
Abstract
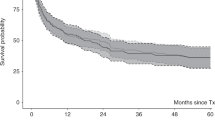
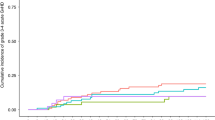
Allogeneic haematopoietic stem-cell transplantation (HSCT) is the only curative treatment for myelofibrosis. We retrospectively analyzed the outcome of patients who underwent allogeneic HSCT, 1994–2008, and the potential risk factors affecting non-relapse mortality (NRM), OS and relapse-free survival (RFS). A total of 39 patients, 15–65 (median 49) years old, diagnosed with primary (n=27) or secondary (n=12) myelofibrosis underwent HSCT (25 related and 14 unrelated). In ten patients, disease had transformed into acute leukaemia. Lille prognosis score was low for 9, intermediate for 16 and high for 14 patients. The conditioning regimen was myeloablative (MAC) for 15 and reduced-intensity (RIC) fludarabine-based for 24, with successful engraftment in 38 patients. A total of 31 patients developed grade I–IV GvHD; 19 developed chronic GvHD. The 3-year OS, RFS and NRM rates (95% confidence interval) were 60% (42–74), 54% (37–59) and 30% (30–45), respectively.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A . Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1976-1995. Am J Hematol 1999; 61: 10–15.
Cervantes F, Villamor N, Esteve J, Montoto S, Rives S, Rozman C et al. ‘Lymphoid’ blast crisis of chronic myeloid leukaemia is associated with distinct clinicohaematological features. Br J Haematol 1998; 100: 123–128.
Okamura T, Kinukawa N, Niho Y, Mizoguchi H . Primary chronic myelofibrosis: clinical and prognostic evaluation in 336 Japanese patients. Int J Hematol 2001; 73: 194–198.
Dupriez B, Morel P, Demory JL, Lai JL, Simon M, Plantier I et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood 1996; 88: 1013–1018.
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895–2901.
Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A . Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood 2005; 105: 973–977.
Guardiola P, Anderson JE, Bandini G, Cervantes F, Runde V, Arcese W et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood 1999; 93: 2831–2838.
Kerbauy DM, Gooley TA, Sale GE, Flowers ME, Doney KC, Georges GE et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant 2007; 13: 355–365.
Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008; 112: 3065–3072.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18: 295–304.
Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med 1980; 69: 204–217.
Sullivan KM, Agura E, Anasetti C, Appelbaum F, Badger C, Bearman S et al. Chronic graft-versus-host disease and other late complications of bone marrow transplantation. Semin Hematol 1991; 28: 250–259.
Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 1995; 15: 825–828.
Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005; 106: 2912–2919.
Prentice RL, Kalbfleisch JD, Peterson Jr AV, Flournoy N, Farewell VT, Breslow NE . The analysis of failure times in the presence of competing risks. Biometrics 1978; 34: 541–554.
Barosi G, Ambrosetti A, Centra A, Falcone A, Finelli C, Foa P et al. Splenectomy and risk of blast transformation in myelofibrosis with myeloid metaplasia. Italian Cooperative Study Group on Myeloid with Myeloid Metaplasia. Blood 1998; 91: 3630–3636.
Kroger N, Holler E, Kobbe G, Bornhauser M, Schwerdtfeger R, Baurmann H et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009; 114: 5264–5270.
Patriarca F, Bacigalupo A, Sperotto A, Isola M, Soldano F, Bruno B et al. Allogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Haematologica 2008; 93: 1514–1522.
Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115: 1703–1708.
Deeg HJ, Gooley TA, Flowers ME, Sale GE, Slattery JT, Anasetti C et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003; 102: 3912–3918.
Nichols WG, Corey L, Gooley T, Davis C, Boeckh M . High risk of death due to bacterial and fungal infection among cytomegalovirus (CMV)-seronegative recipients of stem cell transplants from seropositive donors: evidence for indirect effects of primary CMV infection. J Infect Dis 2002; 185: 273–282.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Lissandre, S., Bay, JO., Cahn, JY. et al. Retrospective study of allogeneic haematopoietic stem-cell transplantation for myelofibrosis. Bone Marrow Transplant 46, 557–561 (2011). https://doi.org/10.1038/bmt.2010.276
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bmt.2010.276
Keywords
This article is cited by
-
Allogeneic transplantation for primary myelofibrosis with BM, peripheral blood or umbilical cord blood: an analysis of the JSHCT
Bone Marrow Transplantation (2014)
