
Abstract
Caloric restriction and autophagy-inducing pharmacological agents can prolong lifespan in model organisms including mice, flies, and nematodes. In this study, we show that transgenic expression of Sirtuin-1 induces autophagy in human cells in vitro and in Caenorhabditis elegans in vivo. The knockdown or knockout of Sirtuin-1 prevented the induction of autophagy by resveratrol and by nutrient deprivation in human cells as well as by dietary restriction in C. elegans. Conversely, Sirtuin-1 was not required for the induction of autophagy by rapamycin or p53 inhibition, neither in human cells nor in C. elegans. The knockdown or pharmacological inhibition of Sirtuin-1 enhanced the vulnerability of human cells to metabolic stress, unless they were stimulated to undergo autophagy by treatment with rapamycin or p53 inhibition. Along similar lines, resveratrol and dietary restriction only prolonged the lifespan of autophagy-proficient nematodes, whereas these beneficial effects on longevity were abolished by the knockdown of the essential autophagic modulator Beclin-1. We conclude that autophagy is universally required for the lifespan-prolonging effects of caloric restriction and pharmacological Sirtuin-1 activators.
Similar content being viewed by others


Main
Macroautophagy (hereafter referred to as autophagy) is a phylogenetically conserved catabolic pathway that delivers cytoplasmic components, such as damaged organelles or long-lived proteins, to lysosomes for bulk degradation. Autophagy involves the formation of an isolation membrane (phagophore) that progressively enwraps portions of the cytoplasm until a double-membraned vesicle (autophagosome) is formed. Autophagosomes mature by fusing with lysosomes (thereby generating autophagolysosomes), followed by degradation of their luminal content by lysosomal hydrolases and export of freshly generated substrates into the cytosol for metabolic recycling.1
Although it has been initially thought that autophagy would contribute to cell death,2, 3 recent evidence suggests that autophagy is mostly a cytoprotective mechanism that allow cells to mobilize their energy reserves and to recycle damaged organelles in conditions of lacking nutrients,4 hypoxia,5 endoplasmic reticulum stress,6 accumulation of misfolded proteins,7 or DNA damage.8 Thus, when cells manifest massive autophagic vacuolization as they succumb to stressful conditions, they likewise do not die by autophagy but with (and even in spite of) autophagy, which has been activated as an ultimate tentative to cope with cellular damage.9 Indeed, in multiple adverse conditions including metabolic stress (inflicted for instance by a combination of nutrient depletion and hypoxia), autophagy ameliorates cellular fitness, meaning that pharmacological induction of autophagy improves cellular survival while its inhibition precipitates cell death.10, 11, 12
The notion that autophagy increases longevity is well illustrated by experiments in which the massive induction of autophagy by either pharmacological methods (such as the administration of rapamycin–Rapa–or spermidine) or genetic manipulations (like the knockdown the autophagy inhibitor p53) improves organismal survival and hence reduces age-associated mortality.13, 14 Caloric restriction (which is the most physiological inducer of autophagy), Rapa (which is the best characterized pharmacological inducer of autophagy), or knockdown of the p53 ortholog cep-1, all extend the lifespan of the nematode Caenorhabditis elegans only when the autophagic machinery is intact.13, 14 Thus, knockdown of AuTophaGy-related (atg) genes annihilates the longevity-enhancing effect of caloric restriction, Rapa, and p53 depletion.13, 14 The reduced activity of atg genes also suppresses the inherent lifespan extension that characterize mutant nematodes with congenital dietary restriction, aberrant insulin/insulin growth factor-1 or mammalian target of rapamycin (mTOR) signaling, and lowered mitochondrial respiration.15 Similarly, it has been found that the lifespan-extending properties of the natural polyamine spermidine strictly rely on the activation of the autophagic program in several model organisms including yeast, nematodes, and flies.16
Sirtuin-1 (Sirt-1) is a NAD+-dependent deacetylase whose transgenic overexpression can prolong lifespan in yeast, C. elegans, and flies.17 Sirt-1 (as well as SIR-2.1, its ortholog in C. elegans) can be activated by caloric restriction (presumably by an increase in intracellular NAD concentration), by depletion of its negative regulators (such as nicotinamide, the end product of Sirt-1-mediated NAD+ breakdown), or by pharmacological activators, in particular resveratrol (Resv).18 Sirt-1 has been shown to induce autophagy through the deacetylation of essential autophagic factors such as ATG5 and ATG7.19 On the basis of these findings, we investigated the molecular mechanisms through which Sirt-1 induces lifespan extension.
Results and Discussion
Sirt-1 increases autophagic flux
Transfection of human colorectal (HCT 116) or cervical (HeLa) cancer cells with a cDNA coding for Sirt-1 induced the accumulation of a co-transfected green fluorescent protein (GFP)-LC3 fusion protein into cytoplasmic puncta, which represent bona fide auto(phago)lysosomes (Figure 1a and b).20 Sirt-1 also promoted the conversion of LC3I to LC3II through proteolytic cleavage and lipidation (Figure 1c). Both morphological and biochemical manifestations of autophagy (GFP-LC3 puncta and LC3I → LC3II conversion, respectively) were prevented by the addition of the Sirt-1 inhibitor EX52721 (Figure 1a–c). Accumulation of GFP-LC3 puncta in response to Sirt-1 may derive from enhanced autophagic sequestration (on rate) or reduced autophagic degradation (off rate). To discriminate between these possibilities, we monitored Sirt-1-induced autophagic vacuolization while assessing the colocalization of GFP-LC3 with the lysosomal marker LAMP-2, either in the absence or in the presence of bafilomycin A1 (BafA1), which suppresses the fusion between autophagosomes and lysosomes4 (Figure 1d and e). Sirt-1 overexpression enhanced the formation of GFP-LC3 puncta even in the presence of BafA1 (Figure 1f), indicating that Sirt-1 stimulates the on rate of autophagy. Importantly, transgenic overexpression of sir-2.1 (the C. elegans ortholog of human sirtuin-1) also induced autophagy in nematodes, as indicated by the increased expression and cytoplasmic aggregation of a DsRed::lgg-1 reporter gene product (LGG-1 is the C. elegans ortholog of human LC3) (Figure 1g and h).

Sirtuin-1 (Sirt-1) induces autophagic vacuolization and has no effects on autophagosome–lysosome fusion. (a–c) Detection of autophagic vacuoles induced by Sirt-1 overexpression and modulation by EX527 in wild type (WT) HCT 116. Cells were co-transfected with a plasmid for the expression of GFP-LC3 together with an empty control vector (pcDNA3) or a Sirt-1-encoding plasmid for 24 h, and then cultured for 6 h in the absence or presence of 100 μM EX527. Representative microphotographs are reported in panel a, and the percentage of cells exhibiting the accumulation of GFP-LC3 in puncta (GFP-LC3vac) is reported in panel b (mean±S.E.M., n=3, *P<0.05). The abundance of Sirt-1, p53 acetylated on Lys382 (p53 Ac Lys382), total p53 as well as LC3 maturation was determined by immunoblotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were assessed to ensure equal loading of lanes. (c) Representative of three independent experiments. Numbers next to bands indicate MW (kDa). (d–f) The effect of bafilomycin A1 (BafA1) on Sirt-1-induced autophagic vacuolization. GFP-LC3-expressing HeLa cells were transfected with the pcDNA3 control vector or with a Sirt-1-encoding plasmid for 24 h, treated with 1 nM BafA1 for further 6 h and finally processed to assess the colocalization between GFP-LC3 and LAMP-2. (d) Representative confocal microphotographs and colocalization profiles within the area of interest (defined by the α → ω arrow). (e) Columns depict the percentage of colocalization between GFP-LC3 and LAMP-2, as quantified in at least 50 cells for each experimental condition (mean±S.E.M., n=3, *P<0.05). (f) The percentage of GFP-LC3vac cells (mean±S.E.M., n=3, *P<0.05; n.s, nonsignificant). (g, h). Autophagic effects of SIR2.1 overexpression in C. elegans. (g) Representative images of WT or SIR2.1-overexpressing (geIn3 genotype; see Materials and Methods) C. elegans cells exhibiting DsRed::LGG1 puncta. (h) Columns depict mean pixel intensity of DsRed::LGG1 (mean±S.E.M., n=3, *P< 0.05)
Next, we explored whether activation of endogenous Sirt-1 by pharmacological agents also induces autophagy. Resv, a pharmacological Sirt-1 activator that may have off-target effects,22 induced GFP-LC3 puncta (Figure 2a) and LC3I → LC3II conversion (Figure 2b) in human cancer cells. In order to confirm that Resv enhanced on-rate autophagic sequestration, we assessed Resv-induced autophagic vacuolization while determining the colocalization of GFP-LC3 with LAMP-2, either in the absence or in the presence of BafA1 (Figure 2c and d). BafA1 exacerbated the Resv-triggered accumulation of GFP-LC3 puncta (Figure 2e). Furthermore, the lysosomal protease inhibitor leupeptin23 increased Resv-induced LC3I → LC3II conversion (Figure 2f), in accordance with the hypothesis that Sirt-1 stimulates the on rate of autophagy. Resv-induced autophagy was fully prevented by pharmacological inhibition of Sirt-1 with EX527 or knockdown of Sirt-1 with a small-interfering RNA (siRNA) (Figure 2g and h), indicating that Resv-triggered autophagy entirely proceeds by the activation of endogenous Sirt-1. Resv also induced autophagy in C. elegans, and this effect was lost in sir-2.1 knockout nematodes (Figure 2i).

Resveratrol (Resv) triggers autophagic vacuolization and fails to inhibit the fusion of autophagosomes with lysosomes. (a) Detection of autophagic vacuoles induced by Resv and modulation by EX527 in wild-type (WT) HCT 116. Cells were transfected with a plasmid encoding GFP-LC3 for 24 h and cultured for additional 6 h in the absence or presence of 100 μM Resv and/or 100 μM EX527. The percentage of cells showing the accumulation of GFP-LC3 in cytoplasmic puncta (GFP-LC3vac) is reported (mean±S.E.M., n=3, *P<0.05). (b) The levels of Sirtuin-1 (Sirt-1), p53 acetylated on Lys382 (p53 Ac Lys382), total p53 as well as LC3 maturation were determined by immunoblotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) abundance was monitored to ensure equal loading of lanes. These data are representative of three independent experiments. Numbers next to bands illustrate MW (kDa). (c–e) Effect of bafilomycin A1 (BafA1) on Resv-induced autophagy. GFP-LC3-expressing HeLa cells were treated with Resv and/or BafA1 for 6 h (unless otherwise indicated) and then processed to assess the colocalization between GFP-LC3 and LAMP-2. Representative confocal microphotographs are shown in panel c, together with the colocalization profiles within the area of interest (defined by the α → ω arrow). (d) Columns illustrate the percentage of colocalization between GFP-LC3 and LAMP-2 (mean±S.E.M., *P<0.05), as quantified in at least 50 cells for each experimental condition. (e) The kinetics of GFP-LC3 redistribution was quantified by conventional fluorescence microscopy (mean±S.E.M., n=3, *P<0.05). (f) LC3 maturation in WT HCT 116 treated with Resv and/or leupeptin (Leu) for the indicated time. GAPDH levels were assessed to ensure equal loading of lanes. Data are representative of three independent experiments. Numbers next to bands indicate MW (kDa). (g, h) The effects of Sirt-1 knockdown on Resv-induced autophagy. WT HCT 116 cells transfected with the indicated siRNAs were re-transfected with a plasmid for the expression of GFP-LC3 for 24 h and then cultured in the presence or absence of 100 μM resveratrol for further 6 h. (g) Columns depict the percentage of GFP-LC3vac cells (mean±S.E.M., n=3, *P<0.05). The abundance of Sirt-1, p53 acetylated on Lys382 (Ac Lys382 p53), total p53, and LC3I/II was determined by immunoblotting. GAPDH levels are shown as control for the equal loading of lanes. (h) Representative of three independent experiments. Numbers next to bands depict MW (kDa). (i) Induction of autophagy by Resv and its dependence on SIRT-2.1 in C. elegans. The effects of 100 μg/ml Resv were assessed either on WT animals or on sir-2.1(ok434) deletion mutants expressing the DsRed::LGG-1 transgene. Columns depict mean pixel intensity of DsRed::LGG1 (mean±S.E.M., n=3, *P<0.05)
We conclude that transfection-enforced overexpression as well as pharmacological activation of Sirt-1 stimulates the autophagic flux, both in human and in nematode cells.
Sirt-1 is required for the induction of autophagy by nutrient deprivation or caloric restriction
Driven by the observation that the activation of Sirt-1 can induce autophagy, we determined whether any of the known autophagy-inducing pathways relies on Sirt-1. For this, we inhibited Sirt-1 by pharmacological (i.e., with EX527) or genetic (i.e., with a Sirt-1-specific siRNA) means in HCT 116 (Figure 3a and b) or HeLa cells (data not shown). Then, we examined whether the stimulation of autophagy by mTOR inhibition (with Rapa), p53 inhibition (with cyclic pifithrin-α), nutrient deprivation (by culture in serum-free Earle's balanced salt solution), and endoplasmic reticulum stress (induced by tunicamycin–Tun) depended on Sirt-1. As assessed by the quantification of GFP-LC3 puncta, only nutrient depletion-induced autophagy was reduced by the inhibition/depletion of Sirt-1 (Figure 3a and b). This result was confirmed in C. elegans, in which the loss-of-function mutation of sir-2.1 abrogated caloric restriction-induced autophagy but not the autophagic response to Rapa or Tun (Figure 3c).

Role of Sirtuin-1 (Sirt-1) in starvation-induced autophagy. (a, b) Sirt-1 requirement for the induction of autophagy by nutrient deprivation in cancer cells. Wild-type (WT) HCT 116 cells were transfected with a GFP-LC3-encoding construct for 24 h and then cultured for additional 6 h in nutrient-free (NF) conditions or treated with 1 μM rapamycin (Rapa), 2.5 μM tunicamycin (Tun), 30 μM cyclic pifithrin-α (Pif-α) in the presence or absence of 100 μM EX527 (a). (b) Alternatively, WT HCT 116 cells were transfected with control or a Sirt-1-depleting siRNA and subsequently with a plasmid for the expression of GFP-LC3 for 24 h, followed by culture in autophagy-inducing conditions as in a. The percentage of cells exhibiting the accumulation of GFP-LC3 in cytoplasmic puncta (GFP-LC3vac) is reported (mean±S.E.M., n=3, *P<0.05). (c) Requirement of the C. elegans Sirtuin-1 ortholog, SIR-2.1, for starvation-induced autophagy. Autophagy was assayed in WT animals and sir-2.1(ok434) deletion mutants expressing the DsRed::LGG-1 transgene, which were fed ad libitum (AL), grown under conditions of dietary restriction (DR), or treated with 2 μg/ml Tun or 1 μg/ml Rapa. Columns illustrate mean pixel intensity of DsRed::LGG1 (mean±S.E.M., n=3, *P<0.05). (d) WT and p53−/− HCT 116 cells were transfected with the indicated siRNAs, re-transfected with a GFP-LC3-encoding plasmid for 24 h and then cultured for further 6 h in control or starvation conditions. Columns depict the percentage of GFP-LC3vac cells (mean±S.E.M., n=3, *P<0.05). (e) Immunoprecipitation of endogenous p53 and lysine-acetylated proteins. HCT 116 cells of the indicated genotype were cultured in starvation conditions or treated with 70 nM trichostatin (TSA, positive control for acetylation) for 6 h, followed by immunoprecipitation of p53 or lysine-acetylated proteins and immunoblotting for the detection of p53 acetylated on Lys382 (Ac Lys382 p53), total p53, or ATG7. Alternatively, total proteins were assayed for ATG7 abundance and LC3 maturation. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were determined to monitor the equal loading of lanes. Results are representative of three independent experiments. Numbers next to bands indicate MW (kDa)
In compliance with the idea that Sirt-1 is specifically required for starvation-induced autophagy but not for the autophagic response triggered by the activation of downstream signals, we observed that the increased baseline levels of autophagy that characterize p53−/− HCT 116 (as compared with their wild-type, WT, counterparts) could not be reduced by knockdown of Sirt-1 (Figure 3d). Intriguingly, although Sirt-1 reportedly deacetylates p53,24 we found no signs of p53 deacetylation in cells overexpressing Sirt-1 (Figure 1c) nor in cells in which endogenous Sirt-1 was activated by Resv (Figure 2b) or by nutrient deprivation (Figure 3e). Rather, in starvation conditions, p53 tended to be hyperacetylated. In contrast, the activation of Sirt-1 by nutrient depletion correlated with ATG7 deacetylation (Figure 3e). In conclusion, it appears that starvation-induced autophagy relies on the activation of Sirt-1, which activates autophagy through a novel mechanism involving ATG7.
Sirt-1-dependent autophagy increases stress resistance in human cells
Metabolic stress (nutrient deprivation plus hypoxia for 48 h) is relatively well tolerated in cancer cell lines because of their capacity to activate the autophagic response.25 Consistent with this notion, we found that knockdown of Sirt-1 reduced the resistance of HCT 116 cells to metabolic stress. Thus, the collapse of the mitochondrial transmembrane potential (Δψm, detected with the Δψm-sensitive dye 3,3′-dihexyloxacarbocyanine iodide – DiOC6(3)), a sign of imminent cell death,26, 27 was more pronounced among metabolically stressed cells in which Sirt-1 was depleted (Figure 4a) or inhibited by pharmacological means (Figure 4b) than in control cells. Sirt-1 knockdown was as efficient as ATG7 depletion in sensitizing cells to death induction by metabolic stress (Figure 4a). The inhibition of Sirt-1 per se induced some extent of cell death (Figure 4a and b), suggesting that Sirt-1 may also participate in the maintenance of baseline autophagy levels. Rapa failed to protect HCT 116 cells from metabolic stress (Figure 4b), presumably because in these conditions mTOR was entirely silenced by the endogenous sensor of energy levels AMP-activated protein kinase.28 Conversely, the synthetic lethality of Sirt-1 inhibition plus metabolic stress (as well as the cytotoxic effect of EX527 alone, although to lower extents) was partially reversed by Rapa (Figure 4b), which can induce autophagy in conditions in which Sirt-1 is inactive (see above, Figure 3a and b). Although the precise molecular mechanisms underlying this phenomenon remain elusive, it cannot be excluded that mTOR signaling might be affected by Sirt-1 inhibition. Irrespective of these theoretical considerations, it appears that Sirt-1 increases the fitness of metabolically stressed cells by activating autophagy.

Sirtuin-1-dependent autophagy favors stress resistance in human cancer cells. (a, b) Metabolic stress-induced cell death is increased by the knockdown or inhibition of Sirtuin-1 (Sirt-1). Wild-type (WT) HCT 116 cells were transfected with the indicated siRNAs (a) or treated with 100 μM EX527 and/or 1 μM rapamycin (Rapa) (b) and subjected to metabolic stress (cultured in nutrient-free, hypoxic conditions) for 48 h. Cells were then stained with the mitochondrial transmembrane potential (Δψm)-sensitive dye DiOC6(3) and the vital dye propidium iodide (PI). Black and white columns illustrate the percentage (mean±S.E.M., n=3, *P<0.05) of DiOC6(3)low/PI− (dying) and DiOC6(3)low/PI+ (dead) cells, respectively
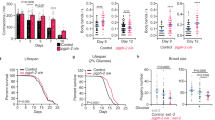
Next, we asked whether autophagy contributes to lifespan-extending effects mediated the C. elegans Sirt-1 ortholog SIR-2.1. Transgenic overexpression of sir-2.1 increased the median and maximum lifespan of nematodes compared with control strains.29 This gain in longevity was diminished by the knockdown of the essential autophagy gene bec-1, the worm ortholog of mammalian beclin-1 (Figure 5a). Transgenic overexpression of the pyrazinamidase/nicotinamidase PNC-1, which activates SIR-2.1 by depleting its negative regulator nicotinamide,30 also increased the lifespan of nematodes, an effect that was lost upon knockdown of sir-2.1 or bec-1 (Figure 5b). The addition of Resv to the culture medium has been previously shown to reduce the aging-associated mortality of C. elegans,31 and this beneficial effect was curtailed both by the knockdown of sirt-2.1 and by the knockdown of bec-1 (Figure 5c). Knockdown of the C. elegans p53 ortholog cep-1 (which is known to increase the longevity of nematodes by stimulating autophagy)14 failed to enhance the gain in longevity mediated by sir-2.1 overexpression (Figure 5d), in line with the hypothesis that p53 depletion and SIR-2.1 accumulation act through similar pathways to prolong survival.

Autophagy is required for the lifespan-extending effects of Sir-2.1 in C. elegans. (a) Longevity conferred by the overexpression of Sir-2.1 (geIn3 genotype) depends on Bec-1 expression (lifespan values: wild type, WT, control mean 14.6±0.7, max 26.0; geIn3 mean 19.5±1.1, max 33.0; bec-1(RNAi) mean 13.1±1.2, max 22.0; geIn3;bec-1(RNAi) mean 14.7±1.1, max 27.0). (b) Depletion of SIR-2.1 or Bec-1 suppresses longevity conferred by overexpression of the pyrazinamidase/nicotinamidase PNC-1 (lifespan values: WT control mean 14.6±0.7, max 26.0; Ex[pnc-1(+)] mean 18.1±1.0, max 29.0; Ex[pnc-1(+)];sir-2.1(ok434) mean 12.9±1.1, max 24.0; Ex[pnc-1(+)];bec-1(RNAi) mean 14.0±1.2, max 23.0). (c) Bec-1 deficiency suppresses longevity conferred by treatment with 100 μg/ml resveratrol (Resv) (lifespan values: WT control mean 14.6±0.7, max 26.0; WT grown on Resv mean 16.2±1.0, max 29.0; bec-1(RNAi) mean 13.1±1.2, max 22.0; bec-1(RNAi) grown on Resv mean 12.9±1.2, max 24.0). (d) Depletion of the C. elegans p53 ortholog Cep-1 does not enhance longevity conferred by Sir-2.1 overexpression (lifespan values: WT control mean 14.6±0.7, max 26.0; geIn3 mean 19.5±1.1, max 33.0; geIn3;cep-1(RNAi) mean 18.8±1.2, max 32.0). In all panels, the percentage of surviving nematodes is plotted against age (days)
In summary, activation of the Sirt-1 ortholog SIR-2.1 by three different methods (overexpression, pharmacological activation with Resv, and depletion of its negative regulator nicotinamide) extends lifespan through the induction of autophagy.
Concluding remarks
Recently, it has been discovered that drugs that induce autophagy such as Rapa and spermidine can prolong the lifespan of rodents.32, 33 Unfortunately, it has not yet been established whether these treatments increase the longevity (and health) of mice through their capacity to induce autophagy or whether they do so by additional effects. In contrast, autophagy is clearly required for the lifespan-extending effect of Rapa and spermidine in C. elegans and Drosophila melanogaster.13, 16 The results presented in this paper, which were obtained in C. elegans, suggest that autophagy is required for the lifespan-extending effects of Rapa as well as for the increase in longevity promoted by caloric restriction and Resv, an agent that has a broad anti-aging activity also in mice.34 Thus, our findings suggest that autophagy induction is a common requirement for lifespan-prolonging measures, while they underscore the importance of autophagy as a protective (rather than destructive) phenomenon.
Our results clearly indicate that caloric restriction/nutrient deprivation stimulates autophagy through the activation of Sirt-1, both in mammals and in nematodes. Sirt-1 functions as a metabolic sensor that detects the increase in NAD+ concentrations resulting from enhanced NADH oxidation. Once activated, Sirt-1 can deacetylate essential autophagic modulators such as ATG5 and ATG7,19 suggesting that a cytoplasmic pool of Sirt-1 may underlie its autophagy-stimulating effects. It remains elusive whether changes in the transcriptome mediated by nuclear Sirt-1 might also contribute to the induction of autophagy. As shown here, the activation of Sirt-1 (which also functions as a histone deacetylase),35 stimulates autophagy, while the inhibition of histone acetyltransferases by spermidine induces autophagy,16 suggesting that both pathways might converge on similar regulators/effectors of autophagy. The precise identity of these proteins remains to be established (Figure 6).

Schematic model of the role of Sirtuin-1 in autophagy. In human cells and C. elegans, Sirtuin-1 activation induced by caloric restriction, resveratrol or by the overexpression of its upstream activator pyrazinamidase/nicotinamidase (pcn-1) stimulates autophagy. This has lifespan-extending effects in nematodes, as do the knockdown of the C. elegans p53 ortholog Cep-1 and rapamycin administration, both of which trigger autophagy by Sirtuin-1-independent pathways
The mechanisms by which enhanced autophagy can improve organismal health and longevity are largely elusive. As a possibility, increased autophagy might improve cellular resistance to stress by augmenting the metabolic buffering capacity of cells. Alternatively, autophagy might enhance organellar turnover and mediate a ‘cleaning effect,’ thereby preventing the accumulation of damaged/old (and hence potentially harmful) mitochondria and lysosomes. Irrespective of these considerations, our data establish the cardinal role of Sirt-1-elicited autophagy in mediating the anti-aging effects of caloric restriction and Resv.
Materials and Methods
Cell lines and culture conditions
HCT 116 colon carcinoma cells (WT or p53−/−, a generous gift from B Vogelstein)36 were cultured in McCoy's 5A medium containing 10% fetal calf serum (FCS), 100 mg/l sodium pyruvate, 10 mM HEPES buffer, 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate (37 °C, 5% CO2). GFP-LC3-expressing HeLa cervical carcinoma cells were cultured in DMEM containing 10% FCS, 1 mM sodium pyruvate, and 10 mM HEPES buffer (37°C, 5% CO2). All media and supplements for cell culture were purchased from Gibco-Invitrogen (Carlsbad, CA, USA). For serum and amino acid deprivation, which we refer to as starvation conditions, cells were cultured in Earle's balanced salt solution (Sigma-Aldrich, St Louis, MO, USA). Cells were seeded in 6- or 12-well plates or in 10 cm dishes and grown for 24 h before treatment with 1 μM Rapa, 100 μM EX527, and 70 nM trichostatin (all from Tocris Bioscience, Ellisville, MO, USA); 30 μM cyclin pifithrin-α (Calbiochem, Nottingham, UK); and 100 μM Resv, 2.5 μM Tun, 1 nM BafA1, and 100 nM leupeptin (all from Sigma-Aldrich), which lasted 6 h unless otherwise stated.
Plasmids, transfection, and RNA interference in human cell cultures
Cells were cultured in six-well plates or 10 cm dishes and transfected at 50% confluence with siRNAs targeting human Sirt-1,37 ATG7 (Thermo Sceintific-Pierce, Whaltam, MA, USA) or with a control unrelated siRNA (Sigma-Aldrich) by means the Oligofectamine (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. siRNA-mediated protein downregulation was assessed by immunoblotting at 48 h after transfection. Transient plasmid transfections were performed with Lipofectamine 2000 (Invitrogen), as suggested by the manufacturer, and cells were used 24 h after transfection. Cells were transfected with the empty vector (pcDNA3) alone or together with a GFP-LC3-encoding plasmid,38 in the presence or the absence of a construct for the overexpression of WT Sirt-1 (Addgene, Cambridge, MA, USA).
Cytofluorometry
The following fluorochromes (Molecular Probes, Carlsbad, CA, USA) were used to determine apoptosis-associated changes by cytofluorometry: DiOC6(3) (40 nM), for measuring the Δψm, and propidium iodide (1 μg/ml), for quantifying plasma membrane integrity.39, 40, 41 Cells were trypsinized and labeled with fluorochromes at 37°C, as previously described,42 followed by analysis with a FACSVantage cytofluorometer (Becton Dickinson, San Jose, CA, USA).
(Immuno)fluorescence microscopy
For (immuno)fluorescence microscopy determinations, cells cultured on coverslips were fixed in paraformaldehyde (4%, wt/vol) for 30 min at room temperature. Cells were then stained with an antibody specific for LAMP-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), revealed by the appropriate Alexa Fluor conjugate (Molecular Probes). Nuclei were counterstained with 2 μM Hoechst 33342 (Molecular Probes). Fluorescence and confocal fluorescence images were captured using an IRE2 microscope equipped with a DC300F camera (both from Leica Microsystems GmbH, Wetzlar, Germany) and an LSM 510 microscope (Carl Zeiss, Jena, Germany) equipped with 63 × /1.15 objective (Olympus America Inc., Center Valley, PA, USA), respectively.43 To determine the percentage of colocalization, images were subjected to software-assisted analysis by means of the Java-based, open-source software ImageJ (freely available at http://rsb.info.nih.gov/ij/).
Immunoblotting and immunoprecipitation
For immunoblotting, cells were collected, washed with cold PBS and lysed as previously described.44 A total of 50 μg of proteins were then separated on 10% SDS-PAGE pre-cast gels (Invitrogen) and electrotransferred to Immobilon membranes (Millipore Corporation, Billerica, MA, USA), according to standard procedures.45, 46 Unspecific binding sites were saturated by incubating membranes for 1 h in 0.05% Tween 20 (in TBS) supplemented with 5% (wt/vol) non-fat powdered milk, followed by the overnight incubation with primary antibodies specific for acetylated lysines (Cell Signaling Technology Inc., Danvers, MA, USA), ATG7 (Sigma-Aldrich), LC3 (Cell Signaling Technology Inc.), Sirt-1 (Santa Cruz Biotechnology), p53 (DO-7; Santa Cruz Biotechnology), and p53 acetylated on Lys382 (Abcam, Cambridge, UK). Revelation was performed with the appropriate horseradish peroxidase (HRP)-labeled secondary antibodies (Southern Biotech, Birmingham, AL, USA) plus the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific-Pierce, Rockford, IL, USA). An anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Chemicon International, Temecula, CA, USA) was used to control equal loading of lanes. For immunoprecipitation, extracts from 8 × 106 WT HCT 116 cells were lysed and 500 μg of proteins were pre-cleaned for 1 h with 15 ml of Protein G Sepharose 4 Fast Flow (GE Healthcare, Piscataway, NJ, USA), followed by incubation for 2 h in the presence of an anti-p53 (DO-7; Santa Cruz Biotechnology) or an anti-acetylated lysine (Cell Signaling Technology Inc.) antibody. Subsequent immunoblotting was carried out by means of TrueBlot-HRP (eBioscience, San Diego, CA, USA) secondary antibodies.
Nematode strains and genetics
We followed standard procedures for C. elegans strain maintenance. Nematode rearing temperature was kept at 20°C. The following strains were used in this study—N2: WT Bristol isolate; LG100: geIn3 [sir-2.1(+)]; VC199: sir-2.1(ok434)IV; and TJ1: cep-1(gk138)I.
C. elegans molecular and cell biology
For engineering pnc-1 dsRNA-producing bacteria, a pnc-1-specific genomic fragment (∼600 bp) was cloned by PCR into the PCRII-TOPO vector (Invitrogen), excised as an EcoRI restriction fragment, and sub-cloned into the pL4440 vector for RNAi. The resulting plasmid was used to transform HT115(DE3) Escherichia coli cells, which lack the dsRNA-specific RNase III.47 Bacteria carrying an empty vector were used in control experiments. For pnc-1 overexpression, a fragment of 9 kb encompassing the pnc-1 coding region and upstream promoter sequences was amplified using the Pfu Ultra DNA polymerase (Stratagene, La Jolla, CA, USA) and co-injected together with the pmyo−2GFP injection marker into the gonads of WT nematodes. For monitoring autophagy, we used a full-length DsRED::LGG-1 fusion protein. LGG-1 is the nematode ortholog of yeast Atg8/Aut7p and of mammalian LC3, a protein involved in autophagosome formation. Construction of the plgg−1DsRED::LGG-1 reporter has been previously described.48 Transgenic embryos expressing DsRED were photographed on an Axioskop 2 Plus epifluorescence microscope (Carl Zeiss). Images were acquired using a 540±15 nm band-pass excitation filter and a 575-nm long-pass emission filter, as previously described.49 Experiments were performed at 20°C and exposure time was kept identical for each embryo. Emission intensity was measured on grayscale images with a pixel depth of 8 bit (256 shades of gray). Mean and maximum pixel intensity for each embryo was calculated from these images by means of the ImageJ software package. For each genotype, we processed at least 15 images over at least three independent trials.
C. elegans lifespan analysis
Lifespan assays were performed at 20°C. Synchronous animal populations were generated by hypochlorite treatment of gravid adults to obtain tightly synchronized embryos, which were allowed to develop into adulthood. For RNAi lifespan experiments, worms were placed on nematode growth media plates containing 1–2 mM isopropyl-β-D-thiogalactopyranoside and seeded with HT115(DE3) bacteria transformed with either the pL4440 empty vector or the test RNAi construct. Progeny was grown through the L4 larval stage and then transferred to fresh plates in groups of 10–20 worms per plate, for a total of 100–150 individuals per experiment. The day of egg harvesting and RNAi initiation was considered as t=0. Thereafter, animals were transferred to fresh plates every 2–4 days and examined daily for touch-provoked movement and pharyngeal pumping until death. Worms that died due to internally hatched eggs, an extruded gonad, or desiccation due to crawling on the edge of the plates were incorporated as such into the data set. Each survival assay was repeated at least three times.
Statistical analysis
Statistical analyses were carried out using the Prism software package (GraphPad Software Inc., San Diego, CA, USA) and the Microsoft Office 2003 Excel software package (Microsoft Corporation, Redmond, WA, USA). In cell culture experiments, values were compared using unpaired Student's t-tests. For multiple comparisons, we used the one-factor analysis of variance corrected by post-hoc Bonferroni test. Survival curves of C. elegans were created using the Kaplan and Meier's product-limit method. Mantel–Cox log-rank statistics were used to evaluate differences between survival curves and to determine P-values.
Conflict of interest
The authors declare no conflict of interest.
Abbreviations
- DiOC6(3):
-
3,3′-dihexyloxacarbocyanine iodide
- atg :
-
AuTophaGy-related gene
- BafA1:
-
bafilomycin A1
- Bec-1:
-
Beclin-1
- Δψm:
-
mitochondrial transmembrane potential
- GFP:
-
green fluorescent protein
- HRP:
-
horseradish peroxidase
- mTOR:
-
mammalian target of rapamycin
- Rapa:
-
rapamycin
- Resv:
-
resveratrol
- siRNA:
-
small-interfering RNA
- Sirt-1:
-
Sirtuin-1
- Tun:
-
tunicamycin
- WT:
-
wild type
References
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 2005; 118: 3091–3102.
Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G . To die or not to die: that is the autophagic question. Curr Mol Med 2008; 8: 78–91.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–239.
Iwata A, Riley BE, Johnston JA, Kopito RR . HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 2005; 280: 40282–40292.
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A . Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 2008; 68: 1485–1494.
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14: 1237–1243.
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–1010.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Jia K, Levine B . Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 2007; 3: 597–599.
Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G . The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy 2008; 4: 870–873.
Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008; 4: 330–338.
Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11: 1305–1314.
Finkel T, Deng CX, Mostoslavsky R . Recent progress in the biology and physiology of sirtuins. Nature 2009; 460: 587–591.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003; 425: 191–196.
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 2008; 105: 3374–3379.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–175.
Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol 2006; 26: 28–38.
Tinhofer I, Bernhard D, Senfter M, Anether G, Loeffler M, Kroemer G et al. Resveratrol, a tumor-suppressive compound from grapes, induces apoptosis via a novel mitochondrial pathway controlled by Bcl-2. FASEB J 2001; 15: 1613–1615.
Mizushima N, Yoshimori T . How to interpret LC3 immunoblotting. Autophagy 2007; 3: 542–545.
van Leeuwen I, Lain S . Sirtuins and p53. Adv Cancer Res 2009; 102: 171–195.
Mathew R, Karantza-Wadsworth V, White E . Role of autophagy in cancer. Nat Rev Cancer 2007; 7: 961–967.
Castedo M, Macho A, Zamzami N, Hirsch T, Marchetti P, Uriel J et al. Mitochondrial perturbations define lymphocytes undergoing apoptotic depletion in vivo. Eur J Immunol 1995; 25: 3277–3284.
Ferri KF, Kroemer G . Mitochondria—the suicide organelles. Bioessays 2001; 23: 111–115.
Feng Z, Zhang H, Levine AJ, Jin S . The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 2005; 102: 8204–8209.
Tissenbaum HA, Guarente L . Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001; 410: 227–230.
Sauve AA . Pharmaceutical strategies for activating sirtuins. Curr Pharm Des 2009; 15: 45–56.
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006; 444: 337–342.
Soda K, Dobashi Y, Kano Y, Tsujinaka S, Konishi F . Polyamine-rich food decreases age-associated pathology and mortality in aged mice. Exp Gerontol 2009; 44: 727–732.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460: 392–395.
Park SK, Kim K, Page GP, Allison DB, Weindruch R, Prolla TA . Gene expression profiling of aging in multiple mouse strains: identification of aging biomarkers and impact of dietary antioxidants. Aging Cell 2009; 8: 484–495.
Gregoretti IV, Lee YM, Goodson HV . Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004; 338: 17–31.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999; 104: 263–269.
Ford J, Jiang M, Milner J . Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res 2005; 65: 10457–10463.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G . Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12: 803–813.
Galluzzi L, Vitale I, Kepp O, Seror C, Hangen E, Perfettini JL et al. Methods to dissect mitochondrial membrane permeabilization in the course of apoptosis. Methods Enzymol 2008; 442: 355–374.
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10: 676–687.
Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16: 1006–1017.
Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14: 1029–1039.
Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L et al. Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res 2008; 68: 5301–5308.
Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One 2007; 2: e1337.
Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J . Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2001; 2: RESEARCH0002.
Samara C, Syntichaki P, Tavernarakis N . Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ 2008; 15: 105–112.
Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N . Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J Cell Biol 2006; 173: 231–239.
Acknowledgements
We thank Dr Andrew Fire for plasmids. Some nematode strains used in this study were provided by the C. elegans Gene Knockout Project at OMRF (http://www.mutantfactory.ouhsc.edu/), which is part of the International C. elegans Gene Knockout Consortium, and the Caenorhabditis Genetics Center, which was funded by the NIH National Center for Research Resources (NCRR). We thank A Jalil and D Metivier for expert assistance. NT was supported by grants from EMBO, the European Research Council (ERC), and the European Commission Coordination Action ENINET (contract number LSHM-CT-2005-19063). GK was supported by the Ligue Nationale contre le Cancer (Equipe labellisée), Agence Nationale pour la Recherche (ANR), European Commission (Apo-Sys, ChemoRes, ApopTrain, Active p53), Fondation pour la Recherche Médicale (FRM), Institut National du Cancer (INCa), and Cancéropôle Ile-de-France. EM was funded by a PhD student grant from ApopTrain. LG was supported by the Apo-Sys consortium of the European Union. SAM received a grant from the Higher Education Commission (HEC) of Pakistan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Melino
Rights and permissions
This article is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Morselli, E., Maiuri, M., Markaki, M. et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 1, e10 (2010). https://doi.org/10.1038/cddis.2009.8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2009.8
Keywords
This article is cited by
-
Dietary restriction reveals sex-specific expression of the mTOR pathway genes in Japanese quails
Scientific Reports (2024)
-
The hunger strikes back: an epigenetic memory for autophagy
Cell Death & Differentiation (2023)
-
An AMP Kinase-pathway dependent integrated stress response regulates ageing and longevity
Biogerontology (2023)
-
Neuroprotective properties of queen bee acid by autophagy induction
Cell Biology and Toxicology (2023)
-
SIN-3 functions through multi-protein interaction to regulate apoptosis, autophagy, and longevity in Caenorhabditis elegans
Scientific Reports (2022)
