Overview of the pathway(s) incriminated in cAMP-induced IPC cell apoptosis. (1) The binding of cAMP to both sites on the regulatory subunits of the PKA holoenzyme leads to release of free, active catalytic subunits. (2) Catalytic PKA subunit translocated to the nucleus can phosphorylate the transcription regulatory proteins of the CREB family (CREB, CREM, ATFs), which bind to available CREs in the promoter region of CREB-regulated genes. The Bim-promoter contains four half CRE sites, two of which is conserved and separated by only 13 bp about 500 bp upstream from the transcription start site. The CREM isoform ICER blocks CRE sites and thereby the action of all CRE-binding transcription factors. Phosphorylated CREB recruits the basal transcriptional machinery via CREB-binding protein (CBP/P300) and other co-factors and adaptors. (3) Transcript elongation is stimulated by phosphorylations of the C-terminal domain of RNA polymerase II by members of the CDK family, especially CDK7 and 9, which are sensitive to inhibition by RCV.39 (4) The translation of Bim-mRNA is inhibited by CHX. (5 and 6) The Bim protein can complex Bcl2 and thereby block Bcl2 prosurvival functions. Bim may (dotted line) also induce apoptosis by stimulating BAX/BAK directly. The dotted arrows from PKA to (5 and 6) is based on the observation that cAMP can synergize with DNR to induce apoptosis in IPCICER cells (Supplementary Figure S3), which do not die in response to cAMP alone (Figure 1a). The dotted arrows from RCV to (5 and 6) are based on the moderate inhibition of cAMP-induced IPC cell death by dominant negative CDK5,6 which might be through inhibition of death processes downstream of Bim mRNA transcription
The induction of Bim by cAMP has some unusual features. It occurs very rapidly, is mediated via CRE/CREB, and depends completely on CDK activity (Figures 2,3,4). The mediation by CRE/CREB is puzzling as the conserved CRE's of the Bim promoter are TATA-less and therefore not expected to be regulated by CREB.17 Antagonists of CDK7,9, like RCV, target the elongation of a subset of gene transcripts.39 The impact of CDK activity on Bim-expression appears not to have been explicitly studied, but is probably not universal. Thus, RCV causes depletion of anti-apoptotic proteins like Mcl-1 in neutrophilic granulocytes, but spares Bim,40 nevertheless our findings suggest some caution in using RCV in tumors depending on Bim expression for eradication.
The strong IPC cell response to anthracyclines is unsurprising in view of their origin from the transplantable BNML model of AML, considered a reliable predictor of the clinical efficacy of antileukemic drugs.5 The IPCBCL2 cells are protected both against cAMP and the first-line anti-leukemic drug DNR. Furthermore, both death inducers promote the cleavage of the HSP90 cochaperone p23 (Figure 6). In spite of these similarities, the cAMP and DNR-induced death did not proceed by a basically common pathway. Firstly, the p23 cleavage has consequence in DNR-induced death only (Figures 6b and e). Although the enforced expression of cleavage-resistant p23 protected and truncated p23 promoted DNR-induced death, they failed to affect cAMP-induced death (Figures 6c and f; Supplementary Figure S2). Similarly, the HSP90/p23-targeting drug GA only promoted the DNR-induced death (Figures 6b and e). Secondly, Bim is induced uniquely in cAMP-induced death (Figure 6a). Thirdly, CDK antagonism inhibits only the cAMP-induced death, whereas GSK3β antagonism only inhibits the DNR-induced death (Supplementary Figure S1).
In conclusion, the IPC cells demonstrate the co-existence of an extremely efficient PKA-CRE-CDK- and Bim-dependent pathway in addition to a p23- and GSK3β enhanced anthracycline-induced death pathway. Studies aimed at exploring the therapeutic usefulness of the components of the PKA-CRE-CDK-Bim-dependent pathway may be warranted. It is of interest that PKA-I selective cAMP analogs can induce IPC AML cell death and that Bim transcription can be mediated through CRE sites. The IPC cell sensitivity to inhibitors of CDK's and GSK3β (Supplementary Figure S1) may be exploited to test the in vivo efficacy of CDK and GSK3β activity modulators. Finally, the new observations regarding the control by cAMP of initiation and elongation of Bim transcript may spur new studies on the expression of this important gene.
Materials and Methods
Reagents and constructs
The cAMP analogs were from BioLog (Bremen, Germany). The new A and B-site-specific cAMP analogs and the paullone analogs for CDK5 and GSK3β inhibition are described in Supplementary Section IV. RCV, GA and DNR were obtained from Sigma (St. Louis, MO, USA). The pMIG-Bim (Addgene plasmid 8786, Cambridge, MA, USA) was kindly made available for the scientific community by Dr. SJ Korsmeyer. The vectors for Cα knockdown are described in Supplementary Section IV. To create expression vectors for shRNAi against Bim, HPLC-purified synthetic oligo DNA (see Supplementary Table S7b for sequences) were annealed and ligated into the Aar1 site of the retroviral shRNAi vector L087 (GenBank Acc: EU424173). Mutated and truncated versions of p2328 were subcloned into vectors for transient transfection of HEK293T cells and stable retroviral transfection of IPC cells (see Supplementary Section IV for details).
Cell culture and retroviral transfection
Wild-type (IPCWT), ICER- and Bcl2-overexpressing (IPCICER, IPCBCL2) clones of the IPC-81 leukemia cell line2, 9, 23 were obtained from Dr. M Lanotte, Hop St. Louis, Paris. They were grown in DMEM (Sigma) supplemented with 10% heat-inactivated horse serum (EuroClone, Milan, Italy), and kept in logarithmic growth phase. Before experiments, the cells were resuspended in fresh growth medium. The Phoenix retroviral producer cell line was a kind gift from Dr. J. Lorens. They were grown in DMEM with 10% fetal bovine serum (EuroClone) and pen/strep as described above. By splitting every 2nd or 3rd day, confluent cells were avoided. The day before transfection, cells were plated at a density of 150 000 cells/cm2. Transfection of phoenix cells was carried out by the calcium phosphate method. For a 10 cm2 well, 2 μg VSV-G plasmid together with 5 μg plasmid of interest was used. Viruses were harvested after 24 (Bim expression) or 48 h (p23 and shRNAi expression), and up-concentrated by centrifugation at 50 000 × g for 3 h, and resuspended in phosphate-buffered saline over night. IPC cells were infected in growth medium with protamine sulfate (5 mg/ml, Sigma) under centrifugation (1500 × g 90 min). For stable expression of Bim shRNAi, puromycin (Sigma) selection was started at 0.1 μg/ml 48 h after infection and gradually increased up to 1 μg/ml within 2 weeks.
Determination of apoptotic morphology and kinase activity
IPC cell apoptosis was assessed in formaldehyde-fixed (2% final concentration) cells. Apoptosis was assessed by phase contrast microscopy of the surface morphology, and fluorescence microscopy of the nuclear morphology after staining of DNA with bisbenzimide, Hoechst 33342 (Sigma). To determine cAMP-dependent protein kinase activity in IPC cells lysate, radioactive phosphate incorporation to kemptide was performed as previously described.32
Microarray analysis
IPC cell RNA was isolated with a combination of TRI reagent (Sigma) and GenElute columns (Sigma). The manufacturer's protocol for TRI reagent was followed but instead of precipitation of the RNA, the aqueous solution was mixed with ethanol (final 35%) and applied to GenElute silica columns and washed and eluted according to the GenElute protocol. The microarray analysis on U230A rat transcriptome chip from Affymetrix (Santa Clara, CA, USA) was according to standard procedures at the Norwegian Microarray consortium Oslo. The processing of the expression data, annotations and cAMP responsive elements in the promoter region is described in the Supplementary Section IV.
Cloning of Bim transcript variants
RNA from 8-CPT-cAMP-stimulated IPC cells was reverse transcribed and amplified by PCR with primers given in Supplementary Table S7a. The PCR product was ligated into the pcDNA5/FRT/V5-His TOPO vector (Invitrogen, Carlsbad, CA, USA), and cloned to E.coli DH5α. Plasmid from ampicillin-resistant clones were isolated and sequenced. The sequences were aligned to known Bim transcripts and the genomic Bim sequence.
Quantitative RT-PCR and western blot
For quantitative RT-PCR, RNA was isolated with TRI reagent (Sigma) according to the protocol from the manufacturer. 1 μg of RNA was reverse transcribed with revert-aid Mu-MLV transcriptase (Fermentas GmbH, St. Leon-Rot, Germany) and oligo-dT primers and a specific reverse primer for 18S ribosomal RNA (18S). qPCR were run with a Lightcycler 480 II (Roche, Basel, Switzerland) using iQ SYBR green supermix (BioRad, Richmond, CA, USA). cDNA corresponding to 25 ng RNA were used in each PCR reaction. Primer sequences are given in Supplementary Table S7a. Bim expression was quantified relative to untreated control and the expression was normalized to the average of succinate dehydrogenase complex subunit alpha and 18S ribosomal RNA.
SDS gel electrophoresis and western blots were done and band intensity evaluated essentially as described previously.28 The membrane were probed with either: rabbit polyclonal anti-Bim (Cat No 202000, EMD Biosciences, San Diego, CA, USA), mouse monoclonal anti-flag (M2, Sigma), anti-Hsp90 (SPA-830, Stressgen, Victoria, Canada), mouse monoclonal anti-actin (ab6276 Abcam, Cambridge, UK) or mouse monoclonal anti-p23 (JJ3, generously supplied by Dr. David O Toft, Mayo Clinic, Rochester, MN, USA). Bands representing the proteins of interest were detected using alkaline-phosphatase-conjugated secondary antibodies (a-3687 and a-3562 Sigma) and CDP-Star substrate (Tropix, Bedford, MA, USA) and detected in a Fuji LAS3000.
Accession codes
Abbreviations
- 2-Cl-8-HA-cAMP:
-
2-chloro-8-hexylamino-adenosine-3′,5′-cyclic monophosphate
- 8-CPT-2′-O-Me-cAMP:
-
8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate
- 8-CPT-cAMP:
-
8-(4-chlorophenylthio)adenosine-3′,5′-cyclic monophosphate
- AML:
-
acute myelogenic leukemia
- ATF1:
-
activating transcription factor 1
- Bcl-2:
-
B-cell lymhoma 2 oncogene protein
- Bim:
-
Bcl-2 interacting mediator of cell death (BCL2L11)
- CDK:
-
cyclin dependent protein kinase
- CHX:
-
cycloheximide
- CRE:
-
cAMP responsive element
- CREB:
-
CRE binding protein
- CREM:
-
CRE modulator
- Epac:
-
exchange factor activated by cAMP (Rap GEF 3,4)
- GA:
-
geldanamycin
- GFP:
-
green fluorescent protein
- GSK3β:
-
glycogen synthase kinase 3β
- ICER:
-
inducible CRE early repressor
- IPC-81:
-
Rat AML cell clone (Institut de Pathologie Cellulaire 81)
- N6-Bnz-8-Pip-cAMP:
-
N6-benzoyl-8-piperdinoadenosine-3′,5′-cyclic monophosphate
- N6−MB-cAMP:
-
N6-monobutyryladenosine-3′,5′-cyclic monophosphate
- PKA-I/II:
-
cAMP-dependent protein kinase (Protein kinase A) type I/II
- RCV:
-
roscovitine
References
Gloerich M, Bos JL . Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 2010; 50: 355–375.
Wiley JM, Yeager AM, Johnson RJ, Lanotte M . A subline of the Brown Norway myeloid leukemia in the Lewis x Brown Norway rat: in vivo growth characteristics and development of an in vitro clonogenic assay. Int J Cell Cloning 1989; 7: 292–302.
Gjertsen BT, Cressey LI, Ruchaud S, Houge G, Lanotte M, Døskeland SO . Multiple apoptotic death types triggered through activation of separate pathways by cAMP and inhibitors of protein phosphatases in one (IPC leukemia) cell line. J Cell Sci 1994; 107: 3363–3377.
Gausdal G, Gjertsen BT, McCormack E, Van Damme P, Hovland R, Krakstad C et al. Abolition of stress-induced protein synthesis sensitizes leukemia cells to anthracycline-induced death. Blood 2008; 111: 2866–2877.
McCormack E, Bruserud O, Gjertsen BT . Animal models of acute myelogenous leukaemia - development, application and future perspectives. Leukemia 2005; 19: 687–706.
Sandal T, Stapnes C, Kleivdal H, Hedin L, Doskeland SO . A novel, extraneuronal role for cyclin-dependent protein kinase 5 (CDK5): modulation of cAMP-induced apoptosis in rat leukemia cells. J Biol Chem 2002; 277: 20783–20793.
McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer 2002; 102: 463–468.
Hallaert DYH, Spijker R, Jak M, Derks IAM, Alves NL, Wensveen FM et al. Crosstalk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual exhaustion of Bcl-2 protection. Cell Death Differ 2007; 14: 1958–1967.
Ruchaud S, Seite P, Foulkes NS, Sassone-Corsi P, Lanotte M . The transcriptional repressor ICER and cAMP-induced programmed cell death. Oncogene 1997; 15: 827–836.
Sassone-Corsi P . Coupling gene expression to cAMP signalling: role of CREB and CREM. Int J Biochem Cell Biol 1998; 30: 27–38.
Pigazzi M, Manara E, Baron E, Basso G . ICER expression inhibits leukemia phenotype and controls tumor progression. Leukemia 2008; 22: 2217–2225.
Pigazzi M, Manara E, Beghin A, Baron E, Tregnago C, Basso G . ICER evokes Dusp1-p38 pathway enhancing chemotherapy sensitivity in myeloid leukemia. Clin Cancer Res 2011; 17: 742–752.
Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell 2005; 7: 351–362.
Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK et al. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 2003; 278: 35394–35402.
Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang X-B, Christensen AE et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods 2008; 5: 277–278.
Gjertsen BT, Mellgren G, Otten A, Maronde E, Genieser HG, Jastorff B et al. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem 1995; 270: 20599–20607.
Conkright MD, Guzmán E, Flechner L, Su AI, Hogenesch JB, Montminy M . Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell 2003; 11: 1101–1108.
Altarejos JY, Montminy M . CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 2011; 12: 141–151.
Kuiperij HB, van der Horst A, Raaijmakers J, Weijzen S, Medema RH, Bos JL et al. Activation of FoxO transcription factors contributes to the antiproliferative effect of cAMP. Oncogene 2005; 24: 2087–2095.
Duan H, Cherradi N, Feige J-J, Jefcoate C . cAMP-dependent posttranscriptional regulation of steroidogenic acute regulatory (STAR) protein by the zinc finger protein ZFP36L1/TIS11b. Mol Endocrinol 2009; 23: 497–509.
Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM et al. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 2004; 119: 1041–1054.
Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P et al. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome 2001; 12: 163–168.
Seite P, Ruchaud S, Hillion J, Gendron MC, Bruland O, Segal-Bendirdjian E et al. Ectopic expression of Bcl-2 switches over nuclear signalling for cAMP-induced apoptosis to granulocytic differentiation. Cell Death Differ 2000; 7: 1081–1089.
Adachi M, Zhao X, Imai K . Nomenclature of dynein light chain-linked BH3-only protein Bim isoforms. Cell Death Differ 2004; 12: 192–193.
Tronstad KJ, Gjertsen BT, Krakstad C, Berge K, Brustugun OT, Døskeland SO et al. Mitochondrial-targeted fatty acid analog induces apoptosis with selective loss of mitochondrial glutathione in promyelocytic leukemia cells. Chem Biol 2003; 10: 609–618.
Laurent G, Jaffrézou JP . Signaling pathways activated by daunorubicin. Blood 2001; 98: 913–924.
Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem 1997; 272: 23843–23850.
Gausdal G, Gjertsen BT, Fladmark KE, Demol H, Vandekerckhove J, Døskeland S-O . Caspase-dependent, geldanamycin-enhanced cleavage of co-chaperone p23 in leukemic apoptosis. Leukemia 2004; 18: 1989–1996.
Bartsch M, Zorn-Kruppa M, Kühl N, Genieser H-G, Schwede F, Jastorff B . Bioactivatable, membrane-permeant analogs of cyclic nucleotides as biological tools for growth control of C6 glioma cells. Biol Chem 2003; 384: 1321–1326.
Lanotte M, Hermouet S, Gombaud-Saintonge G, Dobo I . On growth regulation of the rat promyelocytic leukemia (BNML): growth inhibition and eradication of clonogenic cells by cholera toxin. Leuk Res 1986; 10: 1319–1326.
Kobsar A, Heeg S, Krohne K, Opitz A, Walter U, Böck M et al. Cyclic nucleotide-regulated proliferation and differentiation vary in human hematopoietic progenitor cells derived from healthy persons, tumor patients, and chronic myelocytic leukemia patients. Stem Cells Dev 2008; 17: 81–91.
Duprez E, Gjertsen BT, Bernard O, Lanotte M, Døskeland SO . Antiapoptotic effect of heterozygously expressed mutant RI (Ala336—>Asp) subunit of cAMP kinase I in a rat leukemia cell line. J Biol Chem 1993; 268: 8332–8340.
Wang Y-Y, Deng X, Xu L, Gao F, Flagg T, May WS . Bcl2 enhances induced hematopoietic differentiation of murine embryonic stem cells. Exp Hematol 2008; 36: 128–139.
Zambon AC, Wilderman A, Ho A, Insel PA . Increased expression of the pro-apoptotic protein BIM, a mechanism for cAMP/protein kinase A (PKA)-induced apoptosis of immature T cells. J Biol Chem 2011; 286: 33260–33267.
Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F et al. Proapoptotic Bcl-2 relative bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999; 286: 1735–1738.
Kelly PN, Strasser A . The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 2011; 18: 1414–1424.
Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A et al. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep 2011; 12: 77–83.
Andina N, Conus S, Schneider EM, Fey MF, Simon HU . Induction of Bim limits cytokine-mediated prolonged survival of neutrophils. Cell Death Differ 2009; 16: 1248–1255.
Wesierska-Gadek J, Krystof V . Selective cyclin-dependent kinase inhibitors discriminating between cell cycle and transcriptional kinases: future reality or utopia? Ann NY Acad Sci 2009; 1171: 228–241.
Leitch AE, Riley NA, Sheldrake TA, Festa M, Fox S, Duffin R et al. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur J Immunol 2010; 40: 1127–1138.
Acknowledgements
We are grateful to Nina L Larsen and Anita Wergeland for expert help with cell culturing, to David Micklem and Kai O Skaftnesmo for help with RNAi strategies, to Ursula Havemann for help with synthesis of cAMP analogs, and to Jim Lorens for gifts of retroviral expression vectors. The work received support from the Norwegian Cancer Society, the Western Norway Regional Health Authority; Haukeland University Hospital, Bergen, Norway, and EU Project no. LSHB-CT-2004-503467.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors S Huseby, G Gausdal, TJ Keen, C Krakstad, L Myhren, E Kjærland, K Brønstad, C Kunick, R Kleppe, and S Døskeland declare no conflict of interest. H-G Genieser is CEO and F Shwede is Head of Research and Development of BIOLOG Life Science Institute, which sells cyclic nucleotide analogs for research purposes.
Additional information
Edited by D Bano
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huseby, S., Gausdal, G., Keen, T. et al. Cyclic AMP induces IPC leukemia cell apoptosis via CRE-and CDK-dependent Bim transcription. Cell Death Dis 2, e237 (2011). https://doi.org/10.1038/cddis.2011.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.124
Keywords
This article is cited by
-
Apoptosis-Inducing Active Protein from Marine Clam Donax variabilis on NSCLC Cells
International Journal of Peptide Research and Therapeutics (2021)
-
A drug–biomarker interaction model to predict the key targets of Scutellaria barbata D. Don in adverse-risk acute myeloid leukaemia
Molecular Diversity (2021)
-
Complex roles of cAMP–PKA–CREB signaling in cancer
Experimental Hematology & Oncology (2020)
-
Cyclic AMP can promote APL progression and protect myeloid leukemia cells against anthracycline-induced apoptosis
Cell Death & Disease (2013)
-
CREB-binding protein (CBP) regulates β-adrenoceptor (β-AR)−mediated apoptosis
Cell Death & Differentiation (2013)
