
Abstract
Natural-food-based compounds show substantial promise for prevention and biotherapy of cancers including leukemia. In general, their mechanism of action remains unclear, hampering rational use of these compounds. Herein we show that the common dietary flavonoid apigenin has anticancer activity, but also may decrease chemotherapy sensitivity, depending on the cell type. We analyzed the molecular consequences of apigenin treatment in two types of leukemia, the myeloid and erythroid subtypes. Apigenin blocked proliferation in both lineages through cell-cycle arrest in G2/M phase for myeloid HL60 and G0/G1 phase for erythroid TF1 cells. In both cell lines the JAK/STAT pathway was one of major targets of apigenin. Apigenin inhibited PI3K/PKB pathway in HL60 and induced caspase-dependent apoptosis. In contrast, no apoptosis was detected in TF1 cells, but initiation of autophagy was observed. The block in cell cycle and induction of autophagy observed in this erythroleukemia cell line resulted in a reduced susceptibility toward the commonly used therapeutic agent vincristine. Thus, this study shows that although apigenin is a potential chemopreventive agent due to the induction of leukemia cell-cycle arrest, caution in dietary intake of apigenin should be taken during disease as it potentially interferes with cancer treatment.
Similar content being viewed by others


Main
Apigenin (4′,5,7-trihydroxyflavone), a common dietary flavonoid abundantly present in fruits and vegetables, shows promising biological effects such as prevention and therapy of prostate cancer, suppression of tumorigenesis and angiogenesis in melanoma1 and breast, skin, and colon carcinomas.2 It has been shown to exhibit antitumoral effects in leukemia cells by induction of apoptosis through activation of caspases, inhibition of fatty acid synthase and topoisomerase, and modulation of Bax and Bcl-2 expression,3 and indeed different studies have reported antileukemic effects of apigenin.4, 5, 6, 7 Despite apigenin seems to be promising as a chemopreventive in leukemia, a rational design of trails investigating its potential in this respect awaits further elucidation of its molecular mode of action.
Studies in breast cancer cells have identified some of the potential key protein kinases potentially responsible for apigenin effects, in particular PI3K, PKB and ERK1/2, and other upstream kinases involved in cancer development and progression.8 However, the effects of apigenin on these kinases in leukemia have not been investigated.
The promise of apigenin now urgently calls for investigations as to its molecular mode of action in leukemia cellular physiology. This consideration prompted us analyze the molecular pathways activated by apigenin treatment on the leukemic cell. To this end we established the induction of cell-cycle arrest and cell death in two models of leukemic disease, myeloid (HL60 cells) and erythroid (TF1 cells).
Results
Apigenin as a food-based antileukemia compound
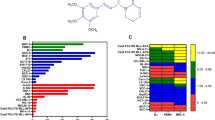
Leukemia cells and human peripheral blood lymphocytes were treated with apigenin (Figure 1a) in concentrations up to 200 μM, for 24 h. As shown in Figure 1b, apigenin caused a dose-dependent reduction in viable leukemia cells. IC50 of apigenin on HL60 was 30 μM, with complete loss of viability at 100 μM. In contrast, erythroleukemic cells were more resistant, 60±6% reduction of viability at 100 μM was observed for both K562 and TF1 cell lines. Importantly, no strong effect of apigenin treatment was observed on normal human peripheral blood lymphocytes, only 20% of cells were sensitive to apigenin at the highest concentrations. These results indicate that apigenin shows promise as a food-based antileukemia compound.

Apigenin reduces cell viability of leukemia cells. (a) Molecular structure of apigenin. (b) Leukemia cells and human peripheral blood lymphocytes were treated with apigenin for 24 h. Cell viability was measured by MTT assay and cell viability of nontreated cells was considered as 100%. (c) Number of HL60 viable cells during different times of treatment with apigenin. After HL60 cells treatment with apigenin for 5, 12, and 24 h, the number of viable cells was quantified using Trypan blue. (d) Number of TF1 viable cells during different times of treatment with apigenin. TF1 cells were treated with apigenin for 18, 24, and 36 h and using the Trypan blue the cell viable was quantified
Apigenin reduces survival due to cell-cycle arrest
As HL60 and TF1 cells showed more sensitivity toward apigenin, we continued our studies with these two cell lines. We further investigated whether the reduced cell numbers were a result of reduced cell cycling or increased cell death, by following the number of viable cells over the hours using vital stain Trypan blue. Apigenin 20, 30, and 40 μM reduced to 60.4±11.7, 79.4±15.5, and 55.8±7%, respectively, the number of viable HL60 cells (Figure 1c) whereas the nontreated cells showed 170% of viability, indicating that apigenin both stops cell proliferation and drives HL60 cells into cell death. TF1 cells showed 175, 125, and 120% of viable cells at 24 h treatment with 25, 50, and 100 μM of apigenin, respectively, whereas nontreated cells showed 225% viability (Figure 1d). Even after 36 h of 50 and 100 μM apigenin, the cell viability remained the same as control at 0 h, and the baseline on 100% was not passed (Figure 1d) indicating that apigenin did not induce cell death in TF1 cells, but blocked cell proliferation.
Through analysis of DNA content by flow cytometry, we observed cell-cycle arrest of HL60 cells in G2/M phase by apigenin (Figure 2a), as the number of cells in G2/M phase increased dose dependently whereas the number of cells in S phase decreased. At 100 μM apigenin, there were 58±8% cells in G2 and M phases compared to 11.2±0.6% in nontreated cells (P<0.05). The percentage of cells in S phase dropped from 42.3±1.3% in nontreated cells to 21.4±2% in treated cells (P<0.01). This was in agreement with an increase in apigenin-induced phosphorylation of Cdc2 at Tyr15 that results in inhibition of this kinase (Figure 2c). Moreover, the expression of c-myc and p21 remained unchanged (Figure 2c).

Apigenin induces cell-cycle arrest. (a) Number of HL60 cells in different phases of cell cycle (G0/G1 phase, S phase and G2/M) as determined by propidium iodide staining followed by flow cytometry. (b) Number of TF1 cells in different phases of cell cycle. Cell-cycle analysis was performed by measuring DNA content by propidium iodide staining after treatment with specified concentrations of apigenin for 24 h. (c) Expression of p21 and c-myc (middle and lower panels) and phosphorylation of CDC2 (upper panel), key proteins in cell-cycle control in HL60 cells was determined using western blotting. (d) Expression CDK4 and 6 in TF1 cells by western blotting. Equal loading was confirmed by reprobing blots for β-actin
TF1 cells were arrested in the G0/G1 phase as shown in Figure 2b. The number of cells in G0/G1 phase increased from 40.7±0.34% cell in untreated cells to 65.12±1.11% on treatment with 50 μM apigenin (P<0.01). The number of cells in S phase was 44.6±3.6% in nontreated cells and decreased to 18.6±0.9% in cells that received 50 μM apigenin. Corroborating these data, results shown in Figure 2d revealed that apigenin led to a complete degradation or inhibition of expression of CDK6, a protein essential for passage through the G1/S phase restriction point. However, no changes were observed in CDK4 protein levels. Hence apigenin is a potent inhibitor of cell-cycle progression in a variety of model systems of leukemia growth.
Downregulation of JAK/STAT pathway in both cells and PI3K/PKB only in HL60 cells by apigenin
We decided to establish the molecular details by which apigenin induces cell-cycle arrest through the analysis of phosphorylation status of some kinases and phosphatases involved in leukemia cell proliferation and survival. Figure 3a shows that treatment of HL60 cells with apigenin resulted in downregulation of PI3K/PKB pathway, possibly due to a decrease in PI3Kp85 subunit expression. We also observed diminished phosphorylation of PKB Ser473 and of PDK-1 Ser241. In agreement with diminished PKB activity, we observed diminished GSK3-β Ser9 phosphorylation. Inhibition of the PI3K/PKB pathway can be a direct consequence of activation of PTEN. As shown in Figure 3c, apigenin treatment decreases phosphorylation of PTEN at Ser380, leading to its activation, providing a mechanistic explanation for this action of apigenin.

Analysis of cell proliferation pathways. Effect of apigenin treatment on key kinases involved in cell proliferation from HL60 (a) and TF1 (b) cells. (c) Effect of apigenin treatment on phosphatases involved in proliferation of HL60 and TF1 cells. Cells were treated with specified concentrations of apigenin for 24 h and the expression or phosphorylation of the proteins was determined using western blot analysis. Equal loading was confirmed by reprobing blots for β-actin. (d) The activity of immunoprecipitated LMWPTP was measured after apigenin treatment of HL60 and TF1 cells by Malachite green assay. (e) Effect of apigenin treatment on proteins involved in cell survival control. HL60 cells were treated with 50 μM apigenin for 5, 12 and 24 h; TF1 cells were treated with 100 μM apigenin for 18, 24 and 36 h; and the phosphorylation of the proteins was determined by western blotting. Equal loading was confirmed by reprobing them for β-actin
In contrast, no effect was observed on PI3K/PKB pathway in TF1 cells treated with apigenin for 24 h (Figure 3b). Phosphorylation levels of PKB at Ser473 and phosphorylation levels of GSK3-β remained unchanged. Unlike HL60 cells, apigenin did not induce activation of PTEN in TF1 cells, which remained strongly phosphorylated at Ser380 (Figure 3c). Thus, apigenin has differential effects depending on the cell type involved.
To obtain more information about the action of apigenin in cell survival, we analyzed MAPKp38, JAK2, and STAT3 and 5. In HL60 cells treated with apigenin an increase in phosphorylation of MAPKp38 was observed at Thr180/182, essential for p38 catalytic action. However, in TF1 cells, the p38 activity remained unchanged after apigenin treatment (Figure 3a). JAK/STAT pathway was downregulated in both cell lines, thus emerging as a general effect of apigenin in leukemia. Apigenin led to decreased phosphorylation of JAK2 and STAT3 in both cell lines and STAT5 in TF1 cells (Figure 3a and b). The strong negative effect of apigenin on STAT3 phosphorylation in TF1 cells can be explained by increase of expression (Figure 3c) and activity of LMWPTP (Figure 3d), one of negative modulators of STATs,9 as well as the strong inhibition of SHP-2; LMWPTP activity was about 4-fold (385±94%), 2-fold (198±28%), and 10-fold higher (1083±47%) in the presence of 25, 50, or 100 μM apigenin, respectively. In HL60 cells, the LMWPTP activity was inhibited about 44, 37, and 36% at 20, 30, and 40 μM apigenin, respectively. Another activator of STATs, the Src protein kinase, was also inhibited by apigenin treatment, showing decrease of phosphorylation at Tyr416, an activation site, in both cell lines, HL60 (Figure 3a) and TF1 (Figure 3b). Hence, this general response toward apigenin across multiple types of leukemia may be important for explaining its broad antileukemic effects.
Both cell lines treated with apigenin showed a decrease of phosphorylation of SHP-2 at activator residue Tyr542 (Figure 3c), which thus may also contribute to the growth inhibition of leukemic cells observed.
TF1 cells show a constitutive activation of mTOR and, consequently, activation of protein synthesis/cell growth pathways (Figure 3e). In the presence of apigenin we observed downregulation of p70S6K phosphorylation and consequently, of S6 protein in TF1 cell. HL60 cells, however, do not show constitutive activation of mTOR and p70S6K, although apigenin decreased S6 protein phosphorylation. Therefore, the functional importance of mTOR pathway regulation in the apigenin response remains unclear.
Apigenin induces caspase-dependent apoptosis in HL60, but not in TF1 cells
In HL60, we observed a significant increase in Annexin-V-positive cells, 31.9±4.1 and 48.13±2.6% for 50 and 100 μM apigenin, respectively (Figure 4a). Consistent with being more resistant to apigenin, only moderate induction of apoptosis was observed at high concentrations of apigenin in TF1 cells; at 100 μM, 14.5±0.1% of cells stained Annexin V positive (P<0.002).

Analysis of apigenin-induced apoptosis in HL60 and TF1 cells. (a) Percentage of apoptotic cells after 24 h treatment with apigenin (25, 50, and 100 μM) was determined using flow cytometric analysis of Annexin V staining. (b) The effect of pan-caspase inhibitor Z-VADfmk on apigenin-induced cell death. Cells were pretreated with 50 μM Z-VADfmk for 1 h before apigenin treatment. HL60 cells were treated with 50 μM apigenin and TF1 cells were treated with 100 μM apigenin for 24 h. (c and d) Effect of PI3K inhibitor Ly294002 on apoptosis induction in HL60 (c) and TF1 (d) cells. Cells were pretreated with 5 μM Ly294002 for 1 h before 24 h apigenin treatment. Afterwards, cells were stained with propidium iodide (PI) and Annexin V. The combined Annexin-V-positive fractions were considered apoptotic (***P<0.001, **<0.01)
To obtain more information about apigenin-induced apoptosis in HL60 and TF1 cells, we pretreated both lineages for 1 h with 50 μM of Z-VADfmk, a pan-caspase inhibitor, before the 24 h apigenin treatment. Figure 3b shows that pretreatment of HL60 cells with Z-VADfmk reduced the number of apoptotic cells 1.3-fold when compared to cells that received apigenin only. No significant difference in apoptosis induction was observed in TF1 cells pretreated with Z-VADfmk (Figure 4b), suggesting that apigenin induces caspase-independent apoptosis in TF1 cells and caspase-dependent apoptosis in HL60 cells.
To understand the role of the PI3K/PKB pathway in apigenin-induced apoptosis, we treated cells for 2 h with 5 μM of LY294002, a PI3K inhibitor, before the 24 h apigenin treatment. As shown in Figure 4c, inhibition of PI3K with LY294002 before apigenin treatment increased the number of apoptotic HL60 cells 1.9-fold when compared with cells that received apigenin only, showing the importance of PI3K/PKB pathways in apoptosis resistance in this lineage. No statistic difference in apoptosis induction was observed in TF1 that received LY294002 before apigenin treatment comparing to cells which received apigenin only (Figure 4c and d), showing that TF1 cells are not dependent on PI3K/PKB pathway activation for their resistance to apigenin-induced apoptosis.
Molecular mechanism of apoptosis induction by apigenin
To further elucidate the apigenin-induced cell death mechanisms in HL60 and TF1 cells, we analyzed the levels of cleaved caspase-3, -7, -8, and PARP. We observed an increased dose- (Figure 5a) and time-dependent effect (Figure 5c) on cleaved caspase-3 levels in HL60 cells. This response was not observed in TF1 cells; only after treatment with 100 μM apigenin a small amount of cleaved caspase-3 (17 kDa fragment) became apparent (Figure 5c). In HL60 cell, caspase-8 cleavage in apigenin-treated cells was observed, indicating that the apoptosis extrinsic pathway was also involved in apigenin cell death induction in this cell line (Figure 5b). In HL60 the levels of full-length caspase-7 decreased when cells were treated with 50 μM apigenin, coinciding with increased presence of the 20 kDa cleaved caspase-7 fragment (Figure 5c).

Analysis of apoptosis pathway mechanism. (a) HL60 and TF1 cells were treated with apigenin for 24 h. The expression of cleaved caspase-3, cleaved PARP, and AIF was evaluated by immunoblotting. (b) Levels of full-length and cleaved caspase-8 (41, 43, and 18 kDa) were determined. (c) Levels of cleaved and uncleaved caspase-7 and cleaved caspase at different periods of apigenin treatment were investigated. HL60 cells were treated with 50 μM apigenin for 5, 12 and 24 h and TF1 cells were treated with 100 μM apigenin for 18, 24, and 36 h. Soluble lysates were matched for protein content and analyzed by western blot analysis. Equal loading was confirmed by reprobing the blots with anti-β-actin antibodies
In TF1 cells, neither cleavage of caspase-8 nor caspase-7 was observed (Figure 5a and b). Furthermore, the levels of cleaved PARP remained unchanged in apigenin-treated cells. We observed an increased of AIF levels at 25 and 50 μM apigenin treatment in TF1 cells (Figure 5a), a strong indication of caspase-independent apoptosis. These results emphasize that despite the generalized leukemia growth inhibition observed, the molecular consequences of apigenin treatment show substantial context dependency.
Apigenin and autophagy
To obtain more insight into the apigenin response in TF1 cells, we analyzed the apoptosis and autophagy pathway activation in the time domain. For apoptosis induction we used TNFα (10 ng/ml) as a control. TF1 cells were slightly induced to undergo apoptosis by TNFα, as was deduced from cytokine-induced cleavage of caspase-7 to its 30 kDa, but not 20 kDa fragment and PARP cleavage. Interestingly, TNFα also induced cleavage of LC3BI into LC3BII after 24 and 36 h of treatment and also induced high expression of the autophagic proteins Atg5 and 12 (Figure 6b). In addition, through electron microscopy (EM) we could observe the signature double-membrane vacuoles, generally accepted as strongly indicative for autophagy (Figure 6d and f). In addition, TNFα activated P70S6K, which led to high phosphorylation of S6 protein, indicative for protein synthesis (Figure 6c), and apparently confirmed by EM, as TNFα-treated TF1 cells showed high numbers of ribosomes on endoplasmic reticulum (ER) surface, indicative of high ER activity (Figure 6e). Apigenin also induced autophagy in TF1 cells, but through inhibition of mTOR and P70S6K. We observed a strong reduction on phosphorylation of S6 protein. No changes in beclin-1 levels were observed, however there was a strong reduction of Atg5, 7, and 12 on TF1 cells treated with apigenin. These data led us to conclude that autophagy is a dominant response to apigenin treatment in at least some types of leukemia.

Effect of apigenin treatment on key kinase molecules involved in the autophagy and apoptosis from TF1 cells. TF1 cells were treated with 10 ng/ml TNFα or 100 μM apigenin for 24 and 36 h. (a) Levels of full-length and cleaved caspase-7 (30 and 20 kDa), full-length and cleaved PARP. (b) Levels autophagy proteins beclin-1, LC3B, Atg5, 7, and 12. (c) Analysis of P70S6K/S6 pathway by western blotting. The expression or phosphorylation of the proteins was determined by western blot analysis. Equal loading was confirmed by reprobing them for β-actin. (d) Electron microscopy of TF1 cells treated with TNFα for 24 h, detail of double-membrane vacuole in formation (original magnification, × 4500). (e) Detail of endoplasmic reticulum, showing intense protein synthesis (original magnification, × 10 000). (f) Detail of double-membrane vacuole (original magnification, × 10 000)
Apigenin leads to apoptosis-like cell death in HL60 and involves autophagy in TF1 as revealed by EM
Further analysis of apigenin effects on HL60 and TF1 cells was performed using EM, emphasizing the differential nature of apigenin responses in leukemia (Figures 7 and 8). In HL60 cells treated with apigenin for 8 h, we observed classical apoptosis structures, chromatin condensation, and reduction of cell volume (Figure 7b). At 24 h of treatment most of cells showed electron-dense cytoplasm with very condensed nucleus and many of non-electron-dense vacuoles (Figure 7c), and some necrotic cells were detected (Figure 7c). In the electron-dense cytosol cell, we identified double-membrane vacuoles, as shown in detail in Figure 7d. TF1 cells showed morphological changes only after 36 h of treatment, where we could find speckles in the nucleus (arrows on the Figure 8c) and electron-dense mitochondria in the cytoplasm (Figure 8c). In addition, a number of cells showed non-electron-dense vacuoles and double-membrane vacuoles, strong evidence of autophagy (Figure 8d). Thus, there is not a common pathway leading to leukemia cell death, despite the general antileukemic activity of apigenin and experiments were initiated to explore the functional consequences of this observation.

Electron microscopy of HL60 cells treated with apigenin for different time points. (a) Control cells, without treatment (original magnification, × 3000). (b) Cells treated with apigenin for 8 h, showing classical apoptotic cells (condensed chromatin, reduction of cell size, and no vacuoles); cells with dense mitochondria and no condensed chromatin and cells with size reduction, many vacuoles and no condensed chromatin (original magnification, × 3000). (c) HL60 cell treated for 24 h (original magnification, × 3000), showing necrotic cells and cells with many vacuoles. (d) Cells treated for 24 h with apigenin, showing cells with dense chromatin, many vacuoles and double membrane, detail in high magnification (original magnification, × 20 000) of vacuoles with cytosol content

Electron microscopy of TF1 cells treated with apigenin for different time points. (a) Control cells, without treatment (original magnification, × 3000). (b) Cells treated with apigenin for 24 h show little morphological difference from nontreated cells (original magnification, × 3000). (c) TF1 cells treated for 36 h (original magnification, × 3000) present dense mitochondria, partly condensed chromatin, and speckles (arrows) (original magnification, × 3000). (d) Detail of vacuoles with double membrane in an authophagic cell (original magnification, × 20 000). (e) Number of viable TF1 cells after vincristine treatment with or without apigenin pretreatment. TF1 cells were treated or not treated with 100 μM apigenin for 24 h, after which cell numbers were normalized and cultured for another 24 h with or without 2 μM vincristine. The number of viable cell was determined using Trypan blue. (f) Determination of percentage of apoptotic TF1 cells by flow cytometric analysis of Annexin V staining. Cells were treated or not treated with apigenin for 24 h, after which cell numbers were normalized and cells were cultured for another 24 h with or without 2 μM vincristine
Apigenin diminishes the cytotoxic effect of the chemotherapeutic vincristine in TF1 cells
The role of autophagy in cells can be of dual consequence either it can prolong cell survival in the absence of nutrients or it can result in cell death. Induction of autophagy is often associated with chemotherapy resistance. Indeed, TF1 cells pretreated with apigenin 24h prior of vincristine showed diminishment of cytotoxic effect of chemotherapy (Figure 8e and f). TF1 cells were treated with 100 μM apigenin for 24 h, after the number of cells of treated and nontreated cells was equalized, and cells were cultured with or without vincristine treatment for an additional 24 h. After the treatment, the viable cells were counted using Trypan blue (Figure 8e). In the absence of apigenin pretreatment, vincristine reduced the number of viable to 39.2±6.1%, compared to cells that did not receive vincristine (100%). However, cells treated with apigenin before vincristine treatment showed 147±7.3% viability, compared to the cells that did not received vincristine (100%).
To obtain more data about the protective effect of apigenin against vincristine, we further analyzed apoptosis induction. TF1 cells were treated with apigenin for 24 h, after which the number of cells of treated and nontreated cultures was equalized followed by the presence or absence of vincristine treatment for an additional 24 h. After the treatment, we analyzed apoptosis induction using flow cytometric analysis of Annexin V binding. Cells that received neither apigenin nor vincristine showed 10.7±0.007% Annexin-V-positive cells. Vincristine treatment alone induced Annexin V staining in 33.6±1.4% cells. Treatment of cells with apigenin alone (for 48 h) showed 22.34±0.4% Annexin-V-positive cells. Strikingly, cells that were pretreated with apigenin followed by vincristine treatment showed a reduction in Annexin-V-positive cells when compared to vincristine treatment alone (19.8±0.2% cells) (Figure 8f). Thus, we showed that apigenin protects TF1 against cell death induced by vincristine. We conclude that although the effects of apigenin are generally chemopreventive regarding leukemia, differential response may include resistance to chemotherapy. Thus, use of such food-based compound in an adjuvant setting when disease is established might not be advisable.
Discussion
Our research group was interested in the possible beneficial biological effects of natural compounds and their derivatives on malignant disease.10, 11, 12, 13, 14 In this work, we explored the effects of apigenin in leukemia, flavonoid commonly present in human diet, which is emerging not only as potent cancer-preventing agent, but also useful for adjuvant therapy.11 Although the induction of apoptosis and cell-cycle arrest, and inhibition of cancer-promoting signaling pathways, certainly supports the chemopreventive action, especially as untransformed lymphocytes do not seem to be really affected by the compound, the observed interaction with therapeutically relevant drugs seems to rule this compound out for adjuvant therapy as it may actually hamper treatment of established disease.
In HL60 cells, apigenin induced cell-cycle arrest in G2/M, whereas TF1 cells were arrested in the G0/G1 phase. The control of cell cycle is mediated by many protein complexes, such as CDK and cyclins. Cdc2, normally driving cells into mitosis, is the ultimate target of pathways that mediate rapid arrest in G2, in response to DNA damage.15 CDK6 forms a complex with cyclin-D and targets the retinoblastoma protein, allowing passage through the G1/S phase restriction point.16 Many studies have reported that apigenin blocks cell cycle in cancer cells,17, 18 however, depending on the cell type, apigenin can arrest the cell cycle in different phases, for example, it can lead G2/M arrest in colon carcinoma,2 breast cancer and pancreatic cancer cells.19 Nevertheless, in human prostate cancer, LNCap and PC3 cells, apigenin induced cell-cycle arrest in G0/G1.20 The results of this study extend these observations that, depending on the cyclins affected, even in the leukemic compartment responses may be substantially different. The outcome of cell-cycle arrest may depend on the phase in which the cell is arrested. Numerous studies, including those using dietary compounds, have linked G2/M arrest with subsequent apoptosis,21, 22 as was observed for HL60 cells in our study. In contrast, G0/G1 arrest is seen on reduction of mTOR activity,23 and has been associated with induction of autophagy,24 reminiscent of apigenin effects in TF1 cells in this study. p70S6K/S6 signaling has been implicated in inhibition of autophagy,25 and inactivation of the mTOR signaling pathway is a prominent consequence of PTEN activation.26 In this study, although apigenin was not able to activate PTEN in TF1 cells, mTOR was strongly downregulated by apigenin. Thus, this flavonoid may be an attractive therapeutic agent for treating tumors carrying inactivated PTEN. Studies with cancer stem cells have shown that inhibition of mTOR pathways by rapamycin can block the generation or maintenance of leukemia-initiating cells in the PTEN-deficient mice.27
The role of autophagy in cell survival is a dual one; it can either help cells to survive during stress or end in apoptosis in the absence of cell rescue.28 The autophagic characteristics observed in HL60 cells suggest that the latter predominates in this cell type following exposure to apigenin, whereas in TF1 cells autophagy is not followed by apoptosis, in apparent agreement with the therapy-resistant phenotype of these cells. Only after 36 h of stringent apigenin treatment did a small fraction of TF1 cells show signs of (a caspase-independent form of) apoptosis. Interestingly, although showing hallmarks of apoptosis on TNFα treatment, for example, cleavage of PARP and caspase activation, EM analysis of TNFα-treated cells showed characteristics of autophagy, confirmed by high levels of sentinel autophagic proteins beclin-1, LC3BII, and Atg5, 7 and 12. In addition, TNFα induced activation of the p70S6K/S6 pathway, indicating that the induction of autophagy in TNFα-treated cells is induced by high expression of autophagic genes related to vacuoles, rather as of inhibition of P70S6K/S6 pathway.
Flavonoids have the potential to bind to the ATP-binding sites of a large number of proteins due to their similarity to ATP structure, which makes flavonoids competitive inhibitors of protein kinases.29 Apigenin had already been reported to be a competitive inhibitor of protein tyrosine kinases.30 In this work we clearly observed that apigenin inhibited PKB/PI3K pathway in HL60 cells, through activation of PTEN and downregulation of PDK-1 and PI3K. Unlike HL60, TF1 showed no changes in PI3K/PKB pathway under apigenin treatment. Furthermore, MAPKp38 was activated in HL60 cells treated with apigenin, but not in TF1 cells. Recently, it has been suggested that p38 and JNK may be involved in the apoptotic process by repression of ERK pathways.31 Thus, the differential effects of apigenin observed on leukemia cell cycle are reflected by differential effects on kinase activities.
Interestingly, the only common target of apigenin in both types of leukemia was the c-Src/JAK/STAT pathway, which constitutes a major mediator of cytokine activity. Constitutively active JAK2 proteins are associated with myeloproliferative disorders, acute lymphoblastic leukemia, acute myeloid leukemia and acute megakaryoblastic leukemia, and inhibition of STAT3 leads to increased apoptosis, decreased proliferation, and decreased tumor size.32 JAK/STAT pathway is regulated by SHP-2, which can stabilize JAK2 protein or induce Src kinase activation. Constitutive activation of SHP-2 in mice cooperates with progression of myeloproliferative disorders.33 In this work we observed that apigenin downregulated SHP-2 and thereby JAK2, which led to a decreased phosphorylation and activation of STAT3 and 5. This effect was more apparent in TF1 cells, which can be explained by the increased level and activity of LMWPTP, another JAK/STAT downregulator, in this cell line. Further experimentation should reveal whether the JAK/STAT signaling cassette is really the principal target for apigenin effect in cancer in general.
Because apigenin induced cell-cycle arrest and autophagy in TF1 cells, we hypothesized that apigenin could inhibit the action of chemotherapeutic agents against leukemic cells. One of the most common chemotherapeutics is vincristine, and indeed on apigenin treatment, cells became more resistant to vincristine-induced cell death. Vincristine binds to tubulin dimers, inhibiting the formation of microtubules. Disruption of the microtubules arrests mitosis in metaphase, therefore, apigenin-induced blockage of TF1 cells in G0/G1 phase might enable these cells to escape the action of vincristine. This action of apigenin clearly disqualifies this compound for adjuvant strategies once disease is established, at least in relation to vincristine. However, it is important to mention that when we combined apigenin with imatinib or mitoxantrone no difference was observed (unpublished data). Therefore, this observation reinforces our hypothesis that the undesirable action of apigenin in the presence of vincristine might be related to the action mechanism of chemotherapeutics.
In conclusion, apigenin is a potential chemopreventive and chemotherapeutic agent in leukemia due to the stimulation on signaling pathways that provoke inhibition of cell proliferation and cell-cycle arrest of fast-cycling cells. This work emphasizes the importance of a deep analysis of molecular mechanism and biochemical details modified by a natural compound with clinically useful properties. Extreme care regarding consumption of such apigenin-like compounds during chemotherapy must be exercised to prevent compromising clinical success, and trails using such compound should not be frivolously initiated. However, the possibility of using apigenin as a model for drug design should not be discarded.
Materials and Methods
Cell lines and reagents
HL60, K562, and TF1 cells were purchased from ATCC (Rockville, MD, USA). Apigenin was from Sigma-Aldrich (Zwijndrecht, The Netherlands). Polyclonal antibodies against AIF, p-PKB (Ser473), p-cdc2 (Thr15), CDK4, CDK6, cleaved caspase-3, cleaved PARP, caspase-8, caspase-7, c-myc, p-GSK-3β (Ser9), p-PI3K p85, p-PDK (Ser241), p-JAK2 (Tyr1007/1008), p-Src (Tyr416), p-STAT3 (Tyr705), p-STAT5 (Tyr-694), p-p38 (Tyr108/182), p-SHP-2 (Tyr542), p-PTEN (Ser380), p-mTOR (Ser2448), p-p70S6K (Thr389), p-S6 (Ser235/236), Beclin-1, LC3BI/II, Atg5, Atg7, Atg12, anti-rabbit and anti-mouse peroxidase-conjugated antibodies were from Cell Signaling Technology (Beverly, MA, USA). p21 and TNFR1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against LMWPTP was purchased from Abcam (Zwijndrecht, The Netherlands). Apigenin and vincristine were purchased from Sigma-Aldrich. Caspase inhibitor Z-VADfmk and apoptosis kit detection (Annexin V-FITC and propidium iodide (PI)) were from BD Biosciences (San Diego, CA, USA). The PI3K inhibitor LY294002 was from Alexis (Läufelingen, Switzerland) and TNFα was from Biovision Inc. (Mountain View, CA, USA)
Cell culture
Leukemia cells were cultured in RPMI 1640 containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum, at 37°C in a 5% CO2 humidified atmosphere. For TF1 cells, 5 ng/ml GM-CSF was added to medium.
Human lymphocytes were obtained from healthy volunteers and isolated by density through Ficoll Paque gradient. Mononuclear cells were harvested and cultured in RPMI 1640 containing 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal bovine serum, and concanavalin A (5 μg/ml) for 48 h when the treatment was carried out.
MTT assay
Cell viability was assessed by MTT reduction assay as described by Mosmann.34 Nontreated cells were taken as 100% of viability and IC50 values were determined from three independent experiments.
Western blotting
Cells were lysed in cell lysis buffer (50 mM Tris-HCl (pH 7.4) containing 1% Tween 20, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM o-vanadate, 1 mM sodium fluoride, 1 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF). After 15 min of centrifugation the cleared lysates were immunoprecipitated and resolved by reduced SDS-polyacrylamide gel electrophoresis, the blots were incubated with the indicated antibodies and the bands were visualized with ECL.
PTP activity
Cells were lysed in cell lysis buffer (20 mM HEPES (pH 7.7), 2.5 mM MgCl2, 0.1 mM EDTA, 1% NP-40, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 1 mM DTT, 10 mg/ml aprotinin, and 10 mg/ml leupeptin). After 15 min of centrifugation, the cleared lysates were immunoprecipitated with GammaBind G Sepharose (GE Healthcare, Hoevelaken, The Netherlands) and the phosphatase activity was determined using the PTP no radioactive assay kit from Sigma.
Flow cytometry for apoptosis analysis
To perform the Annexin and PI assays, the FITC Annexin V Apoptosis Detection Kit II from BD Pharmingen (Breda, The Netherlands) was used. Briefly, control and apigenin-treated cells were collected, washed twice in cold PBS, resuspended in binding buffer (provided in the kit), and stained with Annexin V FITC and PI.
Flow cytometry for cell-cycle analysis
After treatment with apigenin, cells were spun down, washed with PBS, and resuspended in work solution (0.96 g/ml sodium citrate, 1 mg/ml ribonuclease A, 0.02 mg/ml PI, 0.01% Triton X-100). After incubation in the dark for 60 min at room temperature, the samples were analyzed using FACSCalibur flow cytometer (BD Biosciences).
Electron microscopy
HL60 and TF1 treated or nontreated with apigenin for different periods were washed in RPMI, pelleted, and subsequently fixed in 2% glutaraldehyde in 0.1 M phosphate buffer for 24 h at 4°C. Cells were dehydrated, osmicated, and embedded in Epon812 according to routine procedures.33 Semithin sections (1–0.5 μm) were examined by electron microscope (Philips 201; Philips, Eindhoven, the Netherlands).
Statistical analysis
All experiments were performed in triplicate and the results shown in the graphs represent the means and standard errors. Cell viability data were expressed as the means±standard errors of three independent experiments carried out in triplicates. Data from each assay were statistically analyzed by ANOVA. Differences were considered significant when the P-value was <0.05. Western blotting analyses represent three independent experiments.
Conflict of interest
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Proc. 2004/15041-5; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), scholarship to RRRS and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), TI Pharma Grant T3-103.
Abbreviations
- AIF:
-
apoptosis-inducing factor
- Atg:
-
autophagy-related (Atg) genes
- Bax:
-
Bcl-2 associated x protein
- Bcl-2:
-
B-cell lymphoma protein 2
- c-myc:
-
transcriptional regulator protein
- Cdc2:
-
cell division control protein 2
- CDKs:
-
cyclin-dependent kinases
- ERK1/2:
-
extracellular signal-regulated protein kinase 1/2
- GSK-3β:
-
glycogen synthase kinase-3 β
- JAK2:
-
janus family of tyrosine kinase 2
- LC3B:
-
Light Chain 3 isoform B
- LMWPTP:
-
low-molecular-weight protein tyrosine phosphatase
- LY294002:
-
PI3K inhibitor
- MAPKp38:
-
mitogen-activated protein kinase p38
- mTOR:
-
mammalian target of rapamycin, a Ser/Thr protein kinase
- p21:
-
tumor suppressor protein
- p70S6K:
-
70-kDa ribosomal protein S6 kinase
- PARP:
-
poly (ADP-ribose) polymerase
- PDK-1:
-
phosphoinositide-dependent protein kinase 1
- PI3K:
-
phosphoinositide 3-kinase
- PKB:
-
protein kinase B
- PTEN:
-
phosphatase and tensin homolog
- S6:
-
S6 ribosomal protein
- SHP-2:
-
Src homology 2 domain-containing phosphotyrosine phosphatase 2
- Src:
-
Src family of protein tyrosine kinases
- STAT:
-
signal transducer and activator of transcription
- TNF-α:
-
tumor necrosis factor alpha
- TNFR:
-
tumor necrosis factor receptor
- ZVADfmk:
-
pan caspase inhibitor
References
Caltagirone S, Rossi C, Poggi A, Ranelletti F, Natali P, Brunetti M et al. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer 2000; 87: 595–600.
Wang W, Heideman L, Chung C, Pelling J, Koehler K, Birt D . Cell-cycle arrest at G2/M and growth inhibition by apigenin in human colon carcinoma cell lines. Mol Carcinog 2000; 28: 102–110.
Wang I, Lin-Shiau S, Lin JK . Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur J Cancer 1999; 35: 1517–1525.
Chen D, Daniel K, Chen M, Kuhn D, Landis-Piwowar K, Dou Q . Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol 2005; 69: 1421–1432.
Kim J, Eom J, Cheong J, Choi A, Lee J, Yang W et al. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res 2007; 13: 1019–1028.
Monasterio A, Urdaci M, Pinchuk I, López-Moratalla N, Martínez-Irujo J . Flavonoids induce apoptosis in human leukemia U937 cells through caspase- and caspase-calpain-dependent pathways. Nutr Cancer 2004; 50: 90–100.
Vargo M, Voss O, Poustka F, Cardounel A, Grotewold E, Doseff AI . Apigenin-induced-apoptosis is mediated by the activation of PKCdelta and caspases in leukemia cells. Biochem Pharmacol 2006; 72: 681–692.
Way T-D, Kao M-C, Lin J-K . Apigenin induces apoptosis through proteasomal degradation of HER2/neu in HER2/neu-overexpressing breast cancer cells via the phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem 2004; 279: 4479–4489.
Rigacci S, Guidotti V, Parri M, Berti A . Modulation of STAT5 interaction with LMW-PTP during early megakaryocyte differentiation. Biochemistry 2008; 47: 1482–1489.
Bispo de Jesus M, Zambuzzi WF, Ruela de Sousa R, Areche C, Souza A, Aoyama H et al. Ferruginol suppresses survival signaling pathways in androgen-independent human prostate cancer cells. Biochimie 2008; 90: 843–854.
de Sousa RRR, Queiroz K, Souza A, Gurgueira S, Augusto A, Miranda M et al. Phosphoprotein levels, MAPK activities and NFkappaB expression are affected by fisetin. J Enzyme Inhib Med Chem 2007; 22: 439–444.
de Souza A, Kodach L, Gadelha F, Bos C, Cavagis A, Aoyama H et al. A promising action of riboflavin as a mediator of leukaemia cell death. Apoptosis 2006; 11: 1761–1771.
Ferreira C, Bos C, Versteeg H, Justo G, Duran N, Peppelenbosch M . Molecular mechanism of violacein-mediated human leukemia cell death. Blood 2004; 104: 1459–1464.
Queiroz K, Zambuzzi WF, Santos de Souza AC, da Silva R, Machado D, Justo G et al. A possible anti-proliferative and anti-metastatic effect of irradiated riboflavin in solid tumours. Cancer Lett 2007; 258: 126–134.
Stark G, Taylor W . Control of the G2/M transition. Mol Biotechnol 2006; 32: 227–248.
Lukas J, Bartkova J, Bartek J . Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol 1996; 16: 6917–6925.
Choi EJ, Kim G-H . Apigenin causes G(2)/M arrest associated with the modulation of p21(Cip1) and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J Nutr Biochem 2009; 20: 285–290.
Lepley D, Pelling J . Induction of p21/WAF1 and G1 cell-cycle arrest by the chemopreventive agent apigenin. Mol Carcinog 1997; 19: 74–82.
Ujiki M, Ding X-Z, Salabat M, Bentrem D, Golkar L, Milam B et al. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer 2006; 5: 76.
Shukla S, Gupta S . Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic Biol Med 2008; 44: 1833–1845.
Chou L-C, Yang J-S, Huang L-J, Wu H-C, Lu C-C, Chiang J-H et al. The synthesized 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one (CHM-1) promoted G2/M arrest through inhibition of CDK1 and induced apoptosis through the mitochondrial-dependent pathway in CT-26 murine colorectal adenocarcinoma cells. J Gastroenterol 2009; 44: 1055–1063.
Hsu Y-L, Uen Y-H, Chen Y, Liang H-L, Kuo P-L . Tricetin, a dietary flavonoid, inhibits proliferation of human breast adenocarcinoma mcf-7 cells by blocking cell cycle progression and inducing apoptosis. J Agric Food Chem 2009; 57: 8688–8695.
Hidayat S, Yoshino K-i, Tokunaga C, Hara K, Matsuo M, Yonezawa K . Inhibition of amino acid–mTOR signaling by a leucine derivative induces G1 arrest in Jurkat cells. Biochem Biophys Res Commun 2003; 301: 417–423.
Kang KB, Zhu C, Yong SK, Gao Q, Wong MC . Enhanced sensitivity of celecoxib in human glioblastoma cells: induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol Cancer 2009; 8: 66.
Fuhler G, Tyl M, Olthof S, Lyndsay Drayer A, Blom N, Vellenga E . Distinct roles of the mTOR components Rictor and Raptor in MO7e megakaryocytic cells. Eur J Haematol 2009; 83: 235–245.
Rossi DJ, Weissman IL . Pten, tumorigenesis, and stem cell self-renewal. Cell 2006; 125: 229–231.
Yilmaz O, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441: 475–482.
Tan M, Ooi J, Ismail N, Moad A, Muhammad T . Programmed cell death pathways and current antitumor targets. Pharm Res 2009; 26: 1547–1560.
Conseil G, Baubichon-Cortay H, Dayan G, Jault J-M, Barron D, Di Pietro A . Flavonoids: a class of modulators with bifunctional interactions at vicinal ATP- and steroid-binding sites on mouse P-glycoprotein. Proc Natl Acad Sci USA 1998; 95: 9831–9836.
Huang Y, Kuo M, Liu J, Huang S, Lin J . Inhibitions of protein kinase C and proto-oncogene expressions in NIH 3T3 cells by apigenin. Eur J Cancer 1996; 32: 146–151.
Junttila MR, Li S-P, Westermarck J . Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J 2008; 22: 954–965.
Song J, Grandis J . STAT signaling in head and neck cancer. Oncogene 2000; 19: 2489–2495.
Valentino L, Pierre J . JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 2006; 71: 713–721.
Mosmann T . Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55–63.
Diederen J, Peppelenbosch M, Vullings H . Ageing adipokinetic cells in Locusta migratoria: an ultrastructural morphometric study. Cell Tissue Res 1992; 268: 117–121.
Acknowledgements
This work was supported by grant Proc. 2004/15041-5 from FAPESP and by Grant T3-103 from TI Pharma. Scholarship to RRRS was provided by CAPES and CNPq.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by R Mantovani
Rights and permissions
This article is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ruela-de-Sousa, R., Fuhler, G., Blom, N. et al. Cytotoxicity of apigenin on leukemia cell lines: implications for prevention and therapy. Cell Death Dis 1, e19 (2010). https://doi.org/10.1038/cddis.2009.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2009.18
Keywords
This article is cited by
-
Natural Flavonoid Apigenin, an Effective Agent Against Nervous System Cancers
Molecular Neurobiology (2024)
-
The protective effect of apigenin against inorganic arsenic salt-induced toxicity in PC12 cells
Environmental Science and Pollution Research (2023)
-
Antineoplastic activity of plant-derived compounds mediated through inhibition of histone deacetylase: a review
Amino Acids (2023)
-
Chemoprotective and chemosensitizing effects of apigenin on cancer therapy
Cancer Cell International (2021)
-
Apigenin role as cell-signaling pathways modulator: implications in cancer prevention and treatment
Cancer Cell International (2021)
