
Abstract
Background:
It is not clear if family history of cancer increases risk of cancer in children.
Methods:
We followed-up a total of 2 610 937 children born between 1960 and 2001 for cancer risk, and their parents and siblings. In this period, 2477 primary childhood solid tumours (except lymphoma) were diagnosed. The data from the Norwegian Family and Life Course Study and from the Norwegian Cancer Register were used. Classification of hereditary cancer syndromes was based on tumour histology, pedigrees and Chompret’s criteria.
Results:
An association between risk of childhood tumours and first-degree family history of early onset of solid tumours was observed for central nervous system tumours (2.3-fold), neuroblastoma (2.3-fold), retinoblastoma (6.1-fold), hepatic tumours (4.0-fold), and melanomas (8.3-fold). Elevated risk was also seen for osteosarcomas (1.5-fold) when considering first-degree family history of cancer diagnosed at any age. The risk of hepatic tumours, neuroblastomas and melanomas remained elevated even after controlling for probable hereditary cancer syndromes.
Conclusions:
The increased risk for several childhood solid site cancers among those with first-degree relatives diagnosed with solid cancer suggests that genetic or environmental factors are involved. The fact that these associations remained after controlling for hereditary cancer syndromes indicates other genetic mechanisms might be involved.
Similar content being viewed by others

Main
Every year worldwide, 130–140 children per million under the age of 15 are diagnosed with cancer. Cancer incidence within the first 5 years of life is twice as high as in older children (Imbach et al, 2011). Cancer occurring in children has a clinical and biological behaviour that is different from cancer occurring in adults. While in adults, the most frequent cancers are in the respiratory, gastrointestinal and reproductive organs, only 5% of cancerous diseases of children are manifested in these organs. Known cancer predisposition syndromes are found in <10% of childhood cancers (Narod et al, 1991; Strahm and Malkin, 2006). These include the Li-Fraumeni syndrome, neurofibromatosis type I, inherited retinoblastoma mutations, familial Wilms tumour, Beckwith–Wiedemann syndrome, and some disorders of DNA repair (Schiffman et al, 2013). Beyond these, it is not clear if family history of cancer increases the risk of cancer in children.
Family aggregation studies have been helpful in understanding the genetic aetiology of cancer (Ruteshouser and Huff, 2004; Trochet et al, 2004; Bachinski et al, 2005). Few studies investigating family history of cancer and the risk of childhood cancer have been reported. Two studies conducted in Scandinavia, especially for cancers in the central nervous system, show a familial risk for these tumours (Hemminki and Mutanen, 2001; Hemminki et al, 2010). However, other studies show mixed results (Nielsen et al, 2008; Poynter et al, 2010; Lupo et al, 2015). Excess risk among relatives has also been supported by several studies, but the results to date have not been consistent (Draper et al, 1996; Hemminki et al, 2000; Winther et al, 2001; Blumenthal et al, 2008; Neale et al, 2013).
Despite the importance of understanding the aetiology of cancer in children, the rarity of cases has made population-based epidemiologic studies difficult. Our primary goal was to assess the association between family history of solid cancer and solid tumours in children younger than 15 years by histological groups, while controlling for birth defects and probable hereditary cancer syndromes for all children born in Norway between 1960 and 2001.
Material and methods
Population
The Norwegian Family Based Life Course Study is a linkage of several registers (the Cancer Registry, the Medical Birth Registry, Statistics Norway) including genealogy, birth characteristics, sociodemographic indicators and cancer diagnoses and death (Næss and Hoff, 2013). The data for all children born in Norway between 1 January 1960 and 31 December 2001 were available, including the data for their siblings and parents. Case reporting to the cancer registry is close to 100% (Larsen et al, 2009). The current study focused on all solid childhood tumours, except lymphomas, as well as nine specific morphologic types classified according to the International Classification of Diseases for Oncology, 3rd Edition (Steliarova-Foucher et al, 2005).
All solid tumours except lymphomas among the parents and siblings diagnosed at any age were included. Information on cancer site in ICD7 was available from the years 1960–2009 for all solid tumours except lymphomas for both children and relatives. The follow-up of cancer among the relatives started in 1960 (when all citizens in Norway were assigned a personal identification number) and ended in 2009. All solid cancers diagnosed at any age among the relatives diagnosed in the study period (1960–2009) were included in the study. The follow-up period for each child was from birth to the age of cancer diagnosis, or censoring at the age of 14, death, emigration or end of study at 31 December 2001. Children could only be followed until 2001 since information on tumour morphology was available from 1960 to 2001.
The data on birth defects were obtained from The Medical Birth Registry which is a national health registry containing information about all births in Norway since 1967 (Jrgens and The Medical Birth Registry of Norway, 2000). The registry records birth defects that are diagnosed at delivery or by paediatric examination during the initial hospitalisation, around the first 5 days of life. Like other registries based on routine medical birth records, this registry does not capture all birth defects. Estimates of ascertainment vary by defect category. For example, The Medical Birth Registry of Norway captures an estimated 90% of neural tube defects (Lie et al, 1994). Birth defects were defined in accordance with the International Classification of Diseases (ICD-8).
Statistical analysis
Cox regression was used to calculate hazard ratios (HR) and 95% confidence intervals (CIs) for the association between family history of cancer and childhood cancer using age as the timescale. The follow-up period for each child was from birth to the age of cancer diagnosis, or censoring at the age of 14, death, emigration or December 2001.
The follow-up period for parents started before the child’s cancer diagnosis and ended at parent’s age at the end of the study at 2009. Number of years of follow up for parents before the child cancer diagnosis depended on the start of the cancer registry in Norway at 1960. Follow-up for siblings started at birth or at sibling’s age at 1960 for siblings who were born before this year, and ended at December 2009.
We used a dichotomous variable for the presence of solid tumours in a relative (parents and siblings). The analyses, including all first-degree relatives with cancer, were performed by treating the number of relatives with history of solid tumour as a continuous variable in the Cox model. Cancer occurring at young ages is suggestive of a hereditary predisposition (Lynch et al, 1976; Giráldez et al, 2012; Schiffman, 2012). For analysing the effect of age of the relative at cancer diagnosis on the risk of childhood cancer, diagnosis of cancer in a relative before the age of 30 years was considered to be early onset cancer. To minimise potential confounding factors relevant for cancer, we adjusted for child’s sex, child’s birth year, parental educational level and number of family members.
Hereditary cancer syndromes and birth defects
Analyses that excluded children with probable cases of hereditary cancer syndromes, and children with cancer having any birth defect (because of the associated high risk of developing cancer in children with birth defects) were also conducted (Sun et al, 2014). In order to identify potential cases of hereditary cancer syndromes, for all children with cancer and first-degree relatives with cancer, tumour histology was checked, and pedigrees were constructed. Schneider, 2011, describes 36 hereditary syndromes in which cancer is a prominent feature (Schneider, 2011). Chompret’s criteria identifies potential TP53 mutation carriers (Tinat et al, 2009). These criteria were met if the offspring had a tumour from the Li-Fraumeni syndrome (LFS) spectrum (soft tissue sarcoma, osteosarcoma, brain tumour, breast cancer, or adrenocortical carcinoma) and had at least one first-degree relative with an LFS tumour before 46 years. Information on birth defects was obtained from the Medical Birth Registry. We conducted sensitivity analyses for children born between 1980 and 2001 to take into account a longer follow-up time for cancer risk among parents before the child’s cancer diagnosis.
Analyses were run in SPSS version 22 (IBM, Armonk, NY, USA). The project was approved by the Norwegian Data Protection Authority and the Regional Committees for Medical and Health Research Ethics, (REK Sør-Øst: 2010/260). As this is a register-linked study, the approval also covers exemption from informed consent because that would not be feasible to acquire.
Results
A total of 2 610 937 children were included in the study; characteristics of cases and non-cases are summarised in Table 1. The follow-up period comprised 34 031 035 person-years with a mean follow-up time of 11 (SD 4.4) years for cancer cases and 13 (SD 2.7) years for non-cases. During this period, 2477 offspring aged 0–14 years were diagnosed with a first-primary solid tumour. Of these, 1259 (50.8%) children were diagnosed before the age of 5 years. Mean age at cancer diagnosis for children 0-14 years was 5.5 (SD 4.4) years (Table 1).
Supplementary Appendix 1 shows the number of affected family members for the cases and non-cases.
Figure 1 shows the age-standardised incidence rates of the most common tumours in children aged 0–14 years. Children with cancer in the CNS showed the highest incidence rate, 40.9 cases per million in the youngest group (<5 years), 36.7 in the age group 5–9 years and 26.1 in the oldest age group (10–14 years). Hepatic tumours, neuroblastoma and retinoblastoma showed elevated incidence rates in the youngest age group (<5 years), with 7.0, 26.0 and 19.5 affected children per million, respectively. In Supplementary Appendix 2 results for sex ratio for children with cancer—the ratio of the number of male cases to the number of female cases (M/F ratio) are shown. The sex ratio among 2477 individuals with childhood cancer (1339 males and 1138 females) was 1.2. The sex ratio according to type of cancer is presented in Supplementary Appendix 2. The sex ratio for CNS tumours was significantly higher in males than in females, with a ratio of 1.3 (P<0.01), and for hepatic tumours with a sex ratio of 2.5 (P<0.01).

Age-standardised incidence rates of common solid tumours (except lymphoma) in children 0–14 years born 1980–1989 in Norway by diagnostic groups ICCC-3. CNS=central nervous system tumours. Standard world population was used as reference. A full colour version of this figure is available at the British Journal of Cancer journal online.
Table 2 shows the adjusted HRs with confidence intervals and P-values for solid tumours among children aged 0–14 years based on family history of any solid tumour (except lymphoma) in first-degree relatives. Elevated hazard ratio of cancers of the central nervous system (CNS), neuroblastoma, retinoblastoma, hepatic tumours and malignant melanomas in children aged 0–14 years, who had a first-degree family history of early onset of any solid tumour, were observed. Individuals aged 0–14 years having parents with a history of solid cancer showed a 1.5-fold increase in risk for neuroblastoma. The risk was higher (2.3-fold) when the family history was restricted to relatives diagnosed with solid tumours before 30 years of age. Among individuals with a sibling with history of solid tumours, elevated HRs were observed for retinoblastoma (6.1-fold) and hepatoblastoma (5.9-fold). For osteosarcomas, the increased risk was 1.5-fold when considering family history of solid tumours for all relatives. When analysis of germ cell tumours was stratified by sex, no evidence of associations was observed (male HR=0.83, 95% CI=0.46–1.49 and female HR=0.87, 95% CI=0.44–1.70) (data not shown).
Probable hereditary syndromes (data not shown)
We identified 49 (2%) cases of childhood tumours fulfilling the Chompret’s criteria. The majority of those, 33 cases (67%), were found in individuals having a relative with a history of solid tumour before the age 30. More than half of the cases fulfilling the Chompret’s criteria were CNS cases; 18 cases (37%) had relatives with solid tumours before the age of 30. Few osteosarcoma cases were found (n=2). Risk of childhood CNS tumours was increased among individuals with relatives who met the Chompret’s criteria with 3.2-fold increase (95% CI=2.20–4.68).
Birth defects (data not shown)
We found 131 children with birth defects among those with solid tumours; 27 were neural tube birth defects. The cancers observed most commonly in individuals with birth defects were CNS (8%), soft tissue sarcomas (12%) and neuroblastomas (6%). Children with congenital neural tube defects showed an elevated risk for tumour in the CNS compared to children without such defects (HR=39.04, 95% CI=20.24–75.29). We also noted a high number of individuals with these three cancer types who had siblings with birth defects; 27, 15 and 5 siblings with birth defects, respectively. An excess risk for neuroblastoma among individuals having any birth defect was observed (HR=3.21, 95% CI=1.98–5.20). Among the 131 children with cancer and any birth defects, 88 (67.2%) cases were classified as a serious birth defect, 56 (42.7%) had information on type of birth defect distributed as follows: 27 neuronal tube defects (spina bifida=26, encephalocele <5), hydrocephalus=7, cleft palate without cleft lip <5, cleft lip with or without cleft palate=7, oesophageal atresia <5, anorectal atresia <5, hypospadias <5, omfalocele <5, gastroschisis <5 and Downs syndrome <5.
Results after excluding for probable hereditary syndromes and birth defects
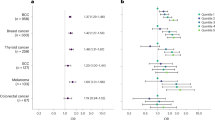
Figure 2 compares the HR for solid tumours based on family history from Table 2 wuth the HR estimated from analysis, which excluded individuals with probable hereditary syndromes. The risk of CNS tumours was markedly elevated among children with relatives with cancer diagnosed at an early age (<30 years) (Table 2). In this first-degree relative group, 18 cases (64%) with probable hereditary syndromes were excluded; two of these children had a history of birth defects. After excluding these cases no evidence of association was observed (Figure 2).

Adjusted HRs (95% confidence interval) for the association between family history of any solid tumours (except lymphoma) in selected first-degree relatives and the risk of solid tumours in children after excluding the data for individuals with probable hereditary cancer syndromes and birth defects. The models are adjusted for sex, birth cohort, parents’ educational level and number of relatives. CNS=central nervous system; HR=hazard ratio. A full colour version of this figure is available at the British Journal of Cancer journal online.
Elevated risk of neuroblastoma was observed among individuals having parents with history of solid tumours and was also observed among individuals with any first-degree relative diagnosed with any solid tumour before the age of 30 years (Table 2). In Figure 2, the results for the risk of neuroblastoma among individuals with parents diagnosed with tumours are shown. After excluding individuals with probable hereditary syndromes and birth defects (n=23), the HR was still elevated. The results for children with neuroblastoma having any first-degree relative diagnosed with any solid tumour before the age of 30 years was no longer statistically significant after exclusion of individuals with hereditary syndromes and birth defects (HR=2.10, 95% CI= 0.87–5.10; data not shown). In this group three cases (37.5%) were excluded with probable hereditary syndromes, two of them were two siblings with probable familial neuroblastoma.
The hazard ratio presented in Figure 2 for retinoblastoma was estimated among individuals with siblings with any diagnosed solid tumour. This group had the most cases and elevated hazards. Six individuals with siblings with familial retinoblastoma were identified and excluded, corresponding to 60% of the cases. The risk was considerably reduced after excluding these cases and was not statistically significant at a 5% level (Figure 2).
The risk for hepatic tumours was high in almost all relative groups analysed (Table 2). Figure 2 shows the analysis, including all first-degree relatives and after excluding for probable hereditary syndromes and birth defects. We found two cases of siblings with hepatoblastoma and three cases with birth defects, and after excluding these cases, a significant association was still observed (HR=1.73, 95% CI=1.07–2.78).
For osteosarcomas, two cases fulfilling the Chompret’s criteria were excluded from the analysis, including all first-degree relatives; no children with birth defects were found. Figure 2 shows a reduction in risk of osteosarcoma after excluding these cases. The HR was no longer statistically significant at a 5% level.
Malignant melanomas and other epithelial tumours showed the highest risk of all tumours analysed in almost all relative groups. We excluded four cases with family history of skin and thyroid tumours as well as three children with birth defects. The HR remained high in both when considering all first-degree relatives (Figure 2), and when only including relatives with cancer diagnosed at a young age (<30 years) with HR=8.76 (95% CI= 3.21–24.32) for epithelial tumours and HR=9.61 (95% CI=2.95–31.27) for melanomas (data not shown).
Results from sensitivity analyses conducted with information for children born between 1980 and 2001 showed similar HRs to those estimated for the whole cohort (Supplementary Appendix 3).
Discussion
This study examined the association between first-degree family history (parents and siblings) of any solid tumour except lymphomas and of such childhood tumours. We found significantly increased risks for childhood cancer in the CNS, neuroblastoma, retinoblastoma, hepatic tumours, osteosarcoma, and malignant melanoma. After controlling for probable hereditary cancer syndromes explaining the family history and restricting the analysis to first-degree relatives diagnosed before the age of 30, the risk for these cancers was not different from the general population except for neuroblastoma, hepatic tumours and malignant melanomas. In accordance with a previous study (Winther et al, 2001), we assume that hereditary cancer syndromes may account for about 40–60% of the occurrence of CNS tumours and retinoblastoma.
Strengths and limitations
This is one of the largest familial studies of childhood solid tumours reported to date, including several tumour subtypes and more than 4 decades of follow-up. In all, 2477 children with solid tumours were included in the study, which allowed us to undertake subgroup analyses by histological types. The large number of cancer cases and long follow-up among relatives may provide more accurate estimates of familial risk than earlier studies with fewer cancer cases. Our study was based on all births in Norway from 1960 to 2001, so bias caused by a skewed study sample is unlikely. Information on the variables of interest was obtained from the linkage of national population-based registers (removing ascertainment and recall bias), and the high quality of cancer case registration allowed for a complete follow-up of the study population. The present study may contribute to knowledge concerning rare tumours in children.
Some limitations must also be noted. Use of total life-time cancer among relatives increased the power of the study; however, because cancer information for family members included cancers diagnosed after the time of cancer in the offspring, this study may have identified significant associations rather than significantly increased risk. Nevertheless, the results certainly suggest that more research is needed on potential genetic contribution to childhood cancers. Multiple comparisons were made, which may have resulted in some chance associations. However, among the main morphologic groups analysed (except for neuroblastomas), all the P-values indicate significance even after a Bonferroni correction (Sterne and Davey Smith, 2001).
The study was limited by lack of details on other potentially important confounders, which were unavailable in the health registries used; examples include infectious disease history and environmental exposures associated with cancer. Information on cancer risk among parents at young ages in the first birth cohort period was not available and could introduce bias by left truncation. However, we conducted sensitivity analysis with data restricted to the most recent part of the cohort, which showed similar hazard ratios. The follow-up for cancer among relatives was until 2009, which could introduce bias, especially among children with young parents. The study is based on children from Norway, primarily a Caucasian population. It is unclear whether these data can be generalised to non-Caucasian populations. Finally, classifications for family history of genetic syndromes were assigned by the data review, not genetic testing and may not be correct. However, earlier studies have confirmed the clinical utility of the Chompret’s criteria reporting a TP53 mutation detection rate of 29–36% (Bougeard et al, 2008; Gonzalez et al, 2009).
Cancer sites in children
Children with cancer in the CNS showed the highest incidence rate. These rates are similar to those reported earlier in the Nordic countries (Schmidt et al, 2011). Hepatic tumours, neuroblastoma and retinoblastoma show elevated incidence rates in children younger than 5 years. These results are also similar to those observed in other Nordic countries (NORDCAN, 2016). The sex ratio for CNS tumours and hepatic tumours was significantly higher in males compared with females. Significant differences were also observed for hepatic tumours. Similar sex ratio differences are also reported in earlier studies (Schmidt et al, 2011; Thompson, 2012).
Our findings agree with Nielsen et al from 2008 regarding CNS tumours (Nielsen et al, 2008). After excluding children with hereditary syndromes, they found no association of these tumours with family history of cancer. Hemminki and Mutanen (2001) found a 2-fold increased risk for childhood CNS, 6.4-fold risk for endocrine tumours and about 60-fold risk for retinoblastoma among children whose parents had the same cancer type (Hemminki and Mutanen, 2001). They reported similar results in a more recent study among individuals aged 0–19 years (Hemminki et al, 2010). Our results also show elevated HR, but no strong evidence of association after excluding children with probable hereditary syndromes.
Elevated risk of germ cell tumours and rhabdomyosarcoma has also been reported by the Children’s Oncology Group (Lupo et al, 2015). For germ cell tumours, a reduced risk among female cases and an elevated risk among male cases of germ cell tumours with a family history of cancer before 40 years was reported (Poynter et al, 2010). Our results stratified by sex found no associations. Nielsen et al (2008) found elevated risk of rhabdomyosarcoma among individuals with a first-degree relative with cancer before age 30. As we did not have enough cases of children with rhabdomyosarcoma having relatives with solid tumours diagnosed at early age; we were no able to conduct similar analysis.
Individuals with Li–Fraumeni syndrome have extraordinarily high risk of developing a diverse array of malignancies, occurring frequently at young ages (Bougeard et al, 2015). In the present study, the risk of childhood CNS tumours was markedly increased among individuals with relatives who met the Chompret’s criteria with 3.2-fold increase. After excluding these cases from the analysis, the risk was considerably reduced, suggesting that hereditary syndromes may play an important role in the aetiology of CNS tumours, when relatives of children affected have a solid tumour at early age. The same analysis conducted among individuals with osteosarcoma showed a similar decrease in risk (Figure 2).
Neuroblastoma has been observed in children with the Beckwith–Wiedemann syndrome, neurofibromatosis, Hirschsprung disease and central hypoventilation syndrome (Coran et al, 2012). It has also been reported in twins and in parents (Pegelow et al, 1975; Tajiri et al, 2010). In our data set, we observed three children with neuroblastoma having relatives with probable hereditary syndromes; a twin sibling with neuroblastoma diagnosed before 1 year of age, and a sibling with a peripheral nerve tumour. After excluding these cases, we did not observe significant association between children with neuroblastoma and family history of solid tumours diagnosed at young age (<30 years). However, for children with neuroblastoma, whose parents were diagnosed with solid tumours, the risk was still elevated after excluding children with probable hereditary syndromes and birth defects. This might indicate that other genetic or environmental factors may be involved in this association. However, in the sensitivity analysis, the risk for neuroblastoma for children having parents with history of solid tumours was elevated, but not statistically significant.
We observed elevated risk of retinoblastoma among individuals with siblings with solid tumours and among individuals with relatives diagnosed with solid tumours at young age (<30 years). Bilateral retinoblastomas are commonly heritable, while unilateral retinoblastomas are commonly non-heritable. Most children with hereditary retinoblastoma do not have an affected parent. But these children can still pass their RB1 gene mutation on to their children. In this analysis, these tumours types could not be distinguished, but a decrease in risk was observed when cases with familial retinoblastoma were excluded and the association was no longer significant (Figure 2).
Hepatic tumours have previously been associated with some genetic conditions such as Beckwith–Wiedemann syndrome, familial adenomatous polyposis and Edwards Syndrome (Spector and Birch, 2012). Beckwith–Wiedemann syndrome represents the most relevant risk factor for hepatoblastoma with a risk 2280 times that of the typical population (DeBaun and Tucker, 1998). There is a well-documented higher risk of hepatoblastoma in children with very low birthweight. Congenital anomalies may also play an important role (Spector et al, 2008). After excluding children with probable hereditary cancer syndromes and congenital malformations, the elevated risk observed among children with these tumours remained, suggesting other factors may affect the association between history of solid tumours and hepatic tumours in children.
The risk for childhood melanoma was elevated in our study and was still significantly elevated after excluding cases of probable hereditary syndromes. Epidemiological studies suggest that interactions between solar exposure, fair skin, immunosuppression and family history of melanoma are the main determinants of childhood melanoma. Familial cases account for ∼10% of malignant melanomas (Pappo, 2003). The incidence of the disease rises steeply with age. These results may indicate that not only family history may play a role in the development of this disease, but also environmental factors.
The diagnosis of a cancer in a child should induce the clinician to investigate the causes of the cancer. In some cases, hereditary syndromes can be detected. It can be a heterogeneous condition and patients can have a wide variety of abnormalities. Furthermore, early diagnosis is crucial to provide appropriate medical management. However, genetic predisposition for childhood cancer is underdiagnosed (Jongmans et al, 2016). This study adds to the body of evidence that inherited genetic susceptibility may be a factor in the development of childhood solid tumours. Identifying children with increased hereditary risk of developing cancer is of high importance, as it may inform a potential cancer therapy protocol. In some predisposition syndromes, a modification in therapy for the cancer is needed, for example, when radiotherapy is contraindicated or when an adjustment of chemotherapy dose is advised (Postema et al, 2017). Furthermore, for several predisposition syndromes, a surveillance program is indicated for the early identification of other malignancies.
Family members of patients with an identified cancer predisposition syndrome may have excesses of some cancers and this may be partly attributable to greater cancer awareness. Establishing the diagnosis of a hereditary syndrome, and beginning surveillance when appropriate can make patients and their families feel more secure. It might also influence family planning and reproductive choices.
Conclusion
The increased associations for several childhood solid site cancers among those with first-degree relatives diagnosed with solid cancer suggests that genetic or environmental factors are involved. The fact that these associations remained after controlling for hereditary cancer syndromes indicates that other genetic mechanisms might be involved.
Change history
20 March 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bachinski LL, Olufemi SE, Zhou X, Wu CC, Yip L, Shete S, Lozano G, Amos CI, Strong LC, Krahe R (2005) Genetic mapping of a third Li-Fraumeni syndrome predisposition locus to human chromosome 1q23. Cancer Res 65: 427–431.
Blumenthal DT, Cannon-Albright LA. Familiality in brain tumors (2008) Familiality in brain tumors. Neurology 71: 1015–1020.
Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, Gauthier-Villars M, Stoppa-Lyonnet D, Consolino E, Brugières L, Caron O, Benusiglio PR, Bressac-de Paillerets B, Bonadona V, Bonaïti-Pellié C, Tinat J, Baert-Desurmont S, Frebourg T (2015) Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 33: 2345–2352.
Bougeard G, Sesboüé R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, Brugières L, Chompret A, de Paillerets BB, Stoppa-Lyonnet D, Bonaïti-Pellié C, Frébourg T French LFS working group (2008) Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45: 535–538.
Coran A, Caldamone A, Adzick N, Krummel T, Laberge J, Shamberger R (2012) Pediatric Surgery 7th ed Elsevier Health Sciences: London.
DeBaun MR, Tucker MA (1998) Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132: 398–400.
Draper GJ, Sanders BM, Lennox EL, Brownbill PA (1996) Patterns of childhood cancer among siblings. Br J Cancer 74: 152–158.
Engholm G, Ferlay J, Christensen N, Kejs AMT, Hertzum-Larsen R, Johannesen TB, Khan S, Leinonen MK, Ólafsdóttir E, Petersen T, Schmidt LKH, Trykker H, Storm HH. NORDCAN: Cancer Incidence, Mortality, Prevalence and Survival in the Nordic Countries, Version 7.3. Association of the Nordic Cancer Registries. Danish Cancer Society. Available at http://www.ancr.nu accessed 08 July 2016.
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, Han JH, Lowstuter K, Longmate J, Sommer SS, Weitzel JN (2009) Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27: 1250–1256.
Hemminki K, Li X, Vaittinen P, Dong C (2000) Cancers in the first-degree relatives of children with brain tumours. Br J Cancer 83: 407–411.
Hemminki K, Mutanen P (2001) Parental cancer as a risk factor for nine common childhood malignancies. Br J Cancer 84: 990–993.
Hemminki K, Tretli S, Olsen JH, Tryggvadottir L, Pukkala E, Sundquist J, Granström C (2010) Familial risks in nervous system tumours: joint Nordic study. Br J Cancer 12: 1786–1790.
Imbach P, Kuhne T, Arceci R SpringerLink (2011) Pediatric Oncology. Berlin, Heidelberg: Springer Berlin Heidelberg.
Jrgens LM The Medical Birth Registry of Norway (2000) Epidemiological research and surveillance throughout 30 years. Acta Obstet Gynecol Scand 79: 435–439.
Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, Hoogerbrugge N (2016) Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur J Med Genet 59: 116–125.
Larsen IK, Småstuen M, Johannesen TB, Langmark F, Parkin DM, Bray F, Møller B (2009) Data quality at the Cancer Registry of Norway: an overview of comparability, completeness, validity and timeliness. Eur J Cancer 45: 1218–1231.
Lie RT, Heuch I, Irgens LM . Maximum likelihood estimation of the proportion of congenital malformations using double registration systems. Biometrics (1994) 50: 433–444.
Lupo PJ, Danysh HE, Plon SE, Curtin K, Malkin D, Hettmer S, Hawkins DS, Skapek SX, Spector LG, Papworth K, Melin B, Erhardt EB, Grufferman S, Schiffman JD (2015) Family history of cancer and childhood rhabdomyosarcoma: a report from the Children's Oncology Group and the Utah Population Database. Cancer Med 4: 781–790.
Lynch HT, Guirgis H, Brodkey F, Maloney K, Lynch PM, Rankin L, Lynch J (1976) Early age of onset in familial breast cancer. Genetic and cancer control implications. Arch Surg 111: 126–131.
Neale RE, Stiller CA, Bunch KJ, Milne E, Mineau GP, Murphy MF (2013) Familial aggregation of childhood and adult cancer in the Utah genealogy. Int J Cancer 133: 2953–2960.
Nielsen S, Mueller BA, Preston-Martin S, Holly EA, Little J, Bracci PM, McCredie M, Peris-Bonet R, Cordier S, Filippini G, Lubin F (2008) Family cancer history and risk of brain tumors in children: results of the SEARCH international brain tumor study. Cancer Causes Control 19: 641–648.
Narod SA, Stiller C, Lenoir GM (1991) An estimate of the heritable fraction of childhood cancer. Br J Cancer 63: 993–999.
Næss Ø, Hoff DA (2013) The Norwegian family based life course (NFLC) study: data structure and potential for public health research. Int J Public Health 58: 57–64.
Pappo AS (2003) Melanoma in children and adolescents. Eur J Cancer 39: 2651–261.
Pegelow CH, Ebbin AJ, Powars D, Towner JW (1975) Familial neuroblastoma. J Pediatr 87: 763–765.
Postema FAM, Hopman SMJ, Hennekam RC, Merks JHM (2017) Consequences of diagnosing a tumor predisposition syndrome in children with cancer: a literature review. Pediatr Blood Cancer Aug 22 doi:10.1002/pbc.26718.
Poynter JN, Radzom AH, Spector LG, Puumala S, Robison LL, Chen Z, Ross JA, Shu XO (2010) Family history of cancer and malignant germ cell tumors in children: a report from the Children's Oncology Group. Cancer Causes Control 21: 181–189.
Ruteshouser EC, Huff V (2004) Familial wilms tumor. Am J Med Genet C Semin Med Genet 129: 29–134.
Schiffman JD, Geller JI, Mundt E, Means A, Means L, Means V (2013) Update on pediatric cancer predisposition syndromes. Pediatr Blood Cancer 60: 1247–1252.
Schiffman JD (2012) Hereditary cancer syndromes: if you look, you will find them. Pediatr Blood Cancer 58: 5–6.
Giráldez MD, López-Dóriga A, Bujanda L, Abulí A, Bessa X, Fernández-Rozadilla C (2012) Susceptibility genetic variants associated with early-onset colorectal cancer. Carcinogenesis 33: 613–619.
Schneider K Hereditary Cancer Syndromes (2011) In Counseling About Cancer pp. 75–150. Hoboken, NJ, USA: John Wiley & Sons.
Schmidt LS, Schmiegelow K, Lahteenmaki P, Träger C, Stokland T, Grell K, Gustafson G, Sehested A, Raashou-Nielsen O, Johansen C, Schüz J (2011) Incidence of childhood central nervous system tumors in the Nordic countries. Pediatr Blood Cancer 56: 65–69.
Spector LG, Birch J (2012) The epidemiology of hepatoblastoma. Pediatr Blood Cancer 59: 776–779.
Spector LG, Johnson KJ, Soler JT, Puumala SE (2008) Perinatal risk factors for hepatoblastoma. Br J Cancer 98: 1570–1573.
Strahm B, Malkin D (2006) Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer 119: 2001–2006.
Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P (2005) International classification of childhood cancer, third edition. Cancer 103: 1457–1467.
Sun Y, Overvad K, Olsen J (2014) Cancer risks in children with congenital malformations in the nervous and circulatory system-A population based cohort study. Cancer Epidemiol 38: 393–400.
Sterne JA, Davey Smith G. (2001) Sifting the evidence-what's wrong with significance tests? BMJ 322: 226–231.
Tajiri T, Souzaki R, Kinoshita Y, Tanaka S, Koga Y, Suminoe A, Hara T, Kohashi K, Oda Y, Masumoto K, Ohira M, Nakagawara A, Taguchi T (2010) Concordance for neuroblastoma in monozygotic twins: case report and review of the literature. J Pediatr Surg 45: 2312–2316.
Thompson P. A. (2012) Pediatric liver tumors, Series. J Gastrointest Oncol 3 (1): 80–81. https://doi.org/10.3978/j.issn.2078-6891.2011.048.
Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frébourg T, Munnich A, Lyonnet S, Delattre O, Amiel J (2004) Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 74: 761–764.
Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, Caron O, Bressac-de Paillerets B, Berthet P, Dugast C, Bonaïti-Pellié C, Stoppa-Lyonnet D, Frébourg T (2009) version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 27: 108–109.
Winther JF, Sankila R, Boice JD, Tulinius H, Bautz A, Barlow L, Glattre E, Langmark F, Möller TR, Mulvihill JJ, Olafsdottir GH, Ritvanen A, Olsen JH (2001) Cancer in siblings of children with cancer in the Nordic countries: a population-based cohort study. Lancet 358: 711–717.
Acknowledgements
This investigation was supported by a research grants from ExtraStiftelsen through the Norwegian Cancer Society, The Research Council of Norway and the Wedel Jarlsbergs Fond. We thank the Norwegian Institute of Public Health for access to family data. Certain data used in this publication were obtained from the Norwegian Cancer registry.
Disclaimer
The authors assume full responsibility for analyses and interpretation of these data.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Del Risco Kollerud, R., Blaasaas, K., Claussen, B. et al. Family history of cancer and the risk of childhood solid tumours: a Norwegian nationwide register-based cohort study. Br J Cancer 118, 905–912 (2018). https://doi.org/10.1038/bjc.2017.493
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.493
Keywords
This article is cited by
-
Correlation between oncological family history and clinical outcome in a large monocentric cohort of pediatric patients with rhabdomyosarcoma
International Journal of Clinical Oncology (2021)
-
Family history of cancer and the risk of childhood brain tumors: a pooled analysis of the ESCALE and ESTELLE studies (SFCE)
Cancer Causes & Control (2019)
-
Germline mutations as potential causes of childhood solid tumours: comments on the Norwegian childhood cancer cohort study
British Journal of Cancer (2018)
