
Abstract
Background:
To determine whether childhood infections were associated with the development of childhood acute lymphoblastic leukaemia (ALL).
Methods:
We included studies that assessed any infection in childhood prior to the diagnosis of ALL in children aged 0–19 years compared to children without cancer. The primary analysis synthesised any infection against the odds of ALL, and secondary analyses assessed the frequency, severity, timing of infections, and specific infectious agents against the odds of ALL. Subgroup analyses by data source were investigated.
Results:
In our primary analysis of 12 496 children with ALL and 2 356 288 children without ALL from 38 studies, we found that any infection was not associated with ALL (odds ratio (OR)=1.10, 95% CI: 0.95–1.28). Among studies with laboratory-confirmed infections, the presence of infections increased the odds of ALL by 2.4-fold (OR=2.42, 95% CI: 1.54–3.82). Frequency, severity, and timing of infection were not associated with ALL.
Conclusions:
The hypothesis put forward by Greaves and others about an infectious aetiology are neither confirmed nor refuted and the overall evidence remains inadequate for good judgement. The qualitative difference in the subgroup effects require further study, and future research will need to address the challenges in measuring infectious exposures.
Similar content being viewed by others


Main
The aetiology of childhood acute lymphoblastic leukaemia (ALL) is largely unknown, and likely arises from interactions between exogenous and/or endogenous exposures, genetic susceptibility, and chance. Genetic causes of ALL account for a small proportion of cases, and while the disease is usually initiated in utero, other promotional exposures are probably necessary for disease emergence (Greaves et al, 2003). There are two key hypotheses on infections and the development of ALL. Kinlen proposed the ‘population mixing’ hypothesis to describe the observed increased rates of childhood ALL following an influx of migrants into rural areas (Kinlen, 1988, 2012). Briefly, the mixing of rural, isolated individuals with the influx of mostly urban individuals into a rural area would create a localised epidemic of an underlying infection due to the increased level of contact between susceptible and infected individuals that may produce the rare response of ALL. Studies from Kinlen and others have found evidence to support the hypothesis (Kinlen, 1988, 2006, 2012; Alexander et al, 1998; Kinlen and Doll, 2004). The hypothesis suggests a direct pathological role of a specific infection, presumed to be viral, in the development of ALL and that a protective effect may be acquired from previous exposure. Currently, there is limited molecular evidence that implicates a specific infection (Martin-Lorenzo et al, 2015; da Conceicao Nunes et al, 2016). Greaves’ ‘delayed infection’ hypothesis for childhood ALL suggests a two-hit model that emphasises the timing of exposure and the child’s immune system (Greaves, 1997, 2006). The first hit occurs in utero through one’s genetic makeup that produces a pre-leukaemic clone. In a small number of pre-leukaemia carriers, it is the absence of exposure to infections in early life, and a postnatal secondary genetic event caused by a delayed, stress-induced infection (second hit) on the developing, ‘unprepared’ immune system that may increase the risk of childhood ALL. Although the mechanisms differ, both hypotheses suggest that ALL is a rare response to one or more common infections acquired through personal contact.
The difficulties in measuring exposure to infectious agents and subsequent responses make it challenging to directly test the hypotheses, especially since no specific leukaemogenic agent has been identified. Several previous epidemiological studies have used a history of infections as an indicator for early exposure to infections. Establishing the timing of the infections is critical to testing the hypotheses; however, birth cohort studies are not feasible given the rarity of childhood ALL. Thus, most studies used a case–control design and interviews to measure infections. Assessing a history of infections through interviews can be problematic due to the potential for recall bias and misclassification of children who had asymptomatic infections (Simpson et al, 2007). Other methods for measuring infections such as using administrative data overcome these limitations, but may lack information on important confounders. Other than narrative summaries (McNally and Eden, 2004; Buffler et al, 2005; Ma et al, 2009; Maia Rda and Wunsch Filho, 2013), no study has attempted to synthesise and quantitatively pool studies examining the relationship using a history of infections, or tried to explain the differences between the studies. The aim of this systematic review and meta-analysis was to assess the relationship between childhood infections, and the development childhood ALL by summarising the findings for an overall measure of infections, the frequency, severity, timing of infections, and examining specific infectious agents and syndromes.
Materials and methods
The Meta-analysis of Observational Studies in Epidemiology (MOOSE) was developed as a guideline for the reporting of meta-analyses of observational studies in epidemiology and was used for the current study (Stroup et al, 2000).
Data sources and searches
We performed electronic searches from inception to 21 February 2017 in Ovid MEDLINE, MEDLINE In-Process and Other Non-Indexed Citations, EMBASE, Web of Science (Science Citation Index Expanded, Social Sciences Citation Index, Conference Proceedings Citation Index for both Science and Social Science & Humanities), and Scopus. Supplementary Table 1 shows the search strategies used. Text words used included acute lymphoblastic leukaemia, acute leukaemia, infection, virus, and bacteria. We limited the search to subjects 0–19 years old, and did not restrict the search by language. References of the included studies were searched, and the first four pages of a Google search using the same keywords were used to search for grey literature.
Study selection
We defined the inclusion and exclusion criteria a priori as studies of any design excluding editorials, reviews, and case reports. Studies were included if: (1) the primary exposure of interest included a prior history of any infection before the diagnosis of childhood ALL; (2) the primary outcome of interest was defined as clinically diagnosed ALL in children aged ⩽19 years; (3) comparisons were made against a control or comparison group; and (4) testing samples must have been collected and assessed prior to treatment, if laboratory investigations were used to determine past infections. Infections must have been reported by the parent or guardian, or obtained through other data sources such as medical records.
We excluded studies based on the following order: (1) definition for infections was not at the individual level, for example, at an ecological level that examines infections aggregated for a region; (2) definition for infections that examined population mixing; (3) infections were not explicitly infections during childhood (e.g., infections during pregnancy); (4) outcomes was not childhood ALL in children aged ⩽19 years; (5) absence of a comparison group; (6) it was a review article; and (7) duplicate publication with the same study population. When more than one publication from a study was available, the most recent version, or the version with the exposure or outcome of interest that was closest to the objectives of this review was included. Studies were not restricted by publication status, and relevant studies in other languages were translated.
Two reviewers (JH and CT) independently evaluated the titles and abstracts of publications identified by the search strategy, and any publication thought to be potentially relevant by either reviewer was retrieved in full. Final inclusion of studies in the systematic review was determined by agreement of both reviewers. Agreement between reviewers was evaluated using the kappa statistic (κ). Strength of agreement was defined as slight (κ=0.00–0.20), fair (κ=0.21–0.40), moderate (κ=0.41–0.60), substantial (κ=0.61–0.80), or almost perfect (κ=0.81–1.00) (Landis and Koch, 1977).
Data extraction and quality assessment
Data extraction was conducted in duplicate (JH and CT) using a standard form, which collected information on: the primary exposure of ‘common infections’, defined as any infection occurring from birth to the diagnosis of ALL; secondary exposures of infection frequency, severity of infections; and study design, region, publication era, and source of controls. In studies that used laboratory investigations for identification of infectious agents, we extracted IgG antibody estimates to represent past infections, and if that was not available, the polymerase chain reactions (PCR) method was extracted to assess for the presence of the agent. We extracted infections occurring in the first year of life or similar time windows in cases with multiple time windows, as we felt this best represented early exposure to infections. We extracted infection frequency levels for common infections, and defined severity based on admission to hospital. The adjusted models that incorporated the most confounders for our primary outcome ALL were extracted. Authors were contacted for further information regarding results that were not presented. Five authors were contacted (Nishi and Miyake, 1989; Schlehofer et al, 1996; Neglia et al, 2000; Rosenbaum et al, 2005; MacArthur et al, 2008), and three responded with no additional information (Nishi and Miyake, 1989; Neglia et al, 2000; Rosenbaum et al, 2005).
Study quality was assessed using the Meta Quality Appraisal Tool (MetaQAT) (Rosella et al, 2015) and the Critical Appraisal Skills Programme (CASP) for case–control (Programme CAS, 2014a), and cohort studies (Programme CAS, 2014b). Two reviewers (JH and CT) assessed each study. For case–control studies, we considered CASP scores of 1–3, 4–6, and 7–9 to be high, moderate, and low-risk of bias, respectively; for cohort studies, we considered CASP scores of 1–4, 5–8, and 9–11 to be high, moderate and low-risk of bias, respectively.
Data synthesis and analysis methods
Our analysis combined data at the study level. Our primary analysis sought to assess exposure to common infections vs no common infections (referent group) on the risk of developing ALL, relying on each study’s definition. The most frequent infection was used when studies did not report a common infection variable. We used the adjusted odds ratio (OR) or rate ratio (RR) to calculate a pooled overall effect, and assumed OR and RR were equivalent due to the rarity of the outcome (Greenland, 1987); ORs or RRs <1 suggest infections are protective against ALL. If a study presented multiple frequency categories, we used the lowest vs the highest category, a method commonly used in meta-analyses (Bae, 2016). The method described by Greenland was used to calculate the variance using the reported 95% confidence intervals (CI) (Greenland, 1987). We calculated a crude OR for studies not reporting one, and to facilitate the calculation we added 0.5 to all cells if one of the four cells reported a zero (Gart and Nam, 1988). In secondary analyses, we used the different exposure levels of infection to compute a regression slope (Greenland and Longnecker, 1992). If an exposure level was defined using a range, we used the midpoint of the range (e.g., 1–3 infections was assigned a frequency of 2), and if the level was ⩾4, we assigned a frequency of 4. For infection severity, a dichotomous variable (yes vs no) was used to determine the relationship with ALL. Post hoc analyses examined the timing of infections in the first year of life compared to infections that occurred after the first year of life, and putative infectious agents was conducted if ⩾3 studies reported the agent.
As we anticipated heterogeneity between the studies, we used an inverse variance weighted average, random-effects model where the Wald-type tests and confidence intervals were estimated under a normal distribution (DerSimonian and Laird, 1986). We investigated potential sources of heterogeneity using subgroup analyses and mixed-effects meta-regression. To examine the association of study-level characteristics and infection effect, we fitted mixed-effects meta-regression models to the natural logarithm of the OR. The natural logarithm of the OR was assumed to have a normal distribution, and a method-of-moments-based estimator to estimate model variables. The mixed-effects model included fixed effects for the covariates, and a random intercept term was specified to model residual heterogeneity not accounted for by the covariates. We corrected for multiple testing using a Bonferroni correction that divides the P-value by the number of tests (Lagakos, 2006). Because of methodological differences (Wiemels, 2012), we tested for interactions to assess the differences between studies that used administrative/medical records, self-reported, and laboratory investigation data (Altman and Bland, 2003). We stratified infections in the first year of life by self-reported data and administrative/medical records data. We explored clinical heterogeneity by conducting a subgroup analysis limiting cases of ALL to B-cell precursor ALL (Wiemels, 2012). We also explored the extent to which region (North America, Europe, Asia, or other), publication era (⩽1999, 2000–2009, ⩾2010), source of controls (general population, general practitioner list, or hospital controls), and risk of bias influenced the magnitude of the average effect estimate in the meta-analysis. Publication bias was assessed by funnel plot and the Egger’s test (Egger et al, 1997; Peters et al, 2008). The meta-analysis was performed using the metafor package in R, version 3.3 (Viechtbauer, 2010).
Results
Titles and abstracts of 9445 records were reviewed and 314 full-text articles were retrieved (Figure 1). There were 39 studies that satisfied the inclusion criteria (Till et al, 1979; van Steensel-Moll et al, 1986; Nishi and Miyake, 1989; Schlehofer et al, 1996; Dockerty et al, 1999; McKinney et al, 1999; Schuz et al, 1999; Neglia et al, 2000; Mackenzie et al, 2001; Petridou et al, 2001; Chan et al, 2002; Perrillat et al, 2002; Salonen et al, 2002; Kerr et al, 2003; Canfield et al, 2004; Jourdan-Da Silva et al, 2004; Ma et al, 2005; Rosenbaum et al, 2005; Surico and Muggeo, 2005; Loutfy et al, 2006; Paltiel et al, 2006; Zaki et al, 2006; Roman et al, 2007; Cardwell et al, 2008; MacArthur et al, 2008; Flores-Lujano et al, 2009; Tesse et al, 2009; Rudant et al, 2010; Zaki and Ashray, 2010; Mahjour et al, 2010; Ahmed et al, 2012; Chang et al, 2012; Vestergaard et al, 2013; Ibrahem et al, 2014; Ajrouche et al, 2015; Lin et al, 2015; Rudant et al, 2015; da Conceicao Nunes et al, 2016; Ateyah et al, 2017), and of those, 38 were included in the meta-analysis. One study did not report infections and the effect estimate could not be calculated (Paltiel et al, 2006). The reviewers had almost perfect agreement on the articles for inclusion (κ=0.85, 95% CI: 0.75, 0.95). Characteristics of the included studies are presented in Table 1. The exposure definitions are presented in Supplementary Table 2. The reviewers had moderate agreement on the judgement of the risk of bias for each study (κ=0.50, 95% CI: 0.28, 0.72). Thirteen studies were judged as being low-risk of bias, 7 as being moderate-risk of bias, and 19 as being high-risk of bias (Supplementary Table 3a and b). We found evidence of publication bias (bias coefficient=1.19, 95% CI: 0.30, 2.08; Supplementary Figure 1).

Study selection flow diagram.
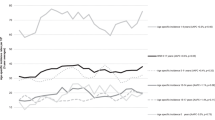
Our analysis included 12 496 children with ALL and 2 356 288 children without ALL. There was no association between infections and ALL, OR=1.10, 95% CI: 0.95, 1.28; P=0.187 (Figure 2). We observed considerable heterogeneity between the studies (I2=76.5%; Q-statistic P<0.001). The trend analysis included 13 studies and we did not find frequency of infections to be associated with ALL (OR=1.00, 95% CI: 0.95, 1.05; P=0.967). In the four studies that assessed the infection severity, the combined average effect of hospitalisations for infections was not associated with ALL (OR=1.22, 95% CI: 0.85, 1.75; P=0.239). Infections that occurred in the first year of life was not associated with ALL (OR=0.99, 95% CI: 0.85, 1.16, P=0.920). Infections that occurred after the first year of life suggested an association with ALL (OR=1.45, 95% CI: 0.71, 2.96, P=0.313), but did not differ compared to infections in the first year of life (interaction effect OR=0.69, 95% CI: 0.32, 1.43, P=0.314) (Supplementary Figure 2). Parvovirus B19 (OR=2.69, 95% CI: 1.16, 6.22, P=0.020) was found to be associated with ALL (Figure 2). No associations were observed for human herpesvirus-6 (OR=0.89, 95% CI: 0.42, 1.87, P=0.752), however Epstein–Barr virus (OR=1.39, 95% CI: 0.83, 2.33, P=0.208), cytomegalovirus (OR=1.95, 95% CI: 0.64, 5.96, P=0.242), influenza (OR=1.97, 95% CI: 0.97, 3.98, P=0.061), and herpes simplex virus (OR=2.04, 95% CI: 0.66, 6.23, P=0.214) showed a strong association to ALL, but lacked precision. Varicella, rubella, mumps, measles, and pertussis were not associated with ALL (Supplementary Figure 3).

Random-effects model examining the association between common infections and risk of childhood acute lymphoblastic leukaemia. CI represents confidence interval. Common infections are reported as a two-class variable, or highest vs lowest in more than two categories. The secondary analysis for frequency of infections is a combined maximum likelihood effect estimate that estimates a trend from summarised dose–response data. The presence of parvovirus B19 was measured as a dichotomous variable, presence of IgG antibodies vs no IgG antibodies for parvovirus B19. All other studies, the reference was no infections.
Subgroup and sensitivity analyses
After applying the Bonferroni correction, the P-value to indicate statistical significance for the additional analyses was <0.005. The data sources for the studies can be found in Table 1. Among the studies that used self-reported data, we found no association between infections and ALL (OR=0.89, 95% CI: 0.79, 1.00, P=0.049; I2=50.5%). Among studies that used administrative/medical record data, we found no association between infections and ALL (OR=1.00, 95% CI: 0.61, 1.63, P=0.994; I2=90.8%). Among studies that used laboratory data, we found infections to be associated with ALL (OR=2.42, 95% CI: 1.54, 3.82, P<0.001, I2=54.2%). The interaction effect showed no difference between self-reported and administrative/medical records data sources (OR=0.89, 95% CI: 0.54, 1.48, P=0.656). Infections identified through laboratory data increased the risk of ALL compared to infections captured through self-reported data (interaction effect OR=2.73, 95% CI: 1.71, 4.36, P<0.001), but not administrative/medical records data sources (interaction effect OR=2.43, 95% CI: 1.24, 4.75, P=0.009). Among studies that used self-reported data, every additional infection reduced the odds of ALL by 4% (OR=0.96, 95% CI: 0.94, 0.98; P<0.001), whereas among studies that used administrative/medical records data, every additional infection increased the odds of ALL by 11% (OR=1.11, 95% CI: 1.07, 1.15; P<0.001). We found self-reported and administrative/medical records data sources qualitatively differed in the frequency of infections (interaction effect OR=0.86, 95% CI: 0.83, 0.90, P<0.001). Severity of infections remained unchanged in studies with self-reported data (OR=1.51, 95% CI: 0.86, 2.65; P=0.158; I2=70.2%). Among self-reported studies, infections in the first year of life suggested a protective effect against ALL (OR=0.88, 95% CI: 0.80, 0.98, P=0.017). No association was found between infections in the first year of life and ALL among administrative/medical records data (OR=0.93, 95% CI: 0.55, 1.56, P=0.775), and did not differ from self-reported studies (interaction effect OR=0.95, 95% CI: 0.56, 1.62, P=0.862).
The results from our primary analysis remained unchanged when we restricted the analysis to B-cell precursor ALL or B-cell common ALL (OR=0.87, 95% CI: 0.77, 0.98, P=0.022). Meta-regression models that assessed study level characteristics included data source, region, publication era, source of controls, and risk of bias. Data source and region accounted for the largest proportion of heterogeneity between the studies (R2=47.2%, see Supplementary Table 4). Stratification by risk of bias indicated studies of low-risk of bias showed similar results to our main analysis (OR=0.92, 95% CI: 0.76, 1.10, P=0.349), whereas studies of moderate-to-high-risk of bias suggested infections increased the risk of ALL (OR=1.45, 95% CI: 1.12–1.86, P=0.005). Compared to studies of moderate-to-high-risk of bias, studies of low-risk of bias were more likely to suggest infections were protective against ALL (OR=0.63, 95% CI: 0.46, 0.87, P=0.004).
Discussion
In this systematic review of 39 studies, we found no association between any common infections, frequency, severity of infections, and timing of infections and childhood ALL. We did however, find a qualitative difference in our subgroup analyses; infections increased the odds of developing ALL by 2.4-fold in studies with laboratory investigations. Further, infections identified through laboratory investigations increased the odds of ALL by 2.7-fold and 2.4-fold compared to infections identified through self-reported and administrative/medical records data, respectively. Among studies that used self-reported data, we found each additional infection reduced the odds of ALL by 4%, and this differed significantly from studies that used administrative/medical records data that suggested each additional infection increased the odds of ALL by 11%. The heterogeneity between the studies remained a challenge and could partly be explained by differences in the data sources.
We failed to demonstrate an association in our primary analysis, but found associations in our secondary and subgroup analyses by data source. There are three plausible explanations for the observed findings. First, the apparent results may be a chance finding from multiple testing. Second, the ascertainment of infections from parental recall has been shown to under-report childhood infections and may be inaccurate in both the timing and occurrence of infections, compared to medical records (McKinney et al, 1991; Simpson et al, 2007). Despite these potential issues, studies that confirmed the self-reported infections with medical records for accuracy and completeness still found an inverse association (Dockerty et al, 1999; Ajrouche et al, 2015). Although studies that used medical records were void of recall bias, they were often unable to include other important confounders, such as ethnicity, parental occupation, maternal age, birth weight, and parity (Dockerty et al, 2001; Hjalgrim et al, 2004; Ma et al, 2005; Lim et al, 2014). Finally, the findings from the laboratory studies must be interpreted with caution due to the study quality, and smaller sample sizes and larger effect sizes as shown by the asymmetry of the funnel plot.
The mutational mechanisms of ALL point to three potential pathways: (1) anomalies in lineage-specific factors (ETV6-RUNX1, IKZF1, and PAX5); (2) flaws in receptor protein tyrosine kinases and their down-stream pathways; and (3) epigenetic modifiers (Whitehead et al, 2016). Recent developments in genome and mouse model studies may change our initial understanding of the aetiology of ALL as new studies have generated new hypotheses with respect to identifying potential infectious candidates (Martin-Lorenzo et al, 2015; Swaminathan et al, 2015). The presence of parvovirus B19 IgG antibodies is associated with the presence of ETV6-RUNX1 (Ibrahem et al, 2014), and is associated with certain class II HLA alleles that are risk factors for the development of childhood ALL. Furthermore, parvovirus B19 has certain characteristics similar to other oncoviruses, that is, its DNA genome persists indefinitely in human tissues following acute infection, causing mild or no disease, and upregulates pro-inflammatory cytokines associated with ALL onset (Kerr and Mattey, 2015). The results from the small laboratory studies will require confirmation in larger population studies. Since half of 15-year-old adolescents have specific antiparvovirus B19 antibodies (Young and Brown, 2004), the measurement of the clinical syndromes caused by parvovirus B19 may be preferred to assess manifestations of the pathogen. Parvovirus B19 infection may provide only a subset of an oncogenic hit in a multistep carcinogenesis process.
The qualitative differences in our findings support the hypothesis of an alternative pathway for ALL development. Recent qualitative reviews have attempted to explain the positive association between infections and ALL and suggested studies that used medical records or administrative data may be capturing children with an earlier than expected altered immune system. These children may respond differently to infections, have a greater propensity to seek medical care when infections are contracted, and/or have a stronger immune response (Wiemels, 2012; Whitehead et al, 2016). The sensitivity to infections may be due to a lack of immunomodulation from lower levels of anti-inflammatory cytokine interleukin-10 in newborns who later go on to develop ALL (Chang et al, 2011).
As in previous reviews, there continues to be substantial heterogeneity among the studies; however, our review focuses on specific objectives and highlights the recent developments of the field (McNally and Eden, 2004; Buffler et al, 2005; Greaves, 2006; Ma et al, 2009; Maia Rda and Wunsch Filho, 2013). There are several limitations of this study. The heterogeneity between the studies in the definition of infections, the time period to observe the infections, and the evidence of publication bias was a challenge. We decided to use any common infection as our main exposure variable in the primary analysis because we felt it to be the most appropriate measure that reflects the hypotheses from Kinlen and Greaves (Kinlen, 1988; Greaves, 2006). The heterogeneity likely stems from the unknown aetiology of ALL, and one that requires further research. The limitation with laboratory investigation studies is the inability to disentangle temporality. The presence of the infectious agent was assessed after a diagnosis of ALL was made and it is unknown if the agent was present before or after the onset of ALL. It is unclear whether the infection occurred before the onset of ALL, or if the potentially reduced immune function because of ALL contributed to the contraction of specific infections. Further, the laboratory studies were appraised as high-risk of bias, often small, and may not be generalisable. Despite the differences in the risk of bias amongst the included studies, our conclusions were unchanged after we stratified the analysis to the 13 studies with a low-risk of bias. Another limitation was the quality of reporting in the studies included in the review. Most studies clearly reported their findings, but studies published earlier tended to have incomplete reporting.
Costs and feasibility are the usual barriers to establishing new large pregnancy and birth cohorts (Riley and Duncan, 2016), research groups have instead combined existing cohorts to study childhood cancers (Brown et al, 2007; Metayer et al, 2013) and other diseases (Larsen et al, 2013). The increased power may help to identify high risk or vulnerable, and understudied populations. The next step should focus on the measurement of infections and infectious exposures. The use of linked administrative data provides a large population for study with accurate information on the timing of physician diagnosed infections, frequency, and severity of infections as answers to these questions remain elusive. Enhancing the administrative data with surveys to obtain other infectious exposures such as day-care attendance, breastfeeding, or by applying emerging technologies that detect and quantify the pathogen burden with greater speed, accuracy, and simplicity (Caliendo et al, 2013) in a subset sample would improve the accuracy and strengthen the measurement of infections. Day-care attendance has been found to increase the risk of exposure to infections, and has been used as a proxy for infections. A meta-analysis found day-care attendance reduced the risk of childhood ALL (Urayama et al, 2010). Breastfeeding has been found to reduce the risk of ALL through its immunologically active components, antibodies, and other elements that influence the development of the infant’s immune system (Kwan et al, 2004; Martin et al, 2005; Amitay and Keinan-Boker, 2015). The challenge will be to disentangle the mechanistic pathways of the infectious aetiology hypothesis by combining different measurements of infectious exposures to determine the total, direct, and indirect effect of infections on the risk of developing childhood ALL.
An infectious aetiology of ALL is suggestive in our study; however, the challenges in measuring infections must be addressed. Parvovirus B19 as a putative causal infectious agent for childhood ALL needs to be tested in larger cohorts and the rather substantial point estimates from influenza, cytomegalovirus, and herpes simplex virus warrant a follow-up in larger studies. Whether children with ALL have a dysregulated immune function present at birth requires further investigation. Only one study conducted an exploratory assessment on a key aspect of Greaves’ hypothesis, the timing of the infections in early life (Crouch et al, 2012). Our future research aims to provide further insight on the timing of infections and the risk of developing childhood ALL. The use of administrative data or medical records with linked laboratory data would overcome the challenges facing studies that used self-reported and laboratory investigation data, and would be ideal to evaluate the association between childhood ALL and the timing and frequency of infections. The review has highlighted knowledge gaps surrounding the relationship between childhood ALL and severity of infections. The causal association of infections will need to be tested in conjunction with other identified risk factors to quantify the direct and indirect interaction and mediated effect of infections on ALL risk. These will be critical research questions in discovering the causes of childhood ALL and will be the foundation for future studies that can combine epidemiologic, genetic, and environmental factors.
Change history
09 January 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ahmed HG, Osman SI, Ashankyty IM (2012) Incidence of Epstein–Barr virus in pediatric leukaemia in the Sudan. Clin Lymphoma Myeloma Leuk 12 (2): 127–131.
Ajrouche R, Rudant J, Orsi L, Petit A, Baruchel A, Lambilliotte A, Gambart M, Michel G, Bertrand Y, Ducassou S, Gandemer V, Paillard C, Saumet L, Blin N, Hemon D, Clavel J (2015) Childhood acute lymphoblastic leukaemia and indicators of early immune stimulation: The Estelle study (SFCE). Br J Cancer 112: 1017–1026.
Alexander FE, Boyle P, Carli PM, Coebergh JW, Draper GJ, Ekbom A, Levi F, McKinney PA, McWhirter W, Michaelis J, Peris-Bonet R, Petridou E, Pompe-Kirn V, Plisko I, Pukkala E, Rahu M, Storm H, Terracini B, Vatten L, Wray N (1998) Spatial clustering of childhood leukaemia: summary results from the EUROCLUS project. Br J Cancer 77 (5): 818–824.
Altman DG, Bland JM (2003) Interaction revisited: the difference between two estimates. BMJ 326 (7382): 219.
Amitay EL, Keinan-Boker L (2015) Breastfeeding and childhood leukaemia incidence: a meta-analysis and systematic review. JAMA Pediatr 169 (6): e151025.
Ateyah ME, Hashem ME, Abdelsalam M (2017) Epstein–Barr virus and regulatory T cells in Egyptian paediatric patients with acute B lymphoblastic leukaemia. J Clin Pathol 70 (2): 120–125.
Bae J-M (2016) Comparison of methods of extracting information for meta-analysis of observational studies in nutritional epidemiology. Epidemiol Health 38: e2016003.
Brown RC, Dwyer T, Kasten C, Krotoski D, Li Z, Linet MS, Olsen J, Scheidt P, Winn DM (2007) Cohort Profile: The International Childhood Cancer Cohort Consortium (I4C). Int J Epidemiol 36 (4): 724–730.
Buffler PA, Kwan ML, Reynolds P, Urayama KY (2005) Environmental and genetic risk factors for childhood leukaemia: appraising the evidence. Cancer Investig 23 (1): 60–75.
Caliendo AM, Gilbert DN, Ginocchio CC, Hanson KE, May L, Quinn TC, Tenover FC, Alland D, Blaschke AJ, Bonomo RA, Carroll KC, Ferraro MJ, Hirschhorn LR, Joseph WP, Karchmer T, MacIntyre AT, Reller LB, Jackson AF Infectious Diseases Society of America (2013) Better tests, better care: improved diagnostics for infectious diseases. Clin Infect Dis 57 (Suppl 3): S139–S170.
Canfield KN, Spector LG, Robison LL, Lazovich D, Roesler M, Olshan AF, Smith FO, Heerema NA, Barnard DR, Blair CK, Ross JA (2004) Childhood and maternal infections and risk of acute leukaemia in children with Down syndrome: a report from the Children’s Oncology Group. Br J Cancer 91 (11): 1866–1872.
Cardwell CR, McKinney PA, Patterson CC, Murray LJ (2008) Infections in early life and childhood leukaemia risk: a UK case–control study of general practitioner records. Br J Cancer 99 (9): 1529–1533.
Chan LC, Lam TH, Lau YL, Li CK, Yuen HL, Lee CW, Ha SY, Yuen PMP, Leung NK, Patheal SL, Greaves MF, Alexander FE (2002) Is the timing of exposure to infection a major determinant of acute lymphoblastic leukaemia in Hong Kong? Paediatr Perinat Epidemiol 16 (2): 154–165.
Chang JS, Tsai CR, Tsai YW, Wiemels JL (2012) Medically diagnosed infections and risk of childhood leukaemia: a population-based case–control study. Int J Epidemiol 41 (4): 1050–1059.
Chang JS, Zhou M, Buffler PA, Chokkalingam AP, Metayer C, Wiemels JL (2011) Profound deficit of IL10 at birth in children who develop childhood acute lymphoblastic leukaemia. Cancer Epidemiol Biomarkers Prev 20 (8): 1736–1740.
Crouch S, Lightfoot T, Simpson J, Smith A, Ansell P, Roman E (2012) Infectious illness in children subsequently diagnosed with acute lymphoblastic leukaemia: modeling the trends from birth to diagnosis. Am J Epidemiol 176 (5): 402–408.
da Conceicao Nunes J, de Araujo GV, Viana MT, Sarinho ES (2016) Association of atopic diseases and parvovirus B19 with acute lymphoblastic leukaemia in childhood and adolescence in the northeast of Brazil. Int J Clin Oncol 21 (5): 989–995.
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7 (3): 177–188.
Dockerty JD, Draper G, Vincent T, Rowan SD, Bunch KJ (2001) Case–control study of parental age, parity and socioeconomic level in relation to childhood cancers. Int J Epidemiol 30 (6): 1428–1437.
Dockerty JD, Skegg DC, Elwood JM, Herbison GP, Becroft DM, Lewis ME (1999) Infections, vaccinations, and the risk of childhood leukaemia. Br J Cancer 80 (9): 1483–1489.
Egger M, Smith GD, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315 (7109): 629–634.
Flores-Lujano J, Perez-Saldivar ML, Fuentes-Panana EM, Gorodezky C, Bernaldez-Rios R, Del Campo-Martinez MA, Martinez-Avalos A, Medina-Sanson A, Paredes-Aguilera R, De Diego-Flores Chapa J, Bolea-Murga V, Rodriguez-Zepeda MC, Rivera-Luna R, Palomo-Colli MA, Romero-Guzman L, Perez-Vera P, Alvarado-Ibarra M, Salamanca-Gomez F, Fajardo-Gutierrez A, Mejia-Arangure JM (2009) Breastfeeding and early infection in the aetiology of childhood leukaemia in down syndrome. Br J Cancer 101 (5): 860–864.
Gart JJ, Nam J (1988) Approximate interval estimation of the ratio of binomial parameters: a review and corrections for skewness. Biometrics 44 (2): 323–338.
Greaves M (2006) Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 6 (3): 193–203.
Greaves MF (1997) Aetiology of acute leukaemia. Lancet 349 (9048): 344–349.
Greaves MF, Maia AT, Wiemels JL, Ford AM (2003) leukaemia in twins: lessons in natural history. Blood 102 (7): 2321–2333.
Greenland S (1987) Quantitative methods in the review of epidemiologic literature. Epidemiol Rev 9: 1–30.
Greenland S, Longnecker MP (1992) Methods for trend estimation from summarised dose–response data, with applications to meta-analysis. Am J Epidemiol 135 (11): 1301–1309.
Hjalgrim LL, Rostgaard K, Hjalgrim H, Westergaard T, Thomassen H, Forestier E, Gustafsson G, Kristinsson J, Melbye M, Schmiegelow K (2004) Birth weight and risk for childhood leukaemia in Denmark, Sweden, Norway, and Iceland. JNCI 96 (20): 1549–1556.
Ibrahem WN, Hasony HJ, Hassan JG (2014) Human parvovirus B19 in childhood acute lymphoblastic leukaemia in Basrah. JPMA 64 (1): 9–12.
Jourdan-Da Silva N, Perel Y, Mechinaud F, Plouvier E, Gandemer V, Lutz P, Vannier JP, Lamagnere JL, Margueritte G, Boutard P, Robert A, Armari C, Munzer M, Millot F, De Lumley L, Berthou C, Rialland X, Pautard B, Hemon D, Clavel J (2004) Infectious diseases in the first year of life, perinatal characteristics and childhood acute leukaemia. Br J Cancer 90 (1): 139–145.
Kerr JR, Barah F, Cunniffe VS, Smith J, Vallely PJ, Will AM, Wynn RF, Stevens RF, Taylor GM, Cleator GM, Eden OB (2003) Association of acute parvovirus B19 infection with new onset of acute lymphoblastic and myeloblastic leukaemia. J Clin Pathol 56 (11): 873–875.
Kerr JR, Mattey DL (2015) The role of parvovirus B19 and the immune response in the pathogenesis of acute leukaemia. Rev Med Virol 25 (3): 133–155.
Kinlen L (1988) Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet 332 (8624): 1323–1327.
Kinlen L (2006) Childhood leukaemia and ordnance factories in west Cumbria during the Second World War. Br J Cancer 95 (1): 102–106.
Kinlen L, Doll R (2004) Population mixing and childhood leukaemia: Fallon and other US clusters. Br J Cancer 91 (1): 1–3.
Kinlen LJ (2012) An examination, with a meta-analysis, of studies of childhood leukaemia in relation to population mixing. Br J Cancer 107 (7): 1163–1168.
Kwan ML, Buffler PA, Abrams B, Kiley VA (2004) Breastfeeding and the risk of childhood leukaemia: a meta-analysis. Public Health Rep 119 (6): 521–535.
Lagakos SW (2006) The challenge of subgroup analyses—reporting without distorting. N Engl J Med 354 (16): 1667–1669.
Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33 (1): 159–174.
Larsen PS, Kamper-Jorgensen M, Adamson A, Barros H, Bonde JP, Brescianini S, Brophy S, Casas M, Charles MA, Devereux G, Eggesbo M, Fantini MP, Frey U, Gehring U, Grazuleviciene R, Henriksen TB, Hertz-Picciotto I, Heude B, Hryhorczuk DO, Inskip H, Jaddoe VW, Lawlor DA, Ludvigsson J, Kelleher C, Kiess W, Koletzko B, Kuehni CE, Kull I, Kyhl HB, Magnus P, Momas I, Murray D, Pekkanen J, Polanska K, Porta D, Poulsen G, Richiardi L, Roeleveld N, Skovgaard AM, Sram RJ, Strandberg-Larsen K, Thijs C, Van Eijsden M, Wright J, Vrijheid M, Andersen AM (2013) Pregnancy and birth cohort resources in europe: a large opportunity for aetiological child health research. Paediatr Perinat Epidemiol 27 (4): 393–414.
Lim JYS, Bhatia S, Robison LL, Yang JJ (2014) Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukaemia. Cancer 120 (7): 955–962.
Lin JN, Lin CL, Lin MC, Lai CH, Lin HH, Yang CH, Sung FC, Kao CH (2015) Risk of leukaemia in children infected with enterovirus: a nationwide, retrospective, population-based, Taiwanese-registry, cohort study. Lancet Oncol 16 (13): 1335–1343.
Loutfy SA, Alam El-Din HM, Ibrahim MF, Hafez MM (2006) Seroprevalence of herpes simplex virus types 1 and 2, Epstein–Barr virus, and cytomegalovirus in children with acute lymphoblastic leukaemia in Egypt. Saudi Med J 27 (8): 1139–1145.
Ma X, Buffler PA, Wiemels JL, Selvin S, Metayer C, Loh M, Does MB, Wiencke JK (2005) Ethnic difference in daycare attendance, early infections, and risk of childhood acute lymphoblastic leukaemia. Cancer Epidemiol Biomarkers Prev 14 (8): 1928–1934.
Ma XM, Urayama K, Chang J, Wiemels JL, Buffler PA (2009) Infection and pediatric acute lymphoblastic leukaemia. Blood Cells Mol Dis 42 (2): 117–120.
MacArthur AC, McBride ML, Spinelli JJ, Tamaro S, Gallagher RP, Theriault GP (2008) Risk of childhood leukaemia associated with vaccination, infection, and medication use in childhood: the Cross-Canada Childhood leukaemia Study. Am J Epidemiol 167 (5): 598–606.
Mackenzie J, Gallagher A, Clayton RA, Perry J, Eden OB, Ford AM, Greaves MF, Jarrett RF (2001) Screening for herpesvirus genomes in common acute lymphoblastic leukaemia. leukaemia 15 (3): 415–421.
Mahjour SB, Ghaffarpasand F, Fattahi MJ, Ghaderi A, Fotouhi Ghiam A, Karimi M (2010) Seroprevalence of human herpes simplex, hepatitis B and Epstein–Barr viruses in children with acute lymphoblastic leukaemia in southern iran. Pathol Oncol Res 16 (4): 579–582.
Maia Rda R, Wunsch Filho V (2013) Infection and childhood leukaemia: review of evidence. Revista de Saude Publica 47 (6): 1172–1185.
Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, Ginzel S, Thiele R, Constantinescu SN, Bartenhagen C, Dugas M, Gombert M, Schafer D, Blanco O, Mayado A, Orfao A, Alonso-Lopez D, De Las Rivas J, Cobaleda C, Garcia-Cenador MB, Garcia-Criado FJ, Sanchez-Garcia I, Borkhardt A (2015) Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukaemia as a result of Pax5-inherited susceptibility. Cancer Discov 5 (12): 1328–1343.
Martin RM, Gunnell D, Owen CG, Smith GD (2005) Breast-feeding and childhood cancer: a systematic review with metaanalysis. Int J Cancer 117 (6): 1020–1031.
McKinney PA, Alexander FE, Nicholson C, Cartwright RA, Carrette J (1991) Mothers’ reports of childhood vaccinations and infections and their concordance with general practitioner records. J Public Health Med 13 (1): 13–22.
McKinney PA, Juszczak E, Findlay E, Smith K, Thomson CS (1999) Pre- and perinatal risk factors for childhood leukaemia and other malignancies: a Scottish case control study. Br J Cancer 80 (11): 1844–1851.
McNally RJ, Eden TO (2004) An infectious aetiology for childhood acute leukaemia: a review of the evidence. Br J Haematol 127 (3): 243–263.
Metayer C, Milne E, Clavel J, Infante-Rivard C, Petridou E, Taylor M, Schuz J, Spector LG, Dockerty JD, Magnani C, Pombo-de-Oliveira MS, Sinnett D, Murphy M, Roman E, Monge P, Ezzat S, Mueller BA, Scheurer ME, Armstrong BK, Birch J, Kaatsch P, Koifman S, Lightfoot T, Bhatti P, Bondy ML, Rudant J, O'Neill K, Miligi L, Dessypris N, Kang AY, Buffler PA (2013) The Childhood leukaemia International Consortium. Cancer Epidemiol 37 (3): 336–347.
Neglia JP, Linet MS, Shu XO, Severson RK, Potter JD, Mertens AC, Wen W, Kersey JH, Robison LL (2000) Patterns of infection and day care utilization and risk of childhood acute lymphoblastic leukaemia. Br J Cancer 82 (1): 234–240.
Nishi M, Miyake H (1989) A case–control study of non-T cell acute lymphoblastic leukaemia of children in Hokkaido, Japan. J Epidemiol Community Health 43 (4): 352–355.
Paltiel O, Laniado DE, Yanetz R, Deutsch L, Calderon-Margalit R, Harlap S, Friedlander Y (2006) The risk of cancer following Hospitalisation for infection in infancy: a Population-Based cohort study. Cancer Epidemiol Biomarkers Prev 15 (10): 1964–1968.
Perrillat F, Clavel J, Auclerc MF, Baruchel A, Leverger G, Nelken B, Philippe N, Schaison G, Sommelet D, Vilmer E, Hemon D (2002) Day-care, early common infections and childhood acute leukaemia: a multicentre French case–control study. Br J Cancer 86 (7): 1064–1069.
Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L (2008) Contour-enhanced meta-analysis funnel plots help distinguish publication bias from other causes of asymmetry. J Clin Epidemiol 61 (10): 991–996.
Petridou E, Dalamaga M, Mentis A, Skalkidou A, Moustaki M, Karpathios T, Trichopoulos D (2001) Evidence on the infectious etiology of childhood leukaemia: the role of low herd immunity (Greece). Cancer Causes Control 12 (7): 645–652.
Programme CAS (2014a) CASP Case Control Checklist. CASP: Oxford.
Programme CAS (2014b) CASP Cohort Study Checklist. CASP: Oxford.
Riley AW, Duncan GJ (2016) Completing a national birth cohort in the united states. JAMA Pediatr 170 (9): 829–830.
Roman E, Simpson J, Ansell P, Kinsey S, Mitchell C, McKinney P, Birch J, Greaves M, Eden T (2007) Childhood acute lymphoblastic leukaemia and infections in the first year of life: a report from the United Kingdom Childhood Cancer Study. Am J Epidemiol 165 (5): 496–504.
Rosella L, Bowman C, Pach B, Morgan S, Fitzpatrick T, Goel V (2015) The development and validation of a meta-tool for quality appraisal of public health evidence: Meta Quality Appraisal Tool (MetaQAT). Public Health 136: 57–65.
Rosenbaum PF, Buck GM, Brecher ML (2005) Allergy and infectious disease histories and the risk of childhood acute lymphoblastic leukaemia. Paediatr Perinat Epidemiol 19 (2): 152–164.
Rudant J, Lightfoot T, Urayama KY, Petridou E, Dockerty JD, Magnani C, Milne E, Spector LG, Ashton LJ, Dessypris N, Kang AY, Miller M, Rondelli R, Simpson J, Stiakaki E, Orsi L, Roman E, Metayer C, Infante-Rivard C, Clavel J (2015) Childhood acute lymphoblastic leukaemia and indicators of early immune stimulation: a Childhood leukaemia International Consortium study. Am J Epidemiol 181 (8): 549–562.
Rudant J, Orsi L, Menegaux F, Petit A, Baruchel A, Bertrand Y, Lambilliotte A, Robert A, Michel G, Margueritte G, Tandonnet J, Mechinaud F, Bordigoni P, Hemon D, Clavel J (2010) Childhood acute leukaemia, early common infections, and allergy: The ESCALE Study. Am J Epidemiol 172 (9): 1015–1027.
Salonen MJH, Siimes MA, Salonen EM, Vaheri A, Koskiniemi M (2002) Antibody status to HHV-6 in children with leukaemia. leukaemia 16 (4): 716–719.
Schlehofer B, Blettner M, Geletneky K, Haaf HG, Kaatsch P, Michaelis J, MuellerLantzsch N, Niehoff D, Winkelspecht B, Wahrendorf J, Schlehofer JR (1996) Sero-epidemiological analysis of the risk of virus infections for childhood leukaemia. Int J Cancer 65 (5): 584–590.
Schuz J, Kaletsch U, Meinert R, Kaatsch P, Michaelis J (1999) Association of childhood leukaemia with factors related to the immune system. Br J Cancer 80 (3-4): 585–590.
Simpson J, Smith A, Ansell P, Roman E (2007) Childhood leukaemia and infectious exposure: a report from the United Kingdom Childhood Cancer Study (UKCCS). Eur J Cancer 43 (16): 2396–2403.
Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D, Moher D, Becker BJ, Sipe TA, Thacker SB (2000) Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis of Observational Studies in Epidemiology (MOOSE) group. JAMA 283 (15): 2008–2012.
Surico G, Muggeo P (2005) Epstein–Barr virus infection at the onset of acute lymphoblastic leukaemia in children. Int Cancer J Austral Asia 4 (1): 19–24.
Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke N, Mooster J, Geng H, Schwarz K, Kogan SC, Casellas R, Schatz DG, Lieber MR, Greaves MF, Muschen M (2015) Mechanisms of clonal evolution in childhood acute lymphoblastic leukaemia. Nat Immunol 16 (7): 766–774.
Tesse R, Santoro N, Giordano P, Cardinale F, De Mattia D, Armenio L (2009) Association between DEFB1 gene haplotype and herpes viruses seroprevalence in children with acute lymphoblastic leukaemia. Pediatr Hematol Oncol 26 (8): 573–582.
Till M, Rapson N, Smith PG (1979) Family studies in acute leukaemia in childhood: a possible association with autoimmune disease. Br J Cancer 40 (1): 62–71.
Urayama KY, Buffler PA, Gallagher ER, Ayoob JM, Ma X (2010) A meta-analysis of the association between day-care attendance and childhood acute lymphoblastic leukaemia. Int J Epidemiol 39 (3): 718–732.
van Steensel-Moll HA, Valkenburg HA, van Zanen GE (1986) Childhood leukaemia and infectious diseases in the first year of life: a Register-Based Case–Control Study. Am J Epidemiol 124 (4): 590–594.
Vestergaard TR, Rostgaard K, Grau K, Schmiegelow K, Hjalgrim H (2013) Hospitalisation for infection prior to diagnosis of acute lymphoblastic leukaemia in children. Pediatric Blood Cancer 60 (3): 428–432.
Viechtbauer W (2010) Conducting meta-analyses in R with the metafor Package. J Stat Softw 36 (3): 48.
Whitehead TP, Metayer C, Wiemels JL, Singer AW, Miller MD (2016) Childhood leukaemia and primary prevention. Curr Prob Pediatric Adolesc Health care 46 (10): 317–352.
Wiemels J (2012) Perspectives on the causes of childhood leukaemia. Chemico-Biol Interact 196 (3): 59–67.
Young NS, Brown KE (2004) Parvovirus B19. N Engl J Med 350 (6): 586–597.
Zaki ME, Hassan SA, Seleim T, Lateef RA (2006) Parvovirus B19 infection in children with a variety of hematological disorders. Hematology 11 (4): 261–266.
Zaki MES, Ashray RE (2010) Clinical and hematological study for Parvovirus b19 infection in children with acute leukaemia. Int J Lab Hematol 32 (2): 159–166.
Acknowledgements
We would like to thank Thomasin Adams-Webber for her assistance with the citation searches of the electronic databases. Ms Adams-Webber is a trained librarian with expertise in conducting searches for systematic reviews in clinical research. The study forms part of Jeremiah Hwee’s doctoral dissertation. This work was supported by the Canadian Institutes of Health Research Frederick Banting and Charles Best Canada Graduate Scholarship to Honour Nelson Mandela—Doctoral award (JH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hwee, J., Tait, C., Sung, L. et al. A systematic review and meta-analysis of the association between childhood infections and the risk of childhood acute lymphoblastic leukaemia. Br J Cancer 118, 127–137 (2018). https://doi.org/10.1038/bjc.2017.360
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.360

{kind=link}
{kind=link}
{kind=link}