
Abstract
Insufficient insulin production or action in diabetic states is associated with growth retardation and impaired bone healing, while the underling mechanisms are unknown. In this study, we sought to define the role of insulin signaling in the growth plate. Insulin treatment of embryonic metatarsal bones from wild-type mice increased chondrocyte proliferation. Mice lacking insulin receptor (IR) selectively in chondrocytes (CartIR−/−) had no discernable differences in total femoral length compared to control littermates. However, CartIR−/− mice exhibited an increase in chondrocyte numbers in the growth plate than that of the controls. Chondrocytes lacking IR had elevated insulin-like growth factor (IGF)-1R mRNA and protein levels. Subsequently, IGF-1 induced phosphorylation of Akt and ERK was enhanced, while this action was eliminated when the cells were treated with IGF-1R inhibitor Picropodophyllin. Deletion of the IR impaired chondrogenic differentiation, and the effect could not be restored by treatment of insulin, but partially rescued by IGF-1 treatment. Intriguingly, the size of hypertrophic chondrocytes was smaller in CartIR−/− mice when compared with that of the control littermates, which was associated with upregulation of tuberous sclerosis complex 2 (TSC2). These results suggest that deletion of the IR in chondrocytes sensitizes IGF-1R signaling and action, IR and IGF-1R coordinate to regulate the proliferation, differentiation and hypertrophy of growth plate chondrocytes.
Similar content being viewed by others


Introduction
The insulin-like growth factors (IGFs) evolved in lower animals to enable a wide range of physiological processes, including smell, food consumption, metabolism, growth, reproduction and dormancy.1,2 These functions were accomplished by the actions of multiple-related ligands that activated a common transmembrane receptor protein.3 In higher organisms including mammals, the insulin and IGF ligands and their receptors diverged and evolved more circumscribed functions. The contemporary view is that IGFs serve dominant roles in cell proliferation, survival and organismal growth, whereas insulin’s primary functions include the regulation of fuel accumulation, storage and energy expenditure.1 However, such a simplistic model tends to obscure the fact that insulin and IGF-1 continue to perform overlapping roles in several physiological processes.
A compelling body of circumstantial evidence suggests that IGF/IGF-1R and insulin/insulin receptor (IR) serve overlapping functions in selected tissues including the skeleton.4 In both humans and mice, loss of IGF-1 action is manifested with severe intrauterine growth retardation and delayed bone development.5–7 Mice with conditional deletion of IGF-1 in chondrocytes have reduced bone accretion during postnatal bone growth.8 Humans with mutations in the IR (Leprechaunism) exhibit profound growth retardation,9 which resembles those seen in mice with engineered mutations of the IR.10 The growth plate of the IR mutant mouse embryo exhibits a profound developmental delay in the appearance of ossification centers and severe growth deficiency (30% of normal size at birth).10 Comparison of the phenotype of these IR-deficient mice with those of IGF-1R knockout mice suggests that IR and IGF-1R have distinct, but partially overlapping, physiological functions.11,12 Mice lacking IGF-1R specifically in chondrocytes exhibit a shorter and narrower growth plate, and the size of individual hypertrophic chondrocytes appeared smaller compared to that of control littermates.13 Both the IR and IGF-1R are expressed and appear to be functional in chondrogenic cells.14,15 However, the cellular targets and molecular mechanisms through which insulin/IR signaling functions to regulate chondrogenesis during skeletal development and following injury are unknown.
Chondrogenesis is regulated by multiple mechanisms including systemic or local factors emanating from the surrounding matrix, inflammatory mediators, cytokines and various growth factors. Key among these are transforming growth factor-beta,16 IGF-1,17 fibroblast growth factors18 and PTHrP/Indian hedgehog, which together coordinate the pace of chondrocyte proliferation and differentiation.13 Distinguishing the skeletal effects of insulin from those of IGF-1 in vivo is complicated by the fact that most cell types express both IR and IGF-1R, as well as IR/IGF–IR hybrid receptors, which can be cross-activated by the others receptor’s ligand.19 In addition, because the signaling pathways downstream from IR and IGF-IR share many components, the characterization of these signaling events may not distinguish which receptor is initiating the signal.20 To circumvent these problems, we generated mice selectively lacking IR in chondrocytes through cross-breeding of the mouse strain carrying the IR-floxed allele21 and the mouse strain carrying Cre recombinase under the control of collagen type II promoter.22 In vivo, CartIR−/− mice demonstrated alterations in growth plate development characterized by an increase in the overall numbers of chondrocytes and a reduction in size of cells in the hypertrophic zone. In vitro loss of IR impaired chondrogenic differentiation and reduced proteoglycan synthesis; these abnormalities could not be rescued by treatment of insulin. Interestingly, IR-deficient chondrocytes had increased IGF-1R levels and were more responsive to IGF-1 consistent with the notion that IGF-1/IGF-1R may compensate, at least in part, for loss of IR signaling.
Materials and methods
Reagents
Bovine insulin was obtained from Sigma-Aldrich (St Louis, MO, USA) and dissolved in acidic water (pH 2–3) to make a stock solution. Human IGF-1 was obtained from GroPep (Theberton, SA, Australia). Picropodophyllin (PPP, IGF-1R inhibitor) was purchased from Calbiochem (La Jolla, CA, USA). Antibodies used for Western blotting included anti-insulin receptorα subunit (C-19), anti-IGF-1Rα subunit (C-20) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and non-immune rabbit serum IgG from Alpha Diagnostic (San Antonio, TX, USA); anti-phospho-Akt (Ser473), anti-Akt; anti-phospho-ERK (Thr202/Tyr204), anti-ERK, anti-tuberous sclerosis complex 1 (TSC1), anti-TSC2 and anti-proliferating cell nuclear antigen (PCNA) were from Cell Signaling (Danvers, MA, USA). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were from Pierce (Rockford, IL, USA). Polyvinylidene fluoride (PVDF) membrane and other electrophoresis supplies were from Bio-Rad (Hercules, CA, USA). Assay kits for flow cytometry analysis of cell proliferation and apoptosis were purchased from BD Pharmingen (San Jose, CA, USA). All other reagents not specified here were purchased from Sigma-Aldrich.
Generation of mice
Col2-Cre mice22 were mated with IRflox/flox mice,21 and a breeding colony was maintained by mating IRflox/flox with Col2-Cre-IRflox/flox mice to generate CartIR−/−mice. IRflox/flox mice, initially generated from C57BL/6J and CD-1 background, had been backcrossed for at least five generations on a C57BL6 background, and Col2-Cre mice, initially established on a C57BL6/129sv background, had been backcrossed for 10 generations on a C57BL6 background before being intercrossed with IRflox/flox mice. Only male animals from the same mixed background strain generation were compared to each other. Col2-Cre mice were genotyped by PCR as previously described.22 IRflox/flox mice were genotyped by PCR with primers crossing the loxP site as previously described.23 In the whole study, we used control littermates IRflox/flox mice as controls. Experimental procedures were carried out by following the protocols approved by Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and Animal (Control of Experiments) Ordinance from Department of Health, Hong Kong SAR; and Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and Johns Hopkins University.
Isolation and culture of murine chondrocytes and adenovirus infection
Primary chondrocytes were isolated from the ribs of newborn IRflox/flox mice euthanized by cervical dislocation using previously established protocol.24 The ribs were digested by pronase and collagenase D and chondrocytes were plated onto a 100 mm Petri dish with DMEM (Gibco, Invitrogen) supplemented with 1% penicillin/streptomycin sulfate (Gibco, Invitrogen), 1% L-glutamine (Gibco, Invitrogen) and 10% fetal bovine serum (Gibco, Invitrogen) at 37 °C in a 5% CO2 humidified incubator. On the second day, non-adherent cells were removed and the cultures were gently rinsed with PBS twice and cultured in the fresh medium. Adherent cells were cultured and medium was changed every 2 days until the cells became confluent. Monolayer chondrocytes were infected with control adenovirus containing green fluorescence protein (Ad-GFP) or Cre recombinase virus M1 (Ad-Cre) (Vector Laboratories, Burlingame, CA, USA) at an MOI of 100 for most experiments. Chondrocytes were harvested after 48 h. Total mRNA was extracted from infected chondrocytes for deletion validation by real-time PCR, and proteins were extracted for signal study by Western blot.
Cartilage nodule formation assay in micromass culture
Briefly, chondrocytes were seeded in a 12-well plate at a density of 1.0×105 cells per well in a final volume of 10 μL and incubated for 2 h at 37 °C to allow adherence. One milliliter of fresh media was added to each well. Three days after seeding of micromass cultures and treatment with various concentrations factors of interest, wells were washed once with phosphate-buffered saline and then fixed for 20 min with 1 mL 10% neutral buffered formalin. The pellets were then washed three times with sterile distilled, deionized water, with the last wash being left on the cells for 15 min. Subsequently, 0.5 mL 1% alcian blue (Sigma) in glacial acetic acid (3%) was added to each well for 30 min or 1 h at room temperature. After two washes with 70% ethanol and three washes with sterile distilled, deionized water, photomicrographs of the stained cultures were obtained.
Fetal mouse metatarsal culture
Metatarsal bone culture was performed following the previously published method.25 In brief, E17.5 embryos were removed from timed-pregnant mice, and metatarsals were dissected. The isolated metatarsals were cultured in 24-well culture plates in 150 µL of DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% of penicillin/streptomycin and 1% L-glutamine for 72 h. Two hundred and fifty microliters of fresh medium were then replaced with or without insulin or IGF-1. The metatarsal bones were cultured for 6 days with replacement of medium every 3 days. The total length of metatarsal bones was measured in each specimen. The proliferating, hypertrophic and mineralizing zones were also measured in the H&E-stained sections. The proliferation of chondrocytes was determined by immunostaining for PCNA in the metatarsal bone sections.
Western blot analysis
Western blotting analysis was performed using standard protocol. Briefly, whole-cell lysate was obtained by cell lysis buffer in the presence of a protease inhibitor cocktail (Sigma-Aldrich). Equal amount of protein was loaded onto an SDS mini-PAGE system after concentrations were determined, and transferred onto a PVDF membrane using a Bio-Rad semidry transfer system. Protein transfer efficiency and size determination were verified using prestained protein markers. Membranes were blocked with 5% dry milk in Tris-buffered saline Tween-20 for 1 h at room temperature and subsequently incubated overnight with primary antibodies at 4 °C. Signals were detected using an HRP-conjugated secondary antibody and the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Thermo Fisher Scientific, Inc., Rockford, IL, USA).
Quantitative real-time PCR
Total RNA was extracted from cells using the Trizol method as recommended by the manufacturer (Invitrogen). The RNA concentration was estimated by spectrophoto-meter and only pure RNA (A260/A280 ratio ≥1.8) was used for further analysis. First strand cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad). The cDNA was amplified in the Opticon Continuous Fluorescent Detector (MJ Research, Waltham, MA, USA) using IQTM SYBR Green supermix (Bio-Rad) and sequence specific primers. PCRs were performed in triplicate for each cDNA, averaged, and normalized to endogenous actin reference transcripts. Primer sequences used were listed in Table 1.
Histology, immunohistochemistry and immunofluorescence
Mouse metatarsal treated with different growth factors (insulin or IGF-1) or femurs from 3-week-old mice were fixed in 10% buffered formalin, decalcified in 8% Na2EDTA, paraffin-embedded and stained with H&E using standard methods. For immunohistochemistry, antigen retrieval was performed by boiling in 10 mmol·L−1 sodium citrate (pH 6.0) for 5 min. Sections were incubated with antibodies against PCNA (PC10) (cell signaling, 1∶2 000) and IGF-1R (N-20) (Santa Cruz, 1∶50). Sections incubated with 1% non-immune IgG solution served as negative controls. Quantitative immunostaining assay was performed by determining the percentage of positively stained cells in four random high-power fields (×200) in four adjacent sections. For immunofluorescence, section were incubated with antibody against TSC2 (D93F12) (cell signaling, 1∶100) and with Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) secondary antibody (Molecular Probes; Invitrogen, 1∶1 000) for 30 min and mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories). Fluorescence image was detected by confocal microscopy with a laser scanning microscope (FV10-ASW 1.7; Olympus). Images were obtained with the same confocal settings for each set of experiments.
Chondrocytes proliferation and apoptosis assay
The mutant or control chondrocytes were plated in six-well plates with the density of 1×105, and cultured in medium with 1% serum for 24 h and then were restimulated with insulin (10 nmol·L−1) for an additional 24 h before harvest. For proliferation analysis, BrdU was added to the medium 12 h before harvesting the cells. The cells were then stained for anti-BrdU-APC and 7-amino-actinomycin D following the manufacturer’s instructions (BD Biosciences). For the apoptosis assay, annexin V-PE (BD Biosciences) and 7-amino-actinomycin D were used for staining. Cells were analyzed by FACSCalibur (BD Biosciences) and 20 000 events were collected. The results were analyzed with ModFit LT 3.3 software.
Statistical analysis
For comparison of cellular and tissue parameters between the wild-type and mutant mice, the statistical analysis was performed using the Microsoft Excel data analysis program for Student’s t-test analysis. For comparisons among multiple groups, the analysis of variance was used. All of the experiments were repeated at least three times unless otherwise stated. The values are expressed as the means±standard deviation (s.d.). A significance level was defined as P<0.05.
Results
Insulin promotes growth of fetal metatarsal cultures
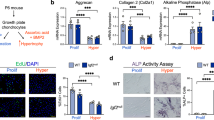
To begin to examine the effects of insulin on chondrocytes, we determined the effects of insulin and IGF-1 on the growth of fetal metatarsal rudiments in vitro. Metatarsal bones from E17.5 wild-type mouse embryos were cultured for 6 days in the absence or presence of 10 nmol·L−1 insulin or 13 nmol·L−1 IGF-1. Both ligands significantly increased the total length (Figure 1a and 1b) primarily by increasing the lengths and numbers of cells in the proliferating zone (Figure 1c and 1d). Immunostaining for PCNA also showed that treatment with insulin and IGF-1 increased PCNA positive cell numbers in the cultured metatarsal bone sections (Figure 1e and 1f). These data demonstrate that insulin exerts mitogenic effects in growth plate chondrocytes that are qualitatively similar to those of IGF-1.

Insulin promotes growth of fetal metatarsal bones in vitro. (a) Metatarsalbones from E17.5 wild-type mouse embryos were cultured for 6 days in the absence or presence of insulin or IGF-1. (b) Quantitation of the actual length of metatarsal bones from (a) as shown. *P<0.05, n=6. (c) Representative histological images of metatarsal bones treated with insulin or IGF-1, with none treatment as control. PZ, proliferating zone; HZ, hypertrophic zone; MZ, mineralizing zone. (d) Quantitation of the proliferating zone of the cartilage rudiments from (c). *P<0.05, n=6. (e) Representative histological sections of metatarsal bones treated with insulin or IGF-1 after staining with antibody against PCNA as described in the section on ‘Materials and methods’. Sections were counterstained with hematoxylin. Scale bar=50 µm. (f) Quantitation for the percentage of PCNA+ cells from (e). *P<0.05, n=6. IGF-1 treatment shows more dramatic effects on the total length in the cultures as positive control. CON, none treatmentcontrol; IGF-1, insulin-like growth factor-1; INS, insulin.
Alterations in growth plate development in CartIR−/− mice
To determine the importance of insulin signaling on growth plate development in vivo, we generated mice deficient in IR in chondrocytes as described in the section on ‘Materials and methods’. Immunostaining for IR showed an efficient elimination of IR in chondrocytes of the growth plates in CartIR−/− mice (Figure 2a). Gross analysis of femurs at 3 and 12 weeks of age showed no significant difference in overall length (Figure 2b and 2c) or organization of the growth plate (Figure 2d) in CartIR−/− mice compared to controls. However, quantitation of the lengths of the different zones of the growth plates indicated that the proliferating zone of the growth plate was modestly increased while the prehypertrophic zone was decreased in CartIR−/− mice compared to controls (Figure 2e and 2f). No significant change was observed at the hypertrophic zone (Figure 2g). Histological analysis revealed that CartIR−/− mice had a significant increase in the total number of chondrocytes in all regions of the growth plate (Figure 2h–j). In addition, compared to controls, CartIR−/− mice showed an increase in the number of PCNA-positive chondrocytes in the proliferating zone (Figure 2k and 2l) and a reduction in size of hypertrophic chondrocytes (Figure 2m and 2n).

Alterations of growth plate development in mice lacking IR in chondrocytes. (a) Immunostaining for IR in chondrocytes of the growth plates of the femurs from WT and IR mutant (ΔIR) mice at the age of 3 weeks. Scale bar=25 µm. Insert scale bar=50 µm. Arrows indicate IR-positive chondrocytes. (b) Representative X-ray images of the femurs from WT and IR mutant (ΔIR) mice at the age of 3 and 12 weeks. (c) Quantitative analysis was performed on femoral length from WT and ΔIR mice at 3 and 12 weeks of age. P>0.05, n=5. (d) Representative histological images of the growth plate of the femur in the 3-week old WT mice and ΔIR mice. P, proliferating zone; Pr, prehypertrophic zone; H, hypertrophic zone. Scale bar=25 µm. (e–g) Quantitation of the length of different zones of growth plates of the femur in the 3-week old WT mice and ΔIR mice. P<0.05, n=5. (h–j) Quantitative analysis of cell numbers of proliferating, prehypertrophic and hypertrophic chondrocytes of growth plates of the femur in the 3-week old ΔIR mice compared with that of the control littermates. P<0.05, n=5. (k) Representative images of immunostaining for PCNA in femoral sections (growth plate) from the ΔIR and the control littermates viewed at lower power (top) and higher power (bottom). Scale bar=25 µm. (l) Quantitation of the number of PCNA-positive cells and the percentage of PCNA-positive cells in growth plates of the femurs in the ΔIR and WT mice. P<0.05, n=5. (m) H&E staining showed decreased hypertrophic cell size in the growth plate of 3-week-old ΔIR mice than that of the control littermates. Double head arrows indicate the diameter of the hypertrophic chondrocytes in the growth plate. Scale bar=25 µm. (n) Quantitation of diameter of the hypertrophic chondrocytes from (m). P<0.05, n=5.
Disruption of IR impairs differentiation of chondrocytes in vitro
We next examined the effect of deletion of IR on chondrogenic differentiation in primary chondrocytes. Monolayers were infected with either Ad-GFP or Ad-Cre (100 MOI) and efficiency of the IR deletion was verified by measuring IR mRNA and protein expression 48 h after infection (Figure 3a and 3b). Chondrocytes lacking IR grown in micromass culture demonstrated a reduction in Alcian Blue staining that persisted even when cells were cultured in the presence of 10 nmol·L−1 insulin, which was partially restored by the treatment of IGF-1 (Figure 3c). These changes were accompanied by marked reductions in expression of Sox9 and collagen type 10 mRNA levels (Figure 3d and 3e).

Disruption of IR impairs differentiation of chondrocytes in vitro. Primary chondrocytes were isolated from IR flox/flox mice, and cultured in chondrogenic medium. The confluent chondrocyte monolayers were infected with either Ad-GFP or Ad-Cre (100 MOI) for 48 h. (a, b) IR mRNA and protein expression in chondrocytes infected with Ad-Cre relative to Ad-GFP as shown by real-time PCR and Western blot . **P<0.01, n=3. (c) The IR mutant (Ad-Cre) and control (Ad-GFP) chondrocytes were processed for cell mass cultures and induced to differentiate in chondrogenic medium in the presence or absence of insulin (10 nmol·L−1) and IGF-1 (13 nmol·L−1). The cell mass was stained with Alcian Blue after 6 days of culture. The chemical staining showed that deletion of the IR decreased chondrogenic differentiation and proteoglycan synthesis. (d, e) Chondrocytes lacking IR and the control cells were cultured and induced to differentiate in chondrogenic medium for 0, 7 and 14 days. Sox 9 and Col-10 mRNA level was detected by real-time PCR in IR mutant chondrocytes compared with that of the control cells. *P<0.05; **P<0.01, n=3.
Upregulation of IGF-1R-dependent signaling in chondrocytes lacking IR
The observation of increased chondrocyte numbers in growth plates from the mice lacking IR suggested the possibility of compensatory upregulation of IGF production and signaling, a similar phenomenon previously observed in osteoblasts lacking IGF-1R12. In accordance with this notion, the expression of IGF-1R increased following disruption of IR (Figure 4a) and increased IGF-1R levels were detected in growth plate chondrocytes from CartIR−/− mice (Figure 4b and 4c). In addition, expression of IGF-1 and IGF-2 mRNA increased following acute deletion of IR in primary chondrocytes (Figure 4d and 4e). Moreover, chondrocytes lacking IR had higher basal and IGF-1 induced Akt and ERK phosphorylation compared to control cells (Figure 4f and 4g). The apparent increase in sensitivity to IGF-1 was dependent on a functional IGF-1R since as IGF-1 induced Akt and ERK phosphorylation was markedly attenuated by the treatment with PPP (Figure 4f and 4g). To determine whether the enhanced responsiveness of IGF-1-generated signals in chondrocytes lacking the IR manifested in a greater biological response, we examined the effect of disruption of IR on chondrocyte proliferation. As shown in Figure 4h, both IGF-1 and insulin increased BrdU incorporation in GFP expressing cells. However, in cells lacking IR, IGF-1 further increased BrdU incorporation, whereas the effect of insulin was diminished as expected. By contrast, deletion of the IR did not significantly affect apoptosis of chondrocytes with or without IGF-1 or insulin treatment (Figure 4i). Collectively, these results suggest the presence of a common mechanism for overlapping functional redundancy in IGF and insulin signaling pathways and may explain our observation of persistent increased proliferation in growth plate chondrocytes from mice lacking IR.

Upregulation of IGF-1R-dependent signaling in chondrocytes lacking IR. (a) Protein expression of IGF-1R was shown by immunoblotting when primary chondrocytes were infected with either Ad-GFP or Ad-Cre (100 MOI). Actin used as loading control. (b) Representative images of immunostaining for IGF-1R in the growth plates of femoral sections from the WT and ΔIR mice. Sections were counterstained with hematoxylin. Red arrows indicate IGF-1R+ chondrocytes. Scale bar=25 µm. (c) The quantification of IGF-1R positive chondrocytes was presented as percentage of total chondrocytes in growth plate from the ΔIR and WT mice. Data represent mean±s.d. *P<0.05, n=5. (d, e) Chondrocytes lacking the IR and the control cells were cultured and induced to differentiate in chondrogenic medium for 0, 7 and 14 days, the mRNA levels of IGF-1 (d) and IGF-2 (e), were measured by real-time PCR in the IR mutant chondrocytes compared with that of the control cells. **P<0.01, n=3. (f, g)The IR mutant (Ad-Cre) and control (Ad-GFP) chondrocytes were serum-starved for 24 h, pre-treated with PPP (10 nmol·L−1) (+PPP) or solvent DMSO only (−PPP) for 4 h, and then treated with IGF-1 (13 nmol·L−1) for 10 minutes. The cell lysates were immunoblotted with antibodies against phospho-Akt and total Akt (f), or phospho-ERK and total ERK (g). Quantitation of the ratios of phospho-Akt to total Akt and phospho-ERK to total ERK was shown. Actin as the loading control. IGF-1, insulin-like growth factor-1; PPP, picropodophyllin. (h, i) Cell proliferation rate was assessed by flow cytometry following BrdU incorporation (h). *P<0.05, n=3. Apoptosis was assessed by flow cytometry using annexin V-PE staining (i). P>0.05, n=3.
Decreased hypertrophic cell size is associated with increased TSC2
To begin to address the mechanisms for the decreased size of the hypertrophic chondrocytes in the CartIR−/− mice, we determined the expression of TSC1 and TSC2 proteins in chondrocytes. In drosophila, TSC1/TSC2 complex acts to antagonize insulin signaling and negatively regulate cell growth and size.26 In primary chondrocytes, basal levels of TSC2 protein were increased following disruption of IR, whereas TSC1 protein levels were unchanged (Figure 5a). Treatment with IGF-1 modestly increased TSC2 levels to a similar extent in control and IR-deficient cells. In accord with these results, TSC2 protein levels determined immunohistochemically were higher in hypertrophic chondrocytes of the CartIR−/− mice compared to controls (Figure 5b). These results indicate that the IR signaling may be necessary to regulate cellular size and growth of hypertrophic chondrocytes during growth plate development at least partially mediated by TSC2.

Disruption of IR decreases size of hypertrophic chondrocytes associated with upregulation of TSC2. (a) TSC2 and TSC1 protein expression in chondrocytes was detected with immunoblotting using TSC2 and TSC1 antibodies in IR mutant (Ad-Cre) or control (Ad-GFP) chondrocytes treated with or without 10 nmol·L−1 insulin or 13 nmol·L−1 IGF-1. Actin as the loading control. (b) TSC2 expression in the sections of growth plate of the ΔIR and WT mice was assessed by immunofluorescence staining as described in the section on ‘Materials and methods’. DAPI was used to stain the nuclei. IgG represents the negative control for immunofluorescence staining. Original magnification, ×40.
Discussion
In the present study, we used the Cre-loxP technique to remove IR from chondrocytes, which has enabled us to determine the contribution of insulin signaling through its cognate receptor. We show that the loss of IR signaling causes upregulation of IGFs and IGF-1R, which appear to act in a compensatory fashion to regulate the proliferation and hypertrophy of chondrocytes.
The present results obtained in using fetal metatarsals and isolated chondrocytes in vitro clearly show that insulin can increase the proliferation of this cell type. Because these experiments used relatively low insulin concentrations that would not be expected to cross-activate the IGF-1R, we attribute these mitogenic actions to signaling through IR. Consistent with this finding, deletion of IR in cultured chondrocytes abolished the mitogenic activity of insulin. The mitogenic activity of insulin is conserved across species and has been previously reported in growth plate chondrocytes from the fetal lamb15. Surprisingly, however, the CartIR−/− mice exhibited increased PCNA-positive chondrocytes in all regions of the developing growth plate. Based on the fact that IR and IGF-1R are abundantly expressed in chondrocytes27,28 and the previous work which showed upregulation of IR signaling in osteoblasts lacking IGF-1R,12 we suspected that the increased proliferation of IR-deficient growth plate chondrocytes could be caused by upregulation of the IGF-1/IGF-1R signaling pathway. In keeping with this idea, IR-deficient chondrocytes demonstrated increased production of IGF-1 and IGF-2, and greater responsibility to IGF-1 the later due in part to increased expression of IGF-1R. These findings suggest the presence of a compensatory pathway present in cells expressing both IR and IGF-1R through which loss of insulin generated signals triggers increased signaling via IGF-1/IGF-1R. Such a mechanism would account for the preservation of relatively normal postnatal growth of the CartIR−/− mice.
Another striking feature of the growth plate in CartIR−/− mice was the decreased size of chondrocytes in the hypertrophic zone, suggesting a normal role for insulin in regulation of chondrocyte size. Previous studies have established a role for the conserved insulin signaling pathway in regulating cell and organ size in a number of species. In Drosophila, inactivation of positive regulators of insulin signaling (e.g., IR, IRS-1-4) decreases cell size, whereas overexpression of these components has the opposite effect.29,30 Insulin induces these effects by controlling the activity of the conserved TOR complex,31 which transduces insulin signals to control cell growth, size, polarity and metabolism32–34 through its ability to sense prevailing nutrient and energy levels. A critical component of this sensor is conferred by the TSC, which inhibits mTOR activity35 and to control the strength of insulin-generated signals according to the energy status of the cell. Thus, loss of TSC2 increases cells size and overexpression of the protein decreases cells size.26 Our finding of increased TSC2 levels in hypertrophic chondrocytes from the CartIR−/− mice is entirely consistent with loss of insulin signaling through mTOR and likely accounts for the smaller size of these chondrocytes. These data support a normal role for insulin signaling, as a determinant of chondrocyte size, which is apparently cannot compensated by local upregulation of IGF-1/IGF-1R. In summary, our studies demonstrate that insulin/IR signaling induces direct, IGF-1R-independent, actions on growth plate chondrocytes, which are necessary for them to proliferate, differentiate and achieve their final size. Our findings expand understanding of physiological role of insulin/IR in the skeleton and suggest the importance of future studies to examine insulin action under pathological conditions such as impaired bone healing associated with diabetes.
References
Fulzele K, Clemens TL . Novel functions for insulin in bone. Bone 2012; 50: 452–456.
Nef S, Parada LF . Hormones in male sexual development. Genes Dev 2000; 14: 3075–3086.
Ebina Y, Ellis L, Jarnagin K et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signaling. Cell 1985; 40: 747–758.
Kim JJ AcciliD . Signalling through IGF-1 and insulin receptors: where is the specificity? Growth Horm IGF Res 2002; 12: 84–90.
Woods KA, Camacho-Hubner C, Savage MO, Clark AJ . Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 1996; 335: 1363–1367.
Wang YM, Nishida S, Sakata T et al. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology 2006; 147: 4753–4761.
Baker J, Liu JP, Robertson EJ, Efstratiadis A . Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993; 75: 73–82.
Govoni KE, Lee SK, Chung YS et al. Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics 2007; 30: 354–362.
Wertheimer E, Lu SP, Backeljauw PF, Davenport ML, Taylor SI . Homozygous deletion of the human insulin receptor gene results in leprechaunism. Nat Genet 1993; 5: 71–73.
Accili D, Drago J, Lee EJ et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet 1996; 12: 106–109.
Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K . Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J 1997; 327 (Pt 1): 209–215.
Fulzele K, Digirolamo DJ, Liu Z, Xu J, Messina JL, Clemens TL . Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J Biol Chem 2007; 282: 25649–25658.
Long F, Joeng KS, Xuan S, Efstratiadis A, McMahon AP . Independent regulation of skeletal growth by Ihh and IGF signaling. Dev Biol 2006; 298: 327–333.
Chan SJ, Plisetskaya EM, Urbinati E, Jin Y, Steiner DF . Expression of multiple insulin and insulin-like growth factor receptor genes in salmon gill cartilage. Proc Natl Acad Sci USA 1997; 94: 12446–12451.
Hill DJ, De SD . Insulin is a mitogen for isolated epiphyseal growth plate chondrocytes from the fetal lamb. Endocrinology 1990; 126: 2661–2670.
Serra R, Chang C . TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res C Embryo Today 2003; 69: 333–351.
Efstratiadis A . Genetics of mouse growth. Int J Dev Biol 1998; 42: 955–976.
Ornitz DM, Marie PJ . FGF signaling pathways inendochondral and intramembranous bone development and human genetic disease. Genes Dev 2002; 16: 1446–1465.
Treadway JL, Frattali AL, Pessin JE . Intramolecular subunit interactions between insulin and insulin-like growth factor 1 alpha beta half-receptors induced by ligand and Mn/MgATP binding. Biochemistry 1992; 31: 11801–11805.
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R . Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 2009; 30: 586–623.
Brüning JC, Michael MD, Winnay JN et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998; 2: 559–569.
Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R . Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol 2004; 276: 124–142.
Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR . Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999; 96: 329–339.
Chen M, Lichtler AC, Sheu TJ et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis 2007; 45: 44–50.
Wang Y, Wan C, Deng L et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest 2007; 117: 1616–1626.
Inoki K, Zhu TQ, Guan KL . TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115: 577–590.
Phornphutkul C, Wu KY, Gruppuso PA . The role of insulin inchondrogenesis. Mol Cell Endocrinol 2006; 249: 107–115.
Phorphutkul C, Wu KY, Yang X, Chen Q, Gruppuso PA . Insulin-like growth factor-1 signaling is modified during chondrocyte differentiation. J Endocrinol 2004; 183: 477–486.
Böhni R, Riesgo-Escovar J, Oldham S et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 1999; 97: 865–875.
Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ . Cell-autonomous regulation of cell and organ growth in Drosophila by AKT/PKB. Nat Cell Biol 1999; 1: 500–506.
Johnston LA, Gallant P . Control of growth and organ size in Drosophila. Bioessays 2002; 24: 54–64.
Jewell JL, Guan KL . Nutrient signaling to mTOR and cell growth. Trends Biochem 2013; 38: 233–242.
Sasaki AT, Firtel RA . Regulation of chemotaxis by the orchestrated activationof Ras, PI3K, and TOR. Eur J Cell Biol 2006; 85: 873–895.
Howell JJ, Manning BD . mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 2011; 22: 94–102.
Inoki K, Li Y, Zhu TQ, Wu J, Guan KL . TSC2 is phosphorylated and inhibited by AKT and suppresses mTOR signaling. Nat Cell Biol 2002; 4: 648–657.
Acknowledgements
We are grateful to Jens Brüning for providing the IRflox/flox mice. We are grateful for the assistance of the Core Laboratories at School of Biomedical Sciences, the Chinese University of Hong Kong, and Histomorphometry and Molecular Analysis Core at the University of Alabama at Birmingham. This work was supported by the Hong Kong Research Grant Council General Research Fund (RGC GRF 475311), National Natural Science Foundation of China (NSFC81171717, 81130034), Shenzhen Strategic Development Fund (GJHS20120702105445379) and the Chinese University of Hong Kong Direct Grant 2041545 to CW.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhang, F., He, Q., Tsang, W. et al. Insulin exerts direct, IGF-1 independent actions in growth plate chondrocytes. Bone Res 2, 14012 (2014). https://doi.org/10.1038/boneres.2014.12
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2014.12
