
Abstract
Transforming growth factor-beta (TGF-β) and bone morphogenic protein (BMP) signaling has fundamental roles in both embryonic skeletal development and postnatal bone homeostasis. TGF-βs and BMPs, acting on a tetrameric receptor complex, transduce signals to both the canonical Smad-dependent signaling pathway (that is, TGF-β/BMP ligands, receptors, and Smads) and the non-canonical-Smad-independent signaling pathway (that is, p38 mitogen-activated protein kinase/p38 MAPK) to regulate mesenchymal stem cell differentiation during skeletal development, bone formation and bone homeostasis. Both the Smad and p38 MAPK signaling pathways converge at transcription factors, for example, Runx2 to promote osteoblast differentiation and chondrocyte differentiation from mesenchymal precursor cells. TGF-β and BMP signaling is controlled by multiple factors, including the ubiquitin–proteasome system, epigenetic factors, and microRNA. Dysregulated TGF-β and BMP signaling result in a number of bone disorders in humans. Knockout or mutation of TGF-β and BMP signaling-related genes in mice leads to bone abnormalities of varying severity, which enable a better understanding of TGF-β/BMP signaling in bone and the signaling networks underlying osteoblast differentiation and bone formation. There is also crosstalk between TGF-β/BMP signaling and several critical cytokines’ signaling pathways (for example, Wnt, Hedgehog, Notch, PTHrP, and FGF) to coordinate osteogenesis, skeletal development, and bone homeostasis. This review summarizes the recent advances in our understanding of TGF-β/BMP signaling in osteoblast differentiation, chondrocyte differentiation, skeletal development, cartilage formation, bone formation, bone homeostasis, and related human bone diseases caused by the disruption of TGF-β/BMP signaling.
Similar content being viewed by others
Introduction
The transforming growth factor-β (TGF-β) superfamily comprises TGF-βs, Activin, bone morphogenetic proteins (BMPs) and other related proteins.1–4 TGF-β superfamily members act through a heteromeric receptor complex, comprised of type I and type II receptors at the cell surface that transduce intracellular signals via Smad complex or mitogen-activated protein kinase (MAPK) cascade.1–4 At least 29 and probably up to 42 TGF-β superfamily members, five type II receptors and seven type I receptors are encoded by the human genome.2 Signals transduced by TGF-β superfamily members regulate the establishment of tissue differentiation through their effects on cell proliferation, differentiation, and migration.1–4
Fetus mammalian skeletal development begins with the condensation of mesenchymal stem cells (MSCs) from neural crest or mesoderm, and is accomplished in two distinct processes: endochondral ossification and intramembranous ossification.5,6 Endochondral ossification takes place in skull base and the posterior part of the skull, the axial skeleton, and the appendicular skeleton. Intramembranous ossification takes place in membranous neuro- and viscerocranium and some parts of the clavicles.5,6 During endochondral ossification, condensed mesenchyme undergoes chondrogenesis and forms cartilage model (anlagen), which is later replaced by mineralized bone. Differentiated chondrocytes were organized into growth plate comprised of different zones, including resting zone, proliferation zone, hypertrophic zone, and calcified zone.5,6 During intramembranous ossification, condensed mesenchyme directly differentiates into osteoblasts.5,6 Several cytokines and growth factors orchestrate skeletogenesis (for example, fibroblast growth factor (FGF), Notch, Wnt, Sonic hedgehog (SHH), Indian hedgehog (IHH), parathyroid hormone-related peptide (PTHrP), TGF-β, and BMP). Among them, TGF-βs and BMPs have diverse functions in skeletogenesis, including mesenchyme condensation, skeleton morphogenesis, growth plate development, and osteoblast differentiation.4 A wide variety of inheritable developmental bone diseases are caused by human genetic mutations related to TGF-β and BMP signaling.
Besides their roles in bone development, TGF-βs and BMPs also regulate the maintenance of postnatal bone and cartilage. TGF-βs have essential roles in coupling bone construction by osteoblast and bone destruction by osteoclast7,8 through osteoclast-mediated Atp6i-specific extracellular acidification9,10 and Cathepsin K-specific extracellular matrix proteins.11,12 Multiple BMPs are also potent osteogenic agents that possess significant clinical implication to accelerate fracture healing in patients.13,14 Furthermore, TGF-βs and BMPs regulate postnatal joint cartilage homeostasis, thus dysregulated TGF-β and BMP signaling are often associated with osteoarthritis in both human disease and mouse models.15–19
In this review, we will focus on current understanding of the mechanisms by which different TGF-βs and BMPs transduce signaling and exert their functions in skeletal development and homeostasis (Figures 1 and 2). We will review mouse models (Table 1) and human bone disorders (Table 2) caused by TGF-β/BMP dysregulation. We will also discuss the crosstalk between TGF-β/BMP signaling and other signaling pathways including Wnt, Hedgehog, Notch, and FGF in bone. Those studies have opened new prospects for generating novel prognostics and therapies against bone diseases.

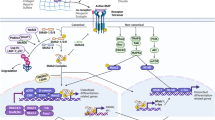
TGF-β signaling in bone. TGF-β is synthesized as a latent protein stored in the ECM, whose activation depends on osteoclastic bone resorption. Active TGF-βs binds to tetrameric receptor complex comprising two TGF-β types I receptors (TβRI) and two type II receptors (TβRII). TβRII transphosphorylases TβRI to induce Smad-dependent and non-Smad-dependent signaling. In the Smad-dependent signaling, phosphorylated R-Smad (Smad2 or 3) complexes with Smad4 and co-translocates into the nuclei, where they recruit co-factors to regulate target gene expression. In the non-Smad-dependent pathway, phosphorylated TAK1 recruit TAB1 to initiate the MKK-p38 MAPK or MKK–ERK1/2 signaling cascade. Smad7 negatively regulate Smad signaling by preventing R-Smad phosphorylation, targeting R-Smad for ubiquitin–proteasome degradation with Smurf2 and inhibiting R-smad/co-Smad complex nuclei translocation. Arkadia positively regulates Smad signaling by targeting Smad7 for ubiquitin–proteasome degradation. MAPK phosphorylates Runx2 to promote its transcriptional activity while Smad2/3 recruits HDACs to antagonize Runx2 activity. TGF-β–Smad signaling promotes proliferation, chemotaxis, and early differentiation of osteoprogenitor. However, it inhibits osteoblast maturation, mineralization, and transition into osteocyte. It also inhibits osteoclast differentiation by decreasing RANKL/OPG secretion ratio, although it promotes osteoclastogenesis via directly binding with receptors on the osteoclast. TGF-β, transforming growth factor-β.

BMP signaling in bone. BMP activity is antagonized by cognate binding proteins, including Noggin, Grem1, Chordin, CHL, and Fellistatin. BMPs bind to homomeric type II receptors, which transphosphorylases homomeric type I receptor to induce Smad-dependent and non-Smad-dependent signaling. In the Smad-dependent signaling, phosphorylated R-Smad (Smad1, 5, or 8) complexes with Smad4 and co-translocates into the nuclei, where they recruit co-factors and Runx2 to regulate osteogenic gene expression, for example, Runx2, Dlx5, and Osx. In the non-Smad-dependent pathway, phosphorylated TAK1 recruit TAB1 to initiate the MKK-p38 MAPK or MKK–ERK1/2 signaling cascade. MAPK phosphorylates Runx2, Dlx5, and Osx to promote their transcriptional activity. MAPK also phosphorylates Runx2 to promote the formation of Smad–Runx2 complex. I-Smad (Smad6 or 7) negatively regulates Smad signaling by preventing R-Smad phosphorylation, targeting R-Smad or type I receptor for ubiquitin–proteasome degradation with Smurf1 and inhibiting R-smad/co-Smad complex nuclei translocation. Arkadia positively regulates Smad signaling by targeting I-Smads for ubiquitin–proteasome degradation. Ubc9/SUMO complex negatively regulates Smad signaling by targeting Smad4 for ubiquitin–proteasome degradation. BMP–Smad signaling promotes almost every step during osteoblast differentiation and maturation. BMP, bone morphogenetic protein.
The role of TGF-β signaling in bone
There are three TGF-βs: TGF-β1, TGF-β2, and TGF-β3 in mammals.2 TGF-βs elicit their cellular response via binding to a tetrameric receptor complex comprising two TGF-β type I receptors (TβRI/ALK5) and two type II kinase receptors (TβRII)2 (Figure 1). TβRII transphosphorylases TβRI resulting in subsequent phosphorylation of receptor-activated Smads (R-Smads), Smad2 and 3 (Figure 1).2 R-Smads then interact with the common Smad (Co-Smad), Smad4, and translocate into the nucleus, where they recruit co-factors to regulate gene (for example, CREB-binding protein (CBP) or p300) transcription.2 Recent studies have revealed that TGF-βs can also activate another group of R-Smad (Smad1, 5, and 8) via binding to ALK1.17,20 As an alternative non-Smad-dependent pathway, TGF-β also activates kinase 1 (TAK1) and TAK1-binding protein 1 (TAB1) which in turn initiates the MKKs (MAPK pathway member-encoding genes kinases)-p38 MAPK or -Erk (extracellular signal-regulated kinase) signaling cascade (Figure 1).4
TGFβs ligands
All TGF-β isoforms are expressed by perichondrium and periosteum21,22 as well as epiphyseal growth plate23. However, only Tgfb2-null mice displayed severe skeletal abnormalities in both endochondral and intramembranous bone,24 while Tgfb1-deficient mice and Tgfb3-null mice had almost normal skeleton.25–27 Thus, TGF-β2, but not TGF-β1 or TGF-β3, is essential for embryonic skeleton development.
TGF-β has an important role in maintaining postnatal bone mass by coupling bone resorption and bone formation (Figure 1). TGF-βs are synthesized as large precursor molecules, composed of mature TGF-β and latency-associated protein (LAP).28 LAP remains noncovalently bound to mature TGF-β as it is secreted, rendering it inactive by masking the ECM (extracellular matrix) of many different tissues.28 Cleaving LAP by osteoclastic bone resorption release active TGF-β1 to induce enrichment of osteoprogenitor in the bone resorption lacunae.7 Tang et al. crossed Tgfb1-null mice with immunodeficient Rag2−/− mice to overcome the early death of Tgfb1-null mice.8,25 The Tgfb1−/−Rag2−/− mice showed a significant loss of trabecular bone density and reduced osteoblast number on the bone surface.8 A gain-of-function mutant of TGF-β1 causes Camurati–Engelmann disease (CED) in humans,19,20 which is characterized by diaphyseal thickening and fluctuating bone volume. This mutation results in a similar bone defect in mice (CED mice).8 However, TGF-β1 is unable to induce osteogenesis in mesenchymal pluripotent cells, but increases the pool of osteoprogenitors by inducing chemotaxis and proliferation.8 Apoptosis of osteoblasts is also blocked by TGF-β1 deletion through maintenance of survival during transdifferentiation into osteocytes.29 In addition, TGF-β treatment blocked osteoblast mineralization in culture,30 indicating its bi-functions in osteoblast differentiation. On the other hand, active TGF-βs regulate bone resorption in a dose-dependent manner. The gradient of TGF-β created during osteoclast bone resorption can limit further osteoclast activity.28 Low concentrations of active TGF-β can induce macrophage migration to the bone resorption pits, whereas high concentrations of active TGF-β inhibit migration of osteoclast precursors.28 TGF-β was also shown to promote osteoclast differentiation at low dose and inhibits osteoclast differentiation at high dose through regulating RANKL/OPG ratio secreted by osteoblasts.7,31 Furthermore, TGF-β1 via binding to its receptor on osteoclasts activates Smad2/3, which associates directly with TRAF6–TAB1–TAK1 complex and favors osteoclast differentiation.32
TGF-β is a double-edged sword in the maintenance of articular cartilage metabolic homeostasis and the pathogenesis of arthritis33. Loss of TGF-β signaling in cartilage induces chondrocyte hypertrophy, ultimately resulting in cartilage degeneration, and pharmacological activation of the TGF-β pathway has therefore been proposed to preserve articular cartilage integrity during osteoarthritis (OA).19,34,35 However, age-dependent switch from TGF-β-ALK5-Smad2/3 to -ALK1-Smad1/5/8 signaling contributes to osteophyte formation and the pathogenesis of osteoarthritis.17,36 At the onset of either OA or rheumatoid arthritis (RA), elevated active TGF-β1 resulted from excessive bone resorption recruits MSCs in the subchondral bone marrow and induces the formation of “osteoid islet”, which leads to the degeneration of the overlying articular cartilage.37,38 Transgenic expression of active TGF-β1 in osteoblastic cells induced OA, whereas inhibition of TGF-β activity in subchondral bone attenuated the degeneration of articular cartilage.37 Similarly, aberrant activation of TGF-β in subchondral bone is involved at the onset of RA joint cartilage degeneration, whereas either systemic or local blockade of TGF-β activity in the subchondral bone attenuated articular cartilage degeneration in RA.38 Furthermore, knockout of the TβRII in nestin-positive MSCs leads to less development of both OA and RA relative to wild-type mice.37,38
Extracellular regulation of TGF-β signaling
ECM proteins regulate TGF-β signaling through controlling ligand availability (Figure 1). LTBPs covalently bind latent TGF-β and modulate tissue levels of TGF-β. Ablation of LTBP-3 reduces both osteogenesis and bone resorption, and resulted in increased bone mass associated with decreased levels of TGF-β.39,40 Binding of TGF-β to small leucine-rich proteoglycans (decorin, biglycan, and lumican) restricts the TGF-β in the ECM so as to inhibit its activity.41 Bgn and Dcn double deficiency results in osteopenia and a striking change in collagen fibril shape.41 Excessive TGF-β signaling, caused by reduced binding of decorin, is also found to be a common mechanism of osteogenesis imperfecta in mouse models.42
TGF-βs receptors
Similarly to TGF-βs, the expression of TGF-β receptors is also detected in the growth plate and perichondrium.21,22,43 Mice lacking either TβRI/ALK5 or TβRII in MSC results in short long bone and defects in joint development fusion.30,44–46 MSC-specific Tgfbr2-deletion and MSC-specific Alk5-deletion both promote chondrocyte hypertrophy and decreased chondrocyte proliferation, which was also observed in DN-TβRII transgenic mice.30,34,44–46 Thus, TGF-β signaling favors chondrocyte proliferation but blocks the transition from proliferative chondrocyte into hypertrophic chondrocyte. Mice lacking TβRII/ALK5 in the chondrocyte can reproduce defects in the skull base and vertebrae as those observed in the Tgfb2-null mice,24,47 but cannot influence normal long bone development.47 This phenotype indicates that TGF-β might regulate growth plate development largely through mediating paracrine cytokines secretion by perichondrium cells.
TGF-β signaling favors bone formation by promoting osteoprogenitor enrichment. Consistently, deletion of Alk5 in MSC resulted in reduced bone collars and trabecular bones associated with decreased osteoblast number.30 The study showed that TGF-β, through TβRI, promotes pre-osteoblast commitment and early differentiation.30 However, deletion of Tgfbr2 in osteoblasts resulted in an unexpected increase in bone mass due to enhanced PTHrP signaling.43
Smad-dependent pathway
Smad2 and Smad3 respond to TGF-β and regulate TGF-β-mediated osteoblast and chondrocyte differentiation (Figure 1). Smad2/3 inhibits Runx2 expression, and activated Smad3 also recruits class II histone deacetylases (HDACs) 4 and 5 to repress the function of Runx2.48–51 TGF-β can no longer inhibit the differentiation of osteoblasts in the absence of Smad3.52,53 Although TGF-β-Smad3 negatively regulates osteoblastogenesis, it also inhibits osteoblast apoptosis and the differentiation into osteocyte. Thus, Smad3-null mice are osteopenic associated with increased osteocyte number and apoptosis.52 Furthermore, TGF-β-induced inhibition of chondrocyte maturation is potentiated by Smad2/3 signaling,54 and abolished by disrupting Smad2/3 activation.54,55 Smad3-null mice also displayed accelerated chondrocyte hypertrophy, at least partially through reduced Noggin level and unopposed BMP signaling.56
In addition, TGF-β-Smad3 signaling is required for postnatal maintenance of articular cartilage homeostasis. Smad3-null mice and chondrocyte-specific Smad3 CKO mice exhibit articular cartilage degeneration and automatically develop OA.57–59 Mutations of Smad3 in human caused aneurysms–osteoarthritis syndrome,15,60,61 which is presenting with aneurysms, mild craniofacial features, and skeletal and cutaneous anomalies. Mechanistically, TGF-β signals through Smad3 to confer a rapid and dynamic repression of Runx2-induced MMP-13 expression, and signals through p38 to promote Runx2-induced MMP-13 expression in the absence of Smad3.58
Non-Smad-dependent pathway
Non-Smad-dependent pathway initiated by TGF-β also contributes to bone formation. Studying MSC-specific Alk5 CKO mouse model revealed that TGF-β promotes osteoblast proliferation and early differentiation through both Smad2/3 and MAPK signaling pathway.30 MAPK signal activated by TGF-β and BMP positively regulates Runx2 expression and function to promote MSC differentiation.62 Furthermore, TGF-β2 stimulates cranial suture closure by promoting osteoprogenitor proliferation via the MKK–ERK signaling.63
Previous studies showed that TGF-β, through Alk5-Smad2/3 or Alk5-MAPK signaling, promotes osteoblast progenitor enrichment and early differentiation, but negatively regulates osteoblast differentiation and mineralization at latter stages. TGF-β is also a molecule coupling bone formation with bone resorption. The relationship between TGF-β and cartilage is more complex. Either loss of TGF-β or excessive active TGF-β contributes to the progress of osteoarthritis. Further studies will elucidate how TGF-β dose-dependently and stage-dependently regulates cartilage integrity. Those findings would facilitate the design of therapeutic approaches for targeting TGF-β in OA treatment.
The role of BMP signaling in bone
BMP signaling is mediated through type I and type II BMP receptors (Figure 2). After binding to BMP ligands, homomeric dimers of the type II receptors form a tetrameric complex with homomeric dimers of the type I receptors, and induce transphosphorylation of the type I receptors. This dynamic interaction leads to signal transduced through either Smads or MAPKs, which further activates the transcription of specific target genes involved in osteoblastic differentiation and bone formation (Figure 2).
BMP ligands
Among the 14 BMPs, BMP-2, 4, 5, 6, 7, and 9 exhibit high osteogenic activity.64,65 Osteogenic capability of BMP-2 and BMP-7 have been vastly studied and the recombinant proteins are currently being investigated in human clinical trials of craniofacial deformities, fracture healing, and spine fusion.13,14 BMP-2 vastly increases osteocalcin expression66 and a short-term expression of the BMP-2 is necessary and sufficient to irreversibly induce bone formation.67 BMP-7 induces the expression of osteoblastic differentiation markers, such as ALP and accelerates calcium mineralization.68–70 BMP-3 is a ‘non-canonical’ BMP molecule that transduces type IIB Activin receptor (AcvrIIB)-Smad2/3 signaling to oppose the osteogenic function of other BMPs.71 In vivo, BMP-3 is mainly produced by osteoblasts and osteocytes,71 and negatively regulates bone mass in vivo, as shown by knockout and transgenic mouse models.72,73 Interestingly, a recent study showed that BMP2 addition to culture media rapidly induced expansion of isolated mouse skeletal stem cell (mSSC).74 Delivery of BMP2 induces de novo formation of the mSSC and bone in extraskeletal locations.74 In addition, co-delivery of BMP2 and sVEGFR1 induces de novo formation of cartilage in extraskeletal locations.74 Expression of the BMP antagonist gremlin 1 defines a population of osteochondroreticular stem cells in the bone marrow, which is self-renewal and is able to generate osteoblasts, chondrocytes, and reticular marrow stromal cells, but not adipocytes.75
BMPs have essential roles at many steps in endochondral bone development. Studies showed that BMP signaling promotes chondrocyte proliferation and differentiation,76–78 by maintaining the expression of Sox9, a transcription factor that is essential for chondrogenic commitment and differentiation.77,79 BMP-7 null mice died shortly after birth and exhibits skeletal patterning defects restricted to the rib cage, skull, and hindlimbs.80 Conditional deletion of BMP-2, BMP-4, or BMP-7 in MSC results in normal skeletongenesis.70,81,82 Only BMP-2 CKO mice have frequent fractures that fail to heal, which is not observed in either Bmp-4 or Bmp-7 CKO mice.70,83,84 The studies indicate that BMPs have duplicate functions in skeleton development, while BMP-2 has a unique role in postnatal bone formation. MSC-specific Bmp-2/-4 DKO mice have extremely malformed limbs and a severe impairment of osteogenesis, which is not observed in Bmp-2/-7 DKO mice.82,85 Chondrogenic condensations in some skeletal elements fail in MSC-specific Bmp-2/-4 DKO mice.82 Furthermore, chondrocyte-specific Bmp-2 CKO and Bmp-2/4 DKO mice exhibits severe disorganization of chondrocytes within the growth plate region.86 Collectively, these studies revealed that coordinated functions of endogenous BMP-2 and BMP-4 are required for osteoblastogenesis as well as chondrocyte proliferation, differentiation and apoptosis during skeletal development.
BMP7 has a pivotal role in postnatal maintenance of articular cartilage. Intra-articular administration of rhBMP7 significantly inhibited articular cartilage degeneration and blocked production of inflammatory cytokines by the synovial membrane.18,87,88 Bmp7f/fPrx1-Cre mice, and Bmpr1af/fGdf5-Cre mice exhibit articular cartilage degeneration and automatically develop OA.89
BMP receptors
There are three type I receptors for BMPs, type IA BMP receptor (BMPR1A/ALK3), type IB BMP receptor (BMPR1B/ALK6), and type I activin receptor (AcvR1/ ALK2). BMPs exert their osteogenic signaling through those receptors. Activating mutants of Acvr1 cause ectopic ossification in mouse90 and in human disease (fibrodysplasia ossificans progressive, FOP).91,92 Mechanically, active AcvR1 induces epithelial-to-mesenchymal transition and promotes ectopic osteoblastogenesis via canonical Smad1/5 signaling.90,93 On the other hand, blocking BMP signaling through overexpression of DN-BMPRII or deletion of Bmpr1a in osteoblast results in low bone mass.94,95 Although deletion of Bmpr1a in preosteoblasts causes an unexpected increase in bone mass due to reduced bone resorption, osteoblast differentiation remains low in this condition.96–98
The three type I BMP receptors (BMPR1A, BMPR1B, and ALK6) have largely redundant roles in skeletal development. BMPR1A and BMPR1B share similar expression pattern in chondrocyte condensations and developing skeletons,99–104 and AcvR1 is expressed throughout the growth plate.105 Activation and dominant-negative experiments proved the BMPRIB is essential for cartilage condensation in chicks.103 Activation experiments showed that BMPR1A, BMPR1B, and AcvR1 are all able to promote chondrogenesis.77,101,103,106–108 However, deletion of each of them alone only results in mild skeletal defects or defects restricted to certain skeletal elements.77,104,105,109,110 In contrast, Bmpr1a/Bmpr1b DKO mice, Acvr1/Bmpr1a DKO mice, and Acvr1/Bmpr1b DKO mice exhibit generalized chondrodysplasia that is much more severe than any of the corresponding mutant strains.104,105,110 The loss of both BMPR1A and BMPR1B blocks chondrocyte condensation, proliferation, differentiation, survival, and function due to impaired Sox proteins (Sox-5, 6, and 9) expression.104,110 In conclusion, those mouse models demonstrated that BMP signaling is essential for almost every step during endochondral bone development (Figure 3).

Crosstalk between BMP, TGF-β, and other signaling during osteoblast differentiation. BMP has dual roles in Wnt signaling. On one hand, BMP inhibits Wnt/β-catenin signaling by increasing Wnt antagonist Dkk1 and Sost expression and by preventing β-catenin nuclei translocation. On the other hand, BMP promotes Wnt/β-catenin signaling by forming co-transcriptional complex with β-catenin/TCF/LEF/Runx2, by increasing Wnt expression, by antagonizing Dvl function and by decreasing b-TrCP expression. BMP signaling promotes FGF, Hh, PTHrP signaling by increasing IHH expression, increasing FGF expression, decreasing Ptch1 expression, respectively. BMP is essential for IHH-induced osteoblast differentiation. FGF, IHH, Wnt, and PTHrP signaling all promotes BMP2 expression so as to enhance BMP signaling. FGF and IHH signaling is essential for BMP-induced osteoblast differentiation. TGF-β antagonizes PTHrP signaling through TGF-β type II receptor complexing and internalizing together with PTHrP receptor (PPR). TGF-β promotes Wnt activity by increasing Wnt ligands expression and decreasing Axin expression. Fibroblast growth factor (FGF) and Wnt all increase TGF-β expression to promote TGF-β signaling. BMP, bone morphogenetic protein; PTHrP, parathyroid hormone-related peptide; TGF-β, transforming growth factor-β.
Smad-depedent pathway
Most BMPs activate Smad1/5/8 as their R-Smad (Figure 2). Smad1/5/8-Smad4 complex transcribed Runx2 expression, as they complex with Runx2 to initiate other osteoblast gene expression.62,111,112 Smad1 is an important mediator of BMP’s osteogenic function. In osteoblast-specific Smad1-CKO mice, BMP signaling was partially inhibited, osteoblast proliferation and differentiation were impaired and mice developed an osteopenic phenotype.113 Smad1 and Smad5 together mediate the role of BMP in endochondral bone development. Chondrocyte-specific deletion of Smad1 results in delayed calvarial bone development and shortening of the growth plate.113,114 The addition of Smad5 haploinsufficiency or insufficiency leads to more severe chondrodysplasia, while addition of Smad8 insufficiency did not worsen the defects.114,115 Thus, Smad8 contributes much less to skeleton development as compared with Smad1 and Smad5. Only BMP3 also activates Smad2/3, which antagonized osteogenic activity of Smad1/5/8 and opposed the function of other BMPs in osteoblast differentiation.116
Both BMPs and TGF-βs signal through Smad4. Smad4 mutation in humans causes Myhre syndrome, a developmental disorder, characterized by short stature and facial dysmorphism.117,118 In vitro Smad4 ablation partially suppressed BMP-4-induced osteoblast differentiation.119 In vivo abrogation of Smad4 in chondrocytes results in dwarfism with a severely disorganized growth plate and ectopic bone collars in perichondrium.120 In the Smad4-deficient growth plate, resting zone was expanded, while chondrocyte proliferation was reduced and hypertrophic differentiation was accelerated.120 Deletion of Smad4 in mature osteoblasts causes lower bone mass and decreased osteoblast proliferation and differentiation up to 6 months of age, while the trabecular bone volume in the mutant mice increased after 7-month-old due to reduced bone resorption.121 Embryonic deletion of Smad4 in pre-osteoblast causes stunted growth, spontaneous fractures and a combination of features seen in osteogenesis imperfecta, cleidocranial dysplasia, and Wnt-deficiency syndromes.122 Postnatal deletion of Smad4 in pre-osteoblasts increases mitosis of cells on trabecular bone surfaces as well as in primary osteoblast cultures, while delays differentiation and matrix mineralization by primary osteoblasts associated with altered β-catenin acvitity.123 Knockout of Smad4 in osteoblast also reveals that BMP/TGFβs coordinate with Wnt signaling to maintain normal bone mass. In summary, Smad4 has multiples roles in growth plate development and postnatal bone homeostasis.
Non-Smad-dependent pathway
TAK1–MKK–MAPK pathway is also involved in the fine-tuning of BMP effects on skeletal development and osteogenic differentiation (Figure 2). Morphological defects of BMP-receptor-deficient mice are found to be associated with a decrease in both p38 MAPK and Smad activity.105 Ectopic ossification of FOP patients is associated with an increase in both p38 MAPK and Smad activity.124 Either chondrocyte- or MSC-specific Tak1-deletion results in disorganized growth plates as well as a failure to maintain interzone cells of the elbow joint.125 Postnatal chondrocyte-specific Tak1 deletion results in severe defects of growth plate and articular cartilage development with reduced expression of SOX protein, proteoglycan and type II collagen.126 Osteoblast-specific deletion of Tak1 results in clavicular hypoplasia and delays fontanelle fusion, which is a phenotype similar to that of cleidocranial dysplasia, a human disease that is caused by Runx2 deficiency.127 Mice with deletion of other MAPK pathway member-encoding genes, MAPK kinase 3 (Mkk3), Mkk6, p38a, or p38b, also display profoundly reduced bone mass secondary to defective osteoblast differentiation.127 Mechanistically, MAPKs phosphorylate a number of osteoblast master transcription factors including Runx2,111,127,128 Dlx-5129 and Osterix130–132 (Figure 2). The phosphorylation facilitates the recruitment of cofactors and promotes their transcriptional activity.111,127–130 MAPKs also positively regulate Runx2 and Osterix expression to promote MSC differentiation.62 Furthermore, MAPK signaling enhances the canonical BMP–Smad signaling by promoting the interaction between Runx2 and Smad complex.111
In conclusion, most BMP ligands are strong osteogenic agents, through both Smad and non-smad signaling pathway, which synergize at osteogenic transcriptional factors (for example, Runx2, Osx). BMPs also have critical roles in multiple stages of endochondral bone development, including mesenchyme condensation, chondrocyte proliferation, and chondrocyte differentiation. In addition, BMP7 and BMPR1A also have roles in postnatal homeostasis of articular cartilage. Recent studies have shown that BMP2 enhances proliferation and enrichment of mouse skeletal stem cell. Thus, it is of full potential to utilize BMP locally in bone and cartilage regeneration.
Regulation of BMP and TGF-β signaling in bone
BMP and TGF-β signaling are delicately controlled by multiple machineries: Extracellular matrix proteins control the availability of the ligands; Inhibitory Smads antagonize various steps in the Smad-dependent signaling; ubiquitin–proteasome machinery controls stability of various signal transducers and inhibitors; miRNAs regulate the signaling at post-translational level; co-repressors and epigenetic factors regulate the signaling at transcriptional level.
Regulators in the ECM
ECM proteins regulate TGF-β signaling through controlling ligand availability (Figure 1). LTBPs covalently bind latent TGF-β and modulate tissue levels of TGF-β. Ablation of LTBP-3 reduces both osteogenesis and bone resorption, and resulted in increased bone mass associated with decreased levels of TGF-β.39,40 Binding of TGF-β to small leucine-rich proteoglycans (decorin, biglycan, and lumican) restricts the TGF-β in the ECM so as to inhibit its activity.41 Bgn and Dcn double deficiency results in osteopenia and a striking change in collagen fibril shape.41 ExcessiveTGF-β signaling caused by reduced binding of decorin is also found to be a common mechanism of osteogenesis imperfecta in mouse models.42
BMP signaling is regulated by a group of cognate binding proteins (for example, Noggin, Chordin, Gremlin, and Follistatin) that competitively bound BMPs to prevent their binding to receptors133–135 (Figure 2). Noggin opposes BMP’s osteogenic function in vivo136 and in vitro.137 Nog-null mice died at birth with severe malformed skeletons due to unopposed BMP signaling,138,139 and addition of deficiency of other BMP antagonists (Grem1 or Follistatin) aggravated the skeleton malformation of Nog−/− mice.140,141 Ectopic expression of Noggin in osteoblast impairs osteoblastogenesis and results in osteopenia in mice.142,143 Ectopic expression of Noggin is also able to block cartilage development, change skeletal morphology, and prevent cranial suture fusion.144,145 Noggin expression is induced by BMPs, and acts in a negative regulatory loop to inhibit BMP activity.133 Other signals including Sox9 and FGFs also regulate BMP signaling via manipulating Noggin expression.145–148 Chordin and Chordin-like proteins (CHL) were identified as a factor dorsalizing the Xenopus embryo.149–152 Chordin and CHL treatment prevented BMP-induced osteoblast differentiation and mineralization,152,153 as well as BMP-induced chondrocyte maturation.149,152,154 Furthermore, Chordin−/−Nog+/− mice exhibit defects restricted to the head associated with increased BMP activity.155
I-Smad
Inhibitory Smads (I-Smad, Smad6, and 7) inhibits BMPs and TGF-βs signal in multiple ways: preventing R-Smad nuclei translocation, competitively binding with type I receptor to prevent R-Smad phosphorylation, and promoting R-smads or receptor degradation by recruiting E3 ubiquitin ligases Smurf1/2.156,157 Smad6 preferentially inhibits BMP signaling (Figure 2), while Smad7 interferes with both BMP and TGFβ signaling157 (Figures 1 and 2). Smad6 and 7 overexpression blocks BMPs-induced osteoblast and chondrocyte differentiation in vitro,107,158–161 and vice versa.160 Chondrocyte-specific Smad6 transgenic mice show postnatal dwarfism, osteopenia, and delayed chondrocyte hypertrophy due to inhibition of Smad1/5/8 signaling.162 Besides its role in BMP signaling, Smad6 and Smurf1 also induce Runx2 degradation in an ubiquitin-proteasome-dependent manner, which directly inhibits osteoblast differentiation.163 Thus, Smad6 expression is also mediated by BMP–Smad1–Runx2 signaling at transcriptional level and regulates BMP and Runx2 activity in a negative feedback loop.160,164 Conditional-overexpression of Smad7 in MSC or stage-specific chondrocytes results in defective mesenchymal condensation, chondrocyte proliferation and chondrocyte maturation, respectively.165
Ubiquitin–proteasomal degradation pathway
Stability of protein transducers within TGFβ and BMP signaling is under control of ubiquitin enzymes and de-ubiquitin enzymes (Figures 1 and 2). Smurf2 is an E3 ubiquitin ligase that targets TGFβ receptor and Smad2/3 for ubiquitin–proteasomal degradation.166,167 Ectopic overexpression of Smurf2 in condensing chondrogenic mesenchyme of chicken wing bud accelerates chondrocyte maturation and ossification.166 Smurf1 is also an E3 ubiquitin ligase with a wide range of a targets, including BMP type I receptors, Smad1/5, Runx2, and MKK2.162,163,167,168 Smad6/Smurf1 double transgenic pups exhibit delayed endochondral bone formation that is more severe than Smad6 transgenic pups.162 Smurf1-deficient mice were born normal but exhibited a temporal increase of bone mass as they aged, due to accumulation of phosphorylated MKK2 and activation of the downstream JNK signaling cascade.167 Arkadia, an E3 ubiquitin ligase that induces ubiquitylation and proteasome-dependent degradation of TGF-β and BMP suppressors, such as Smad6, Smad7, and c-Ski/SnoN, promotes osteoblast mineralization and differentiation induced by BMPs.169 SUMO (small ubiquitin-related modifier) and Ubc9 (ubiquitin conjugating enzyme 9) target Smad4 for degradation and inhibit osteoblastic differentiation induced by BMP2.170,171 Furthermore, deubiquitylating enzyme USP15 mediates K48-linked deubiquitylation of ALK3, to promote ALK3-smad1 signal and BMP-induced osteoblast differentiation.172
Transcriptional repressors
A group of co-repressors controls transcriptional activity of the R-Smad/co-Smad complex (Figure 2). Ski/SnoN interacts with R-Smad proteins to repress their activity. c-Ski/SnoN, evoked by TGF-βsuppresses the BMP signaling and hypertrophic chondrocyte maturation. SnoN is highly expressed in articular cartilage and may involve in TGFβ-mediated cartilage homeostasis. Tob as a co-repressor is another negative regulator of BMP/Smad signaling in osteoblasts.173 BMP2-signal is elevated in the absence of Tob and repressed by overproduction of Tob.174,175 Mice carrying a targeted deletion of the Tob gene had a greater bone mass resulting from increased numbers of osteoblasts and were protected from OVX-induced osteoporosis.174,175
MicroRNAs
Recent researches have raised the importance of microRNA (miRNA) in skeleton homeostasis and in TGF-β and BMP signaling pathway (Figures 1 and 2). BMP treatment regulates multiple miRNA expression during osteoblastogenesis, and a number of those miRNAs feedback to regulate BMP signaling:176–179 miR-133 targets Runx2 and Smad5 to inhibit BMP-induced osteogenesis;176 miR-30 family members negatively regulate BMP-2-induced osteoblast differentiation by targeting Smad1 and Runx2;177,178 miR-322 targets Tob and enhances BMP response.179 In addition, some non-BMP-regulated miRNAs also have regulatory roles in BMP signaling: miR-141 and -200a remarkably modulate the BMP-2-induced pre-osteoblast differentiation through the translational repression of Dlx5;180 miR-542-3p targets BMP7 and represses BMP7-induced osteoblast differentiation and survival;181 miR-20a promotes osteogenic differentiation through upregulation of BMP/Runx2 signaling by targeting PPARγ, Bambi, and Crim1;182 miR-140 targets a mild inhibitor of BMP Dnpep, and loss of miR-140 in mice causes growth defects of endochondral bones and craniofacial deformities.183 Furthermore, several miRNAs are reported to regulate chondrocyte differentiation by targeting TGF-β and BMP signaling. Profiling identified expression of several miRNAs that changed significantly in human OA chondrocyte compared with normal cells targets Smad-signaling, including miR-20b, miR-146a, and miR-345.184–186 miR-146a, a miRNA increased in OA, targets Smad4 and induces chondrocyte apoptosis and cellular responsiveness to TGF-β.187,188 miR-199a(*) targets Smad1 and adversely regulates early chondrocyte differentiation.189
Epigenetic regulation
The Sox9-related transcriptional apparatus activates its target gene expression through p300-mediated histone acetylation on chromatin, and TGF-β has an important role in recruiting p300 to the promoter.190 TGF-β also induces the expression of KDM4B, which removes the silencing H3K9me3 marks on the Sox9 promoter and increased Smad3 occupancy on the Sox9 promoter, so as to enhance Sox9 expression.191 HDAC inhibitor suberoylanilide hydroxamic acid (SAHA; vorinostat) increases the Runx2 promoter acetylation to intrigue Runx2 expression and osteoblastogenesis, in a BMP2-dependent manner.192 The Bmp2 promoter regions of the genes are epigenetically locked with increased CpG methylation and decreased H3K9 acetylation.193 And CpG-demethylating agent or the HDAC inhibitor trichostatin-A renders Bmp2 expression.193 At the protein level, HDACs deacetylate Smad7 to protect it against ubiquitination and degradation.4 BMP-2 signaling stimulates p300-mediated Runx2 acetylation, which increases its stability and transactivation activity.4 HDAC4 and HDAC5 deacetylate Runx2 and renders its degradation through ubiquitin–proteasomal pathway.4
In summary, traditional regulators (for example, ECM, I-Smad, ubiquitin-related proteins, transcriptional co-factors) of BMP and TGF-β signaling have been extensively studied and the regulatory network is finely established. Prosperous progress will be made to discover novel regulation of BMP and TGF-β signaling by miRNA and epigenetic factors, and will provide more clues to design new treatment for bone diseases through targeting BMP and TGF-β signaling.
Crosstalk between TGF-β and BMP signaling with other signaling in bone
Coordinated signaling networks controlled by multiple cytokines regulate skeletogenesis and osteoblast differentiation. To regulate bone homeostasis, TGF-β and BMP signaling dynamically crosstalks with other pathways including Wnt, Hedgehog, FGF, Notch and PTHrP (Figures 3 and 4; Table 3).

Crosstalk between BMP, TGF-β and other signaling in the growth plate. In the epiphyseal growth plate, differentiating chondrocytes are organized into four layers, including resting zone (RZ), proliferation zone (PZ), hypertrophic zone (HZ), and calcified zone (CZ). The calcified zone was gradually replaced by the trabecular bone (TB). Positive regulating cytokines (→) and negative regulating cytokines (--|) of each zone are listed on the left adjacent to them. BMP, bone morphogenetic protein; FGF, fibroblast growth factor; IHH, Indian hedgehog; PTHrP, parathyroid hormone-related peptide; SHH, Sonic hedgehog; TGF-β, transforming growth factor-β.
Crosstalk between TGF-β and BMP signaling with Wnt signaling
TGF-β cooperates with Wnt to stimulate both osteoblast and chondrocyte differentiation in a positive regulatory loop. TGF-β upregulates the expression of Wnts (Wnt-2, -4, -5a, -7a, and -10a) as well as Wnt co-receptor LRP5, and suppresses the expression of β-catenin inhibitor Axin1/2, to promote Wnt/β-catenin signaling.194,195 On the other hand, Wnt signaling induces Runx2-mediated TGFβRI expression in a β-catenin-independent way to promote TGF-β signaling.196 Bosch et al.20 showed that canonical Wnt signaling skews TGF-β signaling towards signaling via ALK1-Smad1/5/8 to promote chondrocyte hypertrophy.
BMPs have dual functions in regulating Wnt signaling. BMP induces Dkk1 and Sost expression through MAPK and Smad signaling, respectively, via BMPRIA to inhibit Wnt signaling in osteoblast, and negatively regulated bone mass.96,98 Smad4 competitively binds β-catenin and prevents its binding with Tcf/Lef transcription complex.123 Thus, ablation of Smad4 either in the crest neuron stem cell or in osteoblast results in upregulation of canonical Wnt signaling so as to promote bone formation.60,123 BMP2-induced formation of Smad1-Dvl1 complex also restricts β-catenin and inhibits its activity.197 On the other hand, BMPs stimulate osteoblast and chondrocyte differentiation synergistically with Wnt signaling. BMP-2 promotes canonical Wnt signaling by inducing Wnt3a, Wnt1, Lrp5 expression and inhibiting the expression of β-TrCP, the F-box E3 ligase was found responsible for β-catenin degradation in osteoblasts.198,199 Ablation of Smad4 in the pre-osteoblasts depletes LRP5 expression and impairs both BMP and Wnt signaling.123 BMP-2-induced LRP-5 expression also contributes to chondrocyte hypertrophy and osteoarthritis progress.200 Osteoblastogenesis is maximized in the presence of both BMP and Wnt signaling.198,201,202 Smad and TCF/LEF/β-catenin cooperatively complex with each other on the promoter to achieve the highest expression of osteoblast genes (Dlx5, Msx2, and Runx2).202 Moreover, activation of Wnt signaling by Wnt3a or overexpression of β-catenin/TCF4 promotes BMP signaling by stimulating BMP2 transcription.203
Crosstalk between TGF-β and BMP signaling with FGF signaling
FGF and BMP signal synergistically promote osteoblast differentiation. BMP signaling is essential for FGF-induced osteoblast differentiation,204 and the osteogenic effect of BMP2 is also repressed in the absence of FGF2.205 FGF2 is responsible for BMP-induced nuclei translocation and accumulation of Runx2 and P-Smad1/5/8.206 FGF2, FGF9, and FGF18 induce while DN-FGFR reduces BMP2 expression.204,207,208 Disruption of the FGF2 in mice impairs the expression of BMP2 and reduces bone mass.205 However, FGF2 has a critical role in osteoblast proliferation but inhibits mineralization while BMP2 is instrumental in stimulating mineralization.209,210 Thus, besides cooperative functions, FGF2 and BMPs have different roles at different stages of osteoblast differentiation.
Antagonistic BMP and FGF pathways control the differentiation and proliferation rate of chondrocytes in the growth plate.76,110 Although BMP signal promotes chondrogenesis, FGF signal is a negative regulator of chondrogenesis and multiple mutations with constitutive activity of FGFR3 result in achondroplasia in humans.211 BMP signaling is required to inhibit activation of FGF signaling (for example, STAT and ERK1/2 activation) at least in part by inhibiting the expression of FGFR1.110 FGF signals are downregulated by ectopic BMP expression or Chordin/Noggin deficiency, and upregulated by CHL1 ectopic expression.153,212 Chondrocyte-specific activation of Fgfr3 in mice induced premature synchondrosis closure and enhanced osteoblast differentiation around synchondrosis associated with promoted BMP signaling, due to increased BMP ligand and decreased BMP antagonist expression.211 Chondrocyte-specific deletion of BMPRIA rescued the bone overgrowth phenotype observed in Fgfr3-deficient mice by reducing chondrocyte differentiation.168
Crosstalk between TGF-β and BMP signaling with Hedgehog signaling
BMPs and Hedgehog signaling cooperatively regulate osteoblastogenesis in long bones.213,214 IHH is required for BMP-induced osteogenesis in vitro.213 Hedgehog-Gli activators direct osteo-chondrogenic function of BMPs toward osteogenesis in the perichondrium.215 SHH-Gli2 stimulates BMP-2 expression at transcription level to enhance osteoblast differentiation.216,217 BMP2 also induces expression of IHH and downregulates expression of patched 1 (PTC1), a negative regulator of Hedgehog signaling, during osteoblast differentiation in a positive feedback loop.213
During growth plate development, IHH-PTHrP regulatory loop keeps chondrocyte in the proliferation pool and favors elongation of long bones. Previous studies have shown that BMP signaling upregulates IHH–PTHrP signaling in vivo, and maintains a normal chondrocyte proliferation rate in parallel with IHH.218,219 Expression of IHH, PTC1, and PTHrP receptor (PPR) were decreased in Smad4-deficient growth plates, but were enhanced in CA-ALK2-, CA-BMPRIA-, or CA-BMPRIB-overexpressed chick limb bud.108,120 In cultured mouse limb explants, BMP2 application induces and Noggin antagonizes IHH expression.218 BMP6 also promotes IHH expression to favor chondrocyte hypertrophy.220 Furthermore, chondrocyte-specific Smad1/5 CKO mice develop chondrodysplasia and exhibit abnormal growth at the end of bones, probably owing to an imbalance in the crosstalk between the BMP, FGF, and IHH/PTHrP pathways.213 On the other hand, IHH also promotes chondrocyte hypertrophy independent of PTHrP via integrating with BMP and Wnt signaling.221 Chondrocyte-specific deletion of PTC1 upregulated BMP expression and BMP–Smad signaling.222 IHH deficiency downregulates BMP expression and reduces cranial bone size and all markers.214
In axial skeleton development, sequential SHH and BMP signals are required for specification of a chondrogenic fate in presomitic tissue.223 In this process, SHH promotes BMP expression, and initiates an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce chondrogenesis.223 On the other hand, BMP activity negatively regulates SHH transcription and a BMP-SHH negative-feedback loop serves to confine SHH expression during limb development.153,212 SHH signal was impaired in the defected craniobone in the absence of Chordin and Noggin, the two BMP cognate binding partners.155
Crosstalk between TGF-β and BMP signaling with Notch signaling
Notch activation favors BMP-induced osteoblast differentiation.224,225 Notch inhibition represses expression of BMP target genes.226 BMP-2 and TGF-β regulates expression of signaling proteins in Notch pathway (for example, Lfng, Hey1, and Hes1).227 Conditional deletion of Jag1, the Notch ligand in CNC, revealed altered collagen deposition, delayed ossification, and reduced expression of early and late determinants of osteoblast development during maxillary ossification, associated with dysregulation of BMP receptor expression in the Maxillary mesenchymal stem cell.
Crosstalk between TGF-β and BMP signaling with PTH signaling
Osteoblast-specific Tgfbr2 CKO mice have increased bone mass which is caused by hyperactivition of PTH type I receptor (PTH1R) and could be rescued by disruption of PTH signaling by injection of PTH (7-34) or ablation of PTH1R.43,228 Mechanistic studies show that Tgfbr2 directly phosphorylates the PTH1R, which modulates PTH-induced endocytosis of both receptors and attenuates both TGF-β and PTH signaling in vivo.43,228 PTH enhances MSC differentiation into the osteoblast lineage through endocytosis of the PTH1R/Lrp6 complex, which enhances the BMPs-receptor binding affinicty.229 Positive regulation of PTHrP expression by TGFβ/BMP signaling can be observed in Smad4 mutant mice, Smad1/5 mutant mice and chick limbs overexpressing constitutively active BMP receptors. It has also been proposed that TGF-β promotes PTHrP expression.22 Both TGF-β and PTH inhibit terminal differentiation of chondrocyte in the growth plate, while BMPs promotes this process.230
Taken together, these studies demonstrate that TGF-βs and BMPs cooperate with other cytokines (Wnt, Hedgehog, FGF, Notch, and PTHrP) to regulate osteoblast and chondrocyte differentiation. Further studies will not only elucidate the specific interactions between those signaling pathways in bone, but also explore the potential of combined treatment for bone diseases by targeting multiple signaling pathways to achieve optimal outcome.
TGF-β and BMP signaling in human diseases and clinical application
Given the important roles of TGF-β and BMP in chondrogenesis and osteogenesis, mutations in TGF-β and BMP signaling cause a wide range of skeletal disorders in human (Table 2). Not only do these studies reveal the roles of TGF-β and BMP signaling in bone formation and homeostasis, but also they decipher the molecular mechanisms and pathogenesis underlying related bone diseases and, most importantly, provide significant insights into the development of novel therapies.
Fibrodysplasia ossificans progressiva (FOP; OMIM 135100)
FOP is a rare autosomal dominant disease characterized by progressive ectopic ossification of connective tissues and skeletal muscles. FOP is caused by activating mutations in BMP type I receptor Acvr1. Among them, R206H is the first characterized and most prevalent mutation, while other site mutations were also reported and contribute to the clinical variability and diverse severity of the disease (for example, R258S, L196P, and G328E).91,92,231–233
Brachydactyly type A2 (BDA2; OMIM 112600)
BDA2 is characterized by hypoplasia of the second middle phalanx of the index finger and sometimes the little finger.234 Inactivating mutations of Bmpr1b gene (for example, I200K, R486K, and R486Q)235,236 or GDF5 gene (for example, L441P and R380Q)237,238 were reported in BDA2 patients. Dathe et al.239 reported another genetic cause of BDA2: duplication of a regulatory element downstream of BMP2 repressed BMP2 expression. GDF5 mutations were also found to be the cause of a number of cartilage disorders, including symphalangsism,238 chondrodysplasia,240 and osteoarthritis.16
Myhre syndrome (MIM 139210)
Inactivated SMAD4 mutation causes Myhre syndrome, which is a developmental disorder characterized by short stature, short hands and feet, facial dysmorphism, muscular hypertrophy, deafness, and cognitive delay.117,118 Myhre syndrome patients carry an I500 mutation at Smad4 that results in a stabilized but unfunctional mutant.118,241
Noggin-mutation-related diseases
As the major BMP antagonist, mutation of Noggin in human causes multiple bone disorders. Missense mutations in BMP antagonist Noggin cause tyrosine kinase-like orphan receptor 2 (ROR2)-negative brachydactyly type B (BDB2; OMIM611377), which is characterized by hypoplasia/aplasia of distal phalanges in combination with distal symphalangism and fusion of carpal/tarsal bones242. Three missense mutations of Noggin were reported to cause Tarsal–carpal coalition syndrome (TCC; OMIM 186570), an autosomal dominant disorder characterized by short first metacarpals causing brachydactyly as well as fusion of the carpals, tarsals, phalanges, and humeroradial.243 Truncating mutation in the NOG gene in two families caused autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies (OMIM 184460).244 Heterozygous noggin mutations were reported to cause segregating proximal symphalangism (SYM1; OMIM 185800), which is characterized by ankylosis of the proximal interphalangeal joints, carpal and tarsal bone fusion or segregating multiple synostoses syndrome (SYNS1; OMIM 186500), which is characterized by multiple joint fusions.138
Other human bone diseases caused by mutations in TGF-β/BMP signaling
Several mutations in the TGF-β1 gene were reported to cause Camurati–Engelmann disease (CED; OMIM131300), a rare autosomal dominant disease characterized by cortical thickening of the diaphyses of long bones, hyperostosis, and bone pain.245,246 Nonsense, missense, and frameshift mutations of Smad3 were reported to cause aneurysms–osteoarthritis syndrome (AOS; OMIM 613795), which is presented with aneurysms, dissections, and tortuosity throughout the arterial tree in association with mild craniofacial features as well as skeletal and cutaneous anomalies.15,60,61
Both BMP and TGF-β possess clinical implications to treat bone diseases including bone healing, rheumatoid arthritis ect. TβRI kinase inhibitor downregulates rheumatoid synoviocytes and prevents the arthritis.247 A small molecule inhibitor of BMP type I receptor activity has been demonstrated to be useful in treating FOP and heterotopic ossification syndromes.248 Currently, two BMP products have been approved by the Food and Drug Administration for clinical applications to treat fractures of long bones and improve intervertebral disk regeneration through a purified collagen matrix, respectively, infused with BMP-2 (Medtronic) or OP-1 BMP-7 (Stryker Biotech) and implanted at the site of the fracture.4 With the use of BMPs increasingly accepted in spinal fusion surgeries, other therapeutic approaches targeting BMP signaling are emerging beyond applications to skeletal disorders.4 In addition, GDF-5 has emerged as a therapeutic target for rheumatic diseases.249 Recent applications graft BMP peptides corresponding to residues 73–92, 89–117, and 68–87 of BMP-2, BMP-7, and BMP-9 as adhesion peptides (GRGDSPC) onto polyethylene terephthatalate surfaces to enhance osteogenic differentiation and mineralization of pre-osteoblastic cells.250 These engineered biomaterials for enhanced bone regeneration are in the initial trial stage of development.
To sum up, mutations in the BMP and TGF-β signaling-related genes result in multiple inheritable bone diseases in humans, including ossification disorders, joint diseases, and skeleton developmental defects. In the future, more molecules or peptides targeting BMP and TGF-β signaling will be developed to treat genetic bone disorders, fracture healing, and osteoarthritis diseases.
Summary and perspective
BMP and TGF-β signaling pathways have important roles in skeletal development and postnatal skeleton homeostasis, by crosstalking with multiple signaling pathways, such as Wnt, Hedgehog, Notch, and FGF. Our understanding of BMP and TGF-β signaling is advanced with the generation of related mouse models, heritable human disease genetic studies and other new technologies including high-throughput screening. Specificity and versatility of BMP and TGF-β signaling are controlled by various ligand-receptor combinations, and transduced by co-Smad/R-Smad complex or MAPK cascade. The signaling is also dedicatedly controlled by factors including extracellular cognate binding proteins, I-Smad, epigenetic factors, and microRNA. Disruption of BMP and TGF-β signaling results in various bone disorders, such as osteoarthritis, FOP and Myhre syndrome. Manipulating BMP and TGF-β signaling pathways possess clinical implications for the treatment of multiple bone diseases including fracture healing, osteoarthritis, osteoporosis, and FOP. So far, BMP-2-and BMP-7-containing osteogenic implants have been used in over one million patients worldwide for the treatment of long bone nonunions, spinal fusions, and acute fractures. More importantly, with the aging population expected to double over the next decade, the number of patients suffering from osteoarthritis and osteoporosis is likely to increase dramatically and so is the cost of Medicare. Thus, it is compelling to elucidate the pathophysiology and the molecular mechanisms underlying skeleton health and bone diseases. Further, TGF and BMP signaling study will provide significant insights into the mechanism underlying how TGF and BMP signaling regulates osteoblast and chondrocyte proliferation, differentiation, maturation and activity in bone and cartilage formation, under both physiological and pathological condition (for example, osteoarthritis, osteoporosis, and bone cancer metastases. This study will also facilitate the design of novel therapeutic approaches for bone diseases.
References
Guo X, Wang XF . Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res 2009; 19: 71–88.
Feng XH, Derynck R . Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 2005; 21: 659–693.
Bernabeu C, Lopez-Novoa JM, Quintanilla M . The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim Biophys Acta 2009; 1792: 954–973.
Chen G, Deng C, Li YP . TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 2012; 8: 272–288.
Berendsen AD, Olsen BR . Bone development. Bone 2015; 80: 14–18.
Long F, Ornitz DM . Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 2013; 5: a008334.
Crane JL, Xian L, Cao X . Role of TGF-β signaling in coupling bone remodeling. Methods Mol Biol 2016; 1344: 287–300.
Tang Y, Wu X, Lei W et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 2009; 15: 757–765.
Li YP, Chen W, Stashenko P . Molecular cloning and characterization of a putative novel human osteoclast-specific 116-kDa vacuolar proton pump subunit. Biochem Biophys Res Commun 1996; 218: 813–821.
Li YP, Chen W, Liang Y et al. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 1999; 23: 447–451.
Chen W, Yang S, Abe Y et al. Novel pycnodysostosis mouse model uncovers cathepsin K function as a potential regulator of osteoclast apoptosis and senescence. Hum Mol Genet 2007; 16: 410–423.
Li YP, Alexander M, Wucherpfenni AL et al. Cloning and complete coding sequence of a novel human cathepsin expressed in giant cells of osteoclastomas. J Bone Miner Res 1995; 8: 1197–1202.
Razzouk S, Sarkis R . BMP-2: biological challenges to its clinical use. N Y State Dent J 2012; 78: 37–39.
Kanakaris NK, Giannoudis PV . Clinical applications of bone morphogenetic proteins: current evidence. J Surg Orthop Adv 2008; 17: 133–146.
Zhang W, Zhou M, Liu C et al. A novel mutation of SMAD3 identified in a Chinese family with aneurysms-osteoarthritis syndrome. BioMed Res Int 2015; 2015: 1–6.
Dodd AW, Syddall CM, Loughlin J . A rare variant in the osteoarthritis-associated locus GDF5 is functional and reveals a site that can be manipulated to modulate GDF5 expression. Eur J Hum Genet 2013; 21: 517–521.
van der Kraan PM, Goumans MJ, Blaney Davidson E et al. Age-dependent alteration of TGF-β signalling in osteoarthritis. Cell Tissue Res 2012; 347: 257–265.
Hayashi M, Muneta T, Ju YJ et al. Weekly intra-articular injections of bone morphogenetic protein-7 inhibits osteoarthritis progression. Arthritis Res Ther 2008; 10: R118.
Blaney Davidson EN, van der Kraan PM, van den Berg WB . TGF-beta and osteoarthritis. Osteoarthritis Cartilage 2007; 15: 597–604.
van den Bosch MH, Blom AB, van Lent PL et al. Canonical Wnt signaling skews TGF-β signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell Signal 2014; 26: 951–958.
Sakou T, Onishi T, Yamamoto T et al. Localization of Smads, the TGF-β family intracellular signaling components during endochondral ossification. J Bone Miner Res 1999; 14: 1145–1152.
Serra R, Karaplis A, Sohn P . Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor beta (TGF-beta) on endochondral bone formation. J Cell Biol 1999; 145: 783–794.
Li TF, O'Keefe RJ, Chen D . TGF-beta signaling in chondrocytes. Front Biosci 2005; 10: 681–688.
Sanford LP, Ormsby I, Gittenberger-de Groot AC et al. TGF beta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997; 124: 2659–2670.
Kulkarni AB, Huh CG, Becker D et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 1993; 90: 770–774.
Kaartinen V, Voncken JW, Shuler C et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 1995; 11: 415–421.
Proetzel G, Pawlowski SA, Wiles MV et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 1995; 11: 409–414.
Crane JL, Cao X . Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J Clin Invest 2014; 124: 466–472.
Karsdal MA, Larsen L, Engsig MT et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J Biol Chem 2002; 277: 44061–44067.
Matsunobu T, Torigoe K, Ishikawa M et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev Biol 2009; 332: 325–338.
Karst M, Gorny G, Galvin RJ et al. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol 2004; 200: 99–106.
Yasui T, Kadono Y, Nakamura M et al. Regulation of RANKL-induced osteoclastogenesis by TGF-β through molecular interaction between Smad3 and Traf6. J Bone Miner Res 2011; 26: 1447–1456.
Leah E . Osteoarthritis: TGF-β overload at bones of cartilage degeneration. Nat Rev Rheumatol 2013; 9: 382.
Serra R, Johnson M, Filvaroff EH et al. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol 1997; 139: 541–552.
Bush JR, Beier F . TGF-β and osteoarthritis--the good and the bad. Nat Med 2013; 19: 667–669.
van der Kraan PM . Age-related alterations in TGF beta signaling as a causal factor of cartilage degeneration in osteoarthritis. Biomed Mater Eng 2014; 24 1 Suppl: 75–80.
Zhen G, Wen C, Jia X et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med 2013; 19: 704–712.
Xu X, Zheng L, Bian Q et al. Aberrant activation of TGF-β in subchondral bone at the onset of rheumatoid arthritis joint destruction. J Bone Miner Res 2015; 30: 2033–2043.
Dabovic B, Levasseur R, Zambuto L et al. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone 2005; 37: 25–31.
Koli K, Ryynänen MJ, Keski-Oja J . Latent TGF-beta binding proteins (LTBPs)-1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone 2008; 43: 679–688.
Nikitovic D, Aggelidakis J, Young MF et al. The biology of small leucine-rich proteoglycans in bone pathophysiology. J Biol Chem 2012; 287: 33926–33933.
Grafe I, Yang T, Alexander S et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med 2014; 20: 670–675.
Qiu T, Wu X, Zhang F et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol 2010; 12: 224–234.
Longobardi L, Li T, Myers TJ et al. TGF-β type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Dev Cell 2012; 23: 71–81.
Spagnoli A, O'Rear L, Chandler RL et al. TGF-beta signaling is essential for joint morphogenesis. J Cell Biol 2007; 177: 1105–1117.
Seo HS, Serra R . Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev Biol 2007; 310: 304–316.
Baffi MO, Slattery E, Sohn P et al. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol 2004; 276: 124–142.
Kang JS, Alliston T, Delston R et al. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J 2005; 24: 2543–2555.
Alliston T, Choy L, Ducy P et al. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J 2001; 20: 2254–2272.
Hjelmeland AB, Schilling SH, Guo X et al. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol Cell Biol 2005; 25: 9460–9468.
Li J, Tsuji K, Komori T et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J Biol Chem 1998; 273: 31009–31015.
Borton AJ, Frederick JP, Datto MB et al. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J Bone Miner Res 2001; 16: 1754–1764.
Kaji H, Naito J, Sowa H et al. Smad3 differently affects osteoblast differentiation depending upon its differentiation stage. Horm Metab Res 2006; 38: 740–745.
Ferguson CM, Schwarz EM, Reynolds PR et al. Smad2 and 3 mediate transforming growth factor-beta1-induced inhibition of chondrocyte maturation. Endocrinology 2000; 141: 4728–4735.
Alvarez J, Serra R . Unique and redundant roles of Smad3 in TGF-beta-mediated regulation of long bone development in organ culture. Dev Dyn 2004; 230: 685–699.
Li TF, Darowish M, Zuscik MJ et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res 2006; 21: 4–16.
Ashcroft GS, Yang X, Glick AB et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1999; 1: 260–266.
Chen CG, Thuillier D, Chin EN et al. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum 2012; 64: 3278–3289.
Yang X, Chen L, Xu X et al. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol 2001; 153: 35–46.
van de Laar IM, Oldenburg RA, Pals G et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011; 43: 121–126.
van de Laar IM, van der Linde D, Oei EH et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet 2012; 49: 47–57.
Lee KS, Hong SH, Bae SC . Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-β and bone morphogenetic protein. Oncogene 2002; 21: 7156–7163.
Lai CF, Cheng SL . Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J Biol Chem 2002; 277: 15514–15522.
Luu HH, Song WX, Luo X et al. Distinct roles of bone morphogenetic proteins in osteogenic differentiation of mesenchymal stem cells. J Orthop Res 2007; 25: 665–677.
Abula K, Muneta T, Miyatake K et al. Elimination of BMP7 from the developing limb mesenchyme leads to articular cartilage degeneration and synovial inflammation with increased age. FEBS Lett 2015; 589: 1240–1248.
Huang Z, Ren PG, Ma T et al. Modulating osteogenesis of mesenchymal stem cells by modifying growth factor availability. Cytokine 2010; 51: 305–310.
Noël D, Gazit D, Bouquet C et al. Short-term BMP-2 expression is sufficient for in vivo osteochondral differentiation of mesenchymal stem cells. Stem Cells 2004; 22: 74–85.
Gu K, Zhang L, Jin T et al. Identification of potential modifiers of Runx2/Cbfa1 activity in C2C12 cells in response to bone morphogenetic protein-7. Cells Tissues Organs (Print) 2004; 176: 28–40.
Shen B, Wei A, Whittaker S et al. The role of BMP-7 in chondrogenic and osteogenic differentiation of human bone marrow multipotent mesenchymal stromal cells in vitro. J Cell Biochem 2010; 109: 406–416.
Tsuji K, Cox K, Gamer L et al. Conditional deletion of BMP7 from the limb skeleton does not affect bone formation or fracture repair. J Orthop Res 2010; 28: 384–389.
Kokabu S, Gamer L, Cox K et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol Endocrinol 2012; 26: 87–94.
Daluiski A, Engstrand T, Bahamonde ME et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet 2001; 27: 84–88.
Gamer LW, Cox K, Carlo JM et al. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev Dyn 2009; 238: 2374–2381.
Chan CK, Seo EY, Chen JY et al. Identification and specification of the mouse skeletal stem cell. Cell 2015; 160: 285–298.
Worthley DL, Churchill M, Compton JT et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015; 160: 269–284.
Minina E, Kreschel C, Naski MC et al. Interaction of FGF, IHH/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell 2002; 3: 439–449.
Kobayashi T, Lyons KM, McMahon AP et al. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA 2005; 102: 18023–18027.
Komatsu Y, Kaartinen V, Mishina Y . Cell cycle arrest in node cells governs ciliogenesis at the node to break left-right symmetry. Development 2011; 138: 3915–3920.
Haas AR, Tuan RS . Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: II. Stimulation by bone morphogenetic protein-2 requires modulation of N-cadherin expression and function. Differentiation 1999; 64: 77–89.
Luo G, Hofmann C, Bronckers AL et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev 1995; 9: 2808–2820.
Selever J, Liu W, Lu MF et al. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev Biol 2004; 276: 268–279.
Bandyopadhyay A, Tsuji K, Cox K et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet 2006; 2: e216.
Tsuji K, Bandyopadhyay A, Harfe BD et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 2006; 38: 1424–1429.
Tsuji K, Cox K, Bandyopadhyay A et al. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J Bone Joint Surg Am 2008; 90 Suppl 1: 14–18.
Long F . Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol 2012; 13: 27–38.
Shu B, Zhang M, Xie R et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci 2011; 124 Pt 20: 3428–3440.
Sekiya I, Tang T, Hayashi M et al. Periodic knee injections of BMP-7 delay cartilage degeneration induced by excessive running in rats. J Orthop Res 2009; 27: 1088–1092.
Takahashi T, Muneta T, Tsuji K et al. BMP-7 inhibits cartilage degeneration through suppression of inflammation in rat zymosan-induced arthritis. Cell Tissue Res 2011; 344: 321–332.
Rountree RB, Schoor M, Chen H et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol 2004; 2: e355.
Medici D, Shore EM, Lounev VY et al. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 2010; 16: 1400–1406.
Shore EM, Xu M, Feldman GJ et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38: 525–527.
Shen Q, Little SC, Xu M et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest 2009; 119: 3462–3472.
van Dinther M, Visser N, de Gorter DJ et al. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J Bone Miner Res 2010; 25: 1208–1215.
Yang C, Yang L, Wan M et al. Generation of a mouse model with expression of bone morphogenetic protein type II receptor lacking the cytoplasmic domain in osteoblasts. Ann N Y Acad Sci 2010; 1192: 286–291.
Mishina Y, Starbuck MW, Gentile MA et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem 2004; 279: 27560–27566.
Kamiya N, Kobayashi T, Mochida Y et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res 2010; 25: 200–210.
Kamiya N, Ye L, Kobayashi T et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res 2008; 23: 2007–2017.
Kamiya N, Ye L, Kobayashi T et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 2008; 135: 3801–3811.
Dewulf N, Verschueren K, Lonnoy O et al. Distinct spatial and temporal expression patterns of two type I receptors for bone morphogenetic proteins during mouse embryogenesis. Endocrinology 1995; 136: 2652–2663.
Francis PH, Richardson MK, Brickell PM et al. Bone morphogenetic proteins and a signalling pathway that controls patterning in the developing chick limb. Development 1994; 120: 209–218.
Kawakami Y, Ishikawa T, Shimabara M et al. BMP signaling during bone pattern determination in the developing limb. Development 1996; 122: 3557–3566.
Yamaji N, Celeste AJ, Thies RS et al. A mammalian serine/threonine kinase receptor specifically binds BMP-2 and BMP-4. Biochem Biophys Res Commun 1994; 205: 1944–1951.
Zou H, Wieser R, Massagué J et al. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev 1997; 11: 2191–2203.
Yoon BS, Ovchinnikov DA, Yoshii I et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA 2005; 102: 5062–5067.
Rigueur D, Brugger S, Anbarchian T et al. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J Bone Miner Res 2015; 30: 733–741.
Enomoto-Iwamoto M, Iwamoto M, Mukudai Y et al. Bone morphogenetic protein signaling is required for maintenance of differentiated phenotype, control of proliferation, and hypertrophy in chondrocytes. J Cell Biol 1998; 140: 409–418.
Fujii M, Takeda K, Imamura T et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell 1999; 10: 3801–3813.
Zhang D, Schwarz EM, Rosier RN et al. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J Bone Miner Res 2003; 18: 1593–1604.
Yi SE, Daluiski A, Pederson R et al. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 2000; 127: 621–630.
Yoon BS, Pogue R, Ovchinnikov DA et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 2006; 133: 4667–4678.
Afzal F, Pratap J, Ito K et al. Smad function and intranuclear targeting share a Runx2 motif required for osteogenic lineage induction and BMP2 responsive transcription. J Cell Physiol 2005; 204: 63–72.
Franceschi RT, Xiao G . Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. J Cell Biochem 2003; 88: 446–454.
Wang M, Jin H, Tang D et al. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage 2011; 19: 751–762.
Keller B, Yang T, Chen Y et al. Interaction of TGFβ and BMP signaling pathways during chondrogenesis. PLoS One 2011; 6: e16421.
Retting KN, Song B, Yoon BS et al. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009; 136: 1093–1104.
Matsumoto Y, Otsuka F, Hino J et al. Bone morphogenetic protein-3b (BMP-3b) inhibits osteoblast differentiation via Smad2/3 pathway by counteracting Smad1/5/8 signaling. Mol Cell Endocrinol 2012; 350: 78–86.
Ishibashi N, Sasaki Y, Asakura Y . Myhre syndrome: a rare craniofacial disorder. Cranio 2014; 32: 300–306.
Le Goff C, Mahaut C, Abhyankar A et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet 2012; 44: 85–88.
Nojima J, Kanomata K, Takada Y et al. Dual roles of smad proteins in the conversion from myoblasts to osteoblastic cells by bone morphogenetic proteins. J Biol Chem 2010; 285: 15577–15586.
Zhang J, Tan X, Li W et al. Smad4 is required for the normal organization of the cartilage growth plate. Dev Biol 2005; 284: 311–322.
Tan X, Weng T, Zhang J et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J Cell Sci 2007; 120: 2162–2170.
Salazar VS, Zarkadis N, Huang L et al. Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-imperfecta-like phenotype. J Cell Sci 2013; 126 (Pt 21): 4974–4984.
Salazar VS, Zarkadis N, Huang L et al. Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with β-catenin. J Cell Sci 2013; 126 (Pt 24): 5598–5609.
Billings PC, Fiori JL, Bentwood JL et al. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res 2008; 23: 305–313.
Gunnell LM, Jonason JH, Loiselle AE et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res 2010; 25: 1784–1797.
Gao L, Sheu TJ, Dong Y et al. TAK1 regulates SOX9 expression in chondrocytes and is essential for postnatal development of the growth plate and articular cartilages. J Cell Sci 2013; 126 (Pt 24): 5704–5713.
Greenblatt MB, Shim JH, Zou W et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest 2010; 120: 2457–2473.
Ge C, Yang Q, Zhao G et al. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res 2012; 27: 538–551.
Ulsamer A, Ortuño MJ, Ruiz S et al. BMP-2 induces Osterix expression through up-regulation of Dlx5 and its phosphorylation by p38. J Biol Chem 2008; 283: 3816–3826.
Ortuno MJ, Ruiz-Gaspa S, Rodriguez-Carballo E et al. p38 Regulates Expression of Osteoblast-specific Genes by Phosphorylation of Osterix. J Biol Chem 2010; 285: 31985–31994.
Celil AB, Hollinger JO, Campbell PG . Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling. J Cell Biochem 2005; 95: 518–528.
Choi YH, Gu YM, Oh JW et al. Osterix is regulated by Erk1/2 during osteoblast differentiation. Biochem Biophys Res Commun 2011; 415: 472–478.
Gazzerro E, Gangji V, Canalis E . Bone morphogenetic proteins induce the expression of noggin, which limits their activity in cultured rat osteoblasts. J Clin Invest 1998; 102: 2106–2114.
Abe E, Yamamoto M, Taguchi Y et al. Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J Bone Miner Res 2000; 15: 663–673.
Wan DC, Pomerantz JH, Brunet LJ et al. Noggin suppression enhances in vitro osteogenesis and accelerates in vivo bone formation. J Biol Chem 2007; 282: 26450–26459.
Glaser DL, Economides AN, Wang L et al. In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg Am 2003; 85-A: 2332–2342.
Wang Y, Hong S, Li M et al. Noggin resistance contributes to the potent osteogenic capability of BMP9 in mesenchymal stem cells. J Orthop Res 2013; 31: 1796–1803.
Gong Y, Krakow D, Marcelino J et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 1999; 21: 302–304.
Wijgerde M, Karp S, McMahon J et al. Noggin antagonism of BMP4 signaling controls development of the axial skeleton in the mouse. Dev Biol 2005; 286: 149–157.
Stafford DA, Brunet LJ, Khokha MK et al. Cooperative activity of noggin and gremlin 1 in axial skeleton development. Development 2011; 138: 1005–1014.
Stafford DA, Monica SD, Harland RM . Follistatin interacts with Noggin in the development of the axial skeleton. Mech Dev 2014; 131: 78–85.
Devlin RD, Du Z, Pereira RC et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology 2003; 144: 1972–1978.
Wu XB, Li Y, Schneider A et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest 2003; 112: 924–934.
Pathi S, Rutenberg JB, Johnson RL et al. Interaction of IHH and BMP/Noggin signaling during cartilage differentiation. Dev Biol 1999; 209: 239–253.
Warren SM, Brunet LJ, Harland RM et al. The BMP antagonist noggin regulates cranial suture fusion. Nature 2003; 422: 625–629.
Zehentner BK, Haussmann A, Burtscher H . The bone morphogenetic protein antagonist Noggin is regulated by Sox9 during endochondral differentiation. Dev Growth Differ 2002; 44: 1–9.
Kalajzic I, Kalajzic Z, Hurley MM et al. Stage specific inhibition of osteoblast lineage differentiation by FGF2 and noggin. J Cell Biochem 2003; 88: 1168–1176.
Reinhold MI, Abe M, Kapadia RM et al. FGF18 represses noggin expression and is induced by calcineurin. J Biol Chem 2004; 279: 38209–38219.
Nakayama N, Han CE, Scully S et al. A novel chordin-like protein inhibitor for bone morphogenetic proteins expressed preferentially in mesenchymal cell lineages. Dev Biol 2001; 232: 372–387.
Fisher S, Halpern ME . Patterning the zebrafish axial skeleton requires early chordin function. Nat Genet 1999; 23: 442–446.
Oren A, Toporik A, Biton S et al. hCHL2, a novel chordin-related gene, displays differential expression and complex alternative splicing in human tissues and during myoblast and osteoblast maturation. Gene 2004; 331: 17–31.
Nakayama N, Han CY, Cam L et al. A novel chordin-like BMP inhibitor, CHL2, expressed preferentially in chondrocytes of developing cartilage and osteoarthritic joint cartilage. Development 2004; 131: 229–240.
Petryk A, Shimmi O, Jia XH et al. Twisted gastrulation and chordin inhibit differentiation and mineralization in MC3T3-E1 osteoblast-like cells. Bone 2005; 36: 617–626.
Zhang D, Ferguson CM, O'Keefe RJ et al. A role for the BMP antagonist chordin in endochondral ossification. J Bone Miner Res 2002; 17: 293–300.
Anderson RM, Lawrence AR, Stottmann RW et al. Chordin and noggin promote organizing centers of forebrain development in the mouse. Development 2002; 129: 4975–4987.
Yan X, Liu Z, Chen Y . Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai) 2009; 41: 263–272.
Moustakas A, Heldin CH . The regulation of TGF signal transduction. Development 2009; 136: 3699–3714.
Scharstuhl A, Diepens R, Lensen J et al. Adenoviral overexpression of Smad-7 and Smad-6 differentially regulates TGF-beta-mediated chondrocyte proliferation and proteoglycan synthesis. Osteoarthritis Cartilage 2003; 11: 773–782.
Valcourt U, Gouttenoire J, Moustakas A et al. Functions of transforming growth factor-beta family type I receptors and Smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J Biol Chem 2002; 277: 33545–33558.
Li X, Ionescu AM, Schwarz EM et al. Smad6 is induced by BMP-2 and modulates chondrocyte differentiation. J Orthop Res 2003; 21: 908–913.
Yano M, Inoue Y, Tobimatsu T et al. Smad7 inhibits differentiation and mineralization of mouse osteoblastic cells. Endocr J 2012; 59: 653–662.
Horiki M, Imamura T, Okamoto M et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J Cell Biol 2004; 165: 433–445.
Shen R, Chen M, Wang YJ et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem 2006; 281: 3569–3576.
Wang Q, Wei X, Zhu T et al. Bone morphogenetic protein 2 activates Smad6 gene transcription through bone-specific transcription factor Runx2. J Biol Chem 2007; 282: 10742–10748.
Iwai T, Murai J, Yoshikawa H et al. Smad7 Inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J Biol Chem 2008; 283: 27154–27164.
Wu Q, Wang M, Zuscik MJ et al. Regulation of embryonic endochondral ossification by Smurf2. J Orthop Res 2008; 26: 704–712.
Yamashita M, Ying SX, Zhang GM et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005; 121: 101–113.
Qi H, Jin M, Duan Y et al. FGFR3 induces degradation of BMP type I receptor to regulate skeletal development. Biochim Biophys Acta 2014; 1843: 1237–1247.
Tsubakihara Y, Hikita A, Yamamoto S et al. Arkadia enhances BMP signalling through ubiquitylation and degradation of Smad6. J Biochem 2015; 158: 61–71.
Shimada K, Suzuki N, Ono Y et al. Ubc9 promotes the stability of Smad4 and the nuclear accumulation of Smad1 in osteoblast-like Saos-2 cells. Bone 2008; 42: 886–893.
Yukita A, Hosoya A, Ito Y et al. Ubc9 negatively regulates BMP-mediated osteoblastic differentiation in cultured cells. Bone 2012; 50: 1092–1099.
Herhaus L, Al-Salihi MA, Dingwell KS et al. USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol 2014; 4: 140065.
Kawamura I, Maeda S, Imamura K et al. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J Biol Chem 2012; 287: 29101–29113.
Usui M, Yoshida Y, Tsuji K et al. Tob deficiency superenhances osteoblastic activity after ovariectomy to block estrogen deficiency-induced osteoporosis. Proc Natl Acad Sci USA 2004; 101: 6653–6658.
Yoshida Y, Tanaka S, Umemori H et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell 2000; 103: 1085–1097.
Li Z, Hassan MQ, Volinia S et al. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc Natl Acad Sci USA 2008; 105: 13906–13911.
Balderman JAF, Lee HY, Mahoney CE et al. Bone morphogenetic protein-2 decreases microRNA-30b and microRNA-30c to promote vascular smooth muscle cell calcification. J Am Heart Assoc 2012; 1: e003905–e003905.
Wu T, Zhou H, Hong Y et al. miR-30 family members negatively regulate osteoblast differentiation. J Biol Chem 2012; 287: 7503–7511.
Gámez B, Rodríguez-Carballo E, Bartrons R et al. MicroRNA-322 (miR-322) and its target protein Tob2 modulate Osterix (Osx) mRNA stability. J Biol Chem 2013; 288: 14264–14275.
Itoh T, Nozawa Y, Akao Y . MicroRNA-141 and -200a are involved in bone morphogenetic protein-2-induced mouse pre-osteoblast differentiation by targeting distal-less homeobox 5. J Biol Chem 2009; 284: 19272–19279.
Kureel J, Dixit M, Tyagi AM et al. miR-542-3p suppresses osteoblast cell proliferation and differentiation, targets BMP-7 signaling and inhibits bone formation. Cell Death Dis 2014; 5: e1050.
Zhang JF, Fu WM, He ML et al. MiRNA-20a promotes osteogenic differentiation of human mesenchymal stem cells by co-regulating BMP signaling. RNA Biol 2011; 8: 829–838.
Nakamura Y, Inloes JB, Katagiri T et al. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol 2011; 31: 3019–3028.
Borgonio Cuadra VM, González-Huerta NC, Romero-Córdoba S et al. Altered expression of circulating microRNA in plasma of patients with primary osteoarthritis and in silico analysis of their pathways. PLoS One 2014; 9: e97690.
Díaz-Prado S, Cicione C, Muiños-López E et al. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet Disord 2012; 13: 144.
Tew SR, Vasieva O, Peffers MJ et al. Post-transcriptional gene regulation following exposure of osteoarthritic human articular chondrocytes to hyperosmotic conditions. Osteoarthritis Cartilage 2011; 19: 1036–1046.
Jin L, Zhao J, Jing W et al. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int J Mol Med 2014; 34: 451–463.
Li J, Huang J, Dai L et al. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res Ther 2012; 14: R75.
Lin EA, Kong L, Bai XH et al. miR-199a, a bone morphogenic protein 2-responsive microRNA, regulates chondrogenesis via direct targeting to Smad1. J Biol Chem 2009; 284: 11326–11335.
Furumatsu T, Asahara H . Histone acetylation influences the activity of Sox9-related transcriptional complex. Acta Med Okayama 2010; 64: 351–357.
Lee HL, Yu B, Deng P et al. Transforming growth factor-β-induced KDM4B promotes chondrogenic differentiation of human mesenchymal. Stem Cells 2016; 34: 711–719.
Lee ZH, Kim HJ, Ryoo HM . A novel osteogenic activity of suberoylanilide hydroxamic acid is synergized by BMP-2. J Bone Metab 2015; 22: 51–56.
Cho YD, Yoon WJ, Kim WJ et al. Epigenetic modifications and canonical wingless/int-1 class (WNT) signaling enable trans-differentiation of nonosteogenic cells into osteoblasts. J Biol Chem 2014; 289: 20120–20128.
Zhou S, Eid K, Glowacki J . Cooperation between TGF-β and Wnt pathways during chondrocyte and adipocyte differentiation of human marrow stromal cells. J Bone Miner Res 2003; 19: 463–470.
Dao DY, Yang X, Chen D et al. Axin1 and Axin2 are regulated by TGF- and mediate cross-talk between TGF- and Wnt signaling pathways. Ann NY Acad Sci 2007; 1116: 82–99.
McCarthy TL, Centrella M . Novel links among Wnt and TGF-beta signaling and Runx2. Mol Endocrinol 2010; 24: 587–597.
Liu Z, Tang Y, Qiu T et al. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J Biol Chem 2006; 281: 17156–17163.
Rawadi G, Vayssière B, Dunn F et al. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res 2003; 18: 1842–1853.
Zhang M, Yan Y, Lim YB et al. BMP-2 modulates beta-catenin signaling through stimulation of Lrp5 expression and inhibition of beta-TrCP expression in osteoblasts. J Cell Biochem 2009; 108: 896–905.
Papathanasiou I, Malizos KN, Tsezou A . Bone morphogenetic protein-2-induced Wnt/β-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res Ther 2012; 14: R82.
Tang N, Song WX, Luo J et al. BMP-9-induced osteogenic differentiation of mesenchymal progenitors requires functional canonical Wnt/beta-catenin signalling. J Cell Mol Med 2009; 13: 2448–2464.
Rodríguez-Carballo E, Ulsamer A, Susperregui AR et al. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J Bone Miner Res 2011; 26: 718–729.
Zhang R, Oyajobi BO, Harris SE et al. Wnt/β-catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone 2013; 52: 145–156.
Jiang T, Ge S, Shim YH et al. Bone morphogenetic protein is required for fibroblast growth factor 2-dependent later-stage osteoblastic differentiation in cranial suture cells. Int J Clin Exp Pathol 2015; 8: 2946–2954.
Naganawa T, Xiao L, Coffin JD et al. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J Cell Biochem 2008; 103: 1975–1988.
Agas D, Sabbieti MG, Marchetti L et al. FGF-2 enhances Runx-2/Smads nuclear localization in BMP-2 canonical signaling in osteoblasts. J Cell Physiol 2013; 228: 2149–2158.
Nagayama T, Okuhara S, Ota MS et al. FGF18 accelerates osteoblast differentiation by upregulating Bmp2 expression. Congenit Anom (Kyoto) 2013; 53: 83–88.
Fakhry A, Ratisoontorn C, Vedhachalam C et al. Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone 2005; 36: 254–266.
Hughes-Fulford M, Li CF . The role of FGF-2 and BMP-2 in regulation of gene induction, cell proliferation and mineralization. J Orthop Surg Res 2011; 6: 8.
Park JB . Effects of the combination of fibroblast growth factor-2 and bone morphogenetic protein-2 on the proliferation and differentiation of osteoprecursor cells. Adv Clin Exp Med 2014; 23: 463–467.
Matsushita T, Wilcox WR, Chan YY et al. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet 2009; 18: 227–240.
Allen JM, McGlinn E, Hill A et al. Autopodial development is selectively impaired by misexpression of chordin-like 1 in the chick limb. Dev Biol 2013; 381: 159–169.
Long F, Chung UI, Ohba S et al. IHH signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004; 131: 1309–1318.
Lenton K, James AW, Manu A et al. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis 2011; 49: 784–796.
Hojo H, Ohba S, Taniguchi K et al. Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J Biol Chem 2013; 288: 9924–9932.
Zhao M, Ko SY, Liu JH et al. Inhibition of microtubule assembly in osteoblasts stimulates bone morphogenetic protein 2 expression and bone formation through transcription factor Gli2. Mol Cell Biol 2008; 29: 1291–1305.
Zhao M, Qiao M, Harris SE et al. The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Mol Cell Biol 2006; 26: 6197–6208.
Minina E, Wenzel HM, Kreschel C et al. BMP and IHH/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001; 128: 4523–4534.
Kameda T, Koike C, Saitoh K et al. Developmental patterning in chondrocytic cultures by morphogenic gradients: BMP induces expression of indian hedgehog and noggin. Genes Cells 1999; 4: 175–184.
Grimsrud CD, Romano PR, D'Souza M et al. BMP-6 is an autocrine stimulator of chondrocyte differentiation. J Bone Miner Res 1999; 14: 475–482.
Mak KK, Kronenberg HM, Chuang PT et al. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008; 135: 1947–1956.
Mak KK, Chen MH, Day TF et al. Wnt/beta-catenin signaling interacts differentially with IHH signaling in controlling endochondral bone and synovial joint formation. Development 2006; 133: 3695–3707.
Zeng L . SHH establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev 2002; 16: 1990–2005.
Choi KY, Kim HJ, Lee MH et al. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev Dyn 2005; 233: 115–121.
Tezuka K, Yasuda M, Watanabe N et al. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res 2002; 17: 231–239.
Nobta M, Tsukazaki T, Shibata Y et al. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J Biol Chem 2005; 280: 15842–15848.
de Jong DS, Steegenga WT, Hendriks JM et al. Regulation of Notch signaling genes during BMP2-induced differentiation of osteoblast precursor cells. Biochem Biophys Res Commun 2004; 320: 100–107.
Atfi A, Baron R . PTH battles TGF-β in bone. Nat Cell Biol 2010; 12: 205–207.
Yu B, Zhao X, Yang C et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res 2012; 27: 2001–2014.
Kobayashi T, Soegiarto DW, Yang Y et al. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest 2005; 115: 1734–1742.
Bocciardi R, Bordo D, Di Duca M et al. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet 2009; 17: 311–318.
Gregson CL, Hollingworth P, Williams M et al. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011; 48: 654–658.
Petrie KA, Lee WH, Bullock AN et al. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS One 2009; 4: e5005.
Su P, Ding H, Huang D et al. A 4.6 kb genomic duplication on 20p12.2-12.3 is associated with brachydactyly type A2 in a Chinese family. J Med Genet 2011; 48: 312–316.
Lehmann K, Seemann P, Boergermann J et al. A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type C/symphalangism-like phenotype or brachydactyly type A2. Eur J Hum Genet 2006; 14: 1248–1254.
Lehmann K, Seemann P, Stricker S et al. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci USA 2003; 100: 12277–12282.
Plöger F, Seemann P, Schmidt-von Kegler M et al. Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum Mol Genet 2008; 17: 1222–1233.
Seemann P, Schwappacher R, Kjaer KW et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest 2005; 115: 2373–2381.
Dathe K, Kjaer KW, Brehm A et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet 2009; 84: 483–492.
Thomas JT, Kilpatrick MW, Lin K et al. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 1997; 17: 58–64.
Lindor NM, Gunawardena SR, Thibodeau SN . Mutations of SMAD4 account for both LAPS and Myhre syndromes. Am J Med Genet 2012; 158A: 1520–1521.
Lehmann K, Seemann P, Silan F et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet 2007; 81: 388–396.
Dixon ME, Armstrong P, Stevens DB et al. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet Med 2001; 3: 349–353.
Hirshoren N, Gross M, Banin E et al. P35S mutation in the NOG gene associated with Teunissen-Cremers syndrome and features of multiple NOG joint-fusion syndromes. Eur J Med Genet 2008; 51: 351–357.
Campos-Xavier B, Saraiva JM, Savarirayan R et al. Phenotypic variability at the TGF-beta1 locus in Camurati-Engelmann disease. Hum Genet 2001; 109: 653–658.
Kinoshita A, Saito T, Tomita H et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet 2000; 26: 19–20.
Sakuma M, Hatsushika K, Koyama K et al. TGF- type I receptor kinase inhibitor down-regulates rheumatoid synoviocytes and prevents the arthritis induced by type II collagen antibody. Int Immunol 2006; 19: 117–126.
Yu PB, Deng DY, Lai CS et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med 2008; 14: 1363–1369.
Lories RJ, Luyten FP . Bone morphogenetic protein signaling in joint homeostasis and disease. Cytokine Growth Factor Rev 2005; 16: 287–298.
Zouani OF, Chollet C, Guillotin B et al. Differentiation of pre-osteoblast cells on poly(ethylene terephthalate) grafted with RGD and/or BMPs mimetic peptides. Biomaterials 2010; 31: 8245–8253.
Dünker N, Krieglstein K . Tgfbeta2 -/- Tgfbeta3 -/- double knockout mice display severe midline fusion defects and early embryonic lethality. Anat Embryol 2002; 206: 73–83.
Gu Z, Reynolds EM, Song J et al. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development 1999; 126: 2551–2561.
Nomura M, Li E . Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 1998; 393: 786–790.
Zhou S . TGF-β regulates β-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J Cell Biochem 2011; 112: 1651–1660.
Tachi K, Takami M, Sato H et al. Enhancement of bone morphogenetic protein-2-induced ectopic bone formation by transforming growth factor-β1. Tissue Eng Part A 2011; 17: 597–606.
Sasaki T, Ito Y, Bringas P Jr et al. TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development 2006; 133: 371–381.
van der Horst G, Farih-Sips H, Löwik CW et al. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone 2003; 33: 899–910.
Zhang R, Edwards JR, Ko SY et al. Transcriptional regulation of BMP2 expression by the PTH-CREB signaling pathway in osteoblasts. PLoS One 2011; 6: e20780.
Acknowledgements
We thank Dr Joel Jules for his extensive reading and discussion of our manuscript. We apologize to the many researchers whose work could not be cited due to space limitations. Work in our laboratory is supported by grants by NIH grant AR-044741 (Y-PL) and R01DE023813 (Y-PL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wu, M., Chen, G. & Li, YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4, 16009 (2016). https://doi.org/10.1038/boneres.2016.9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2016.9
This article is cited by
-
Molecular landscape of congenital vertebral malformations: recent discoveries and future directions
Orphanet Journal of Rare Diseases (2024)
-
Nerve growth factor receptor limits inflammation to promote remodeling and repair of osteoarthritic joints
Nature Communications (2024)
-
Noggin promotes osteogenesis in human adipose-derived mesenchymal stem cells via FGFR2/Src/Akt and ERK signaling pathway
Scientific Reports (2024)
-
FSTL1 Accelerates Nucleus Pulposus Cell Senescence and Intervertebral Disc Degeneration Through TLR4/NF-κB Pathway
Inflammation (2024)
-
Circular RNA-Mediated Regulation of Oral Tissue-Derived Stem Cell Differentiation: Implications for Oral Medicine and Orthodontic Applications
Stem Cell Reviews and Reports (2024)
