
Abstract
Background:
Patients with BRAFV600E-mutated metastatic colorectal cancer (mCRC) have a poorer prognosis as well as resistance to anti-EGFR antibodies. However, it is unclear whether BRAF mutations other than BRAFV600E (BRAFnon-V600E mutations) contribute to anti-EGFR antibody resistance.
Methods:
This study was composed of exploratory and inference cohorts. Candidate biomarkers identified by whole exome sequencing from super-responders and nonresponders in the exploratory cohort were validated by targeted resequencing for patients who received anti-EGFR antibody in the inference cohort.
Results:
In the exploratory cohort, 31 candidate biomarkers, including KRAS/NRAS/BRAF mutations, were identified. Targeted resequencing of 150 patients in the inference cohort revealed 40 patients with RAS (26.7%), 9 patients with BRAFV600E (6.0%), and 7 patients with BRAFnon-V600E mutations (4.7%), respectively. The response rates in RAS, BRAFV600E, and BRAFnon-V600E were lower than those in RAS/BRAF wild-type (2.5%, 0%, and 0% vs 31.9%). The median PFS in BRAFnon-V600E mutations was 2.4 months, similar to that in RAS or BRAFV600E mutations (2.1 and 1.6 months) but significantly worse than that in wild-type RAS/BRAF (5.9 months).
Conclusions:
Although BRAFnon-V600E mutations identified were a rare and unestablished molecular subtype, certain BRAFnon-V600E mutations might contribute to a lesser benefit of anti-EGFR monoclonal antibody treatment.
Similar content being viewed by others

Main
KRAS exon 2 mutations were the first validated predictive biomarker for primary resistance to anti-epidermal growth factor receptor (EGFR) monoclonal antibodies (cetuximab and panitumumab) in patients with metastatic colorectal cancer (mCRC) (Amado et al, 2008; Van Cutsem et al, 2009). Recently, minor RAS (KRAS exons 3 and 4 or NRAS exons 2, 3, and 4) mutations observed in ∼15–20% of mCRC with wild-type KRAS exon 2 cases have been validated as negative predictive biomarkers for anti-EGFR antibody treatment (Douillard et al, 2013). Therefore, expanded RAS (KRAS and NRAS) testing before administration of anti-EGFR antibody treatment has become essential to maximise the therapeutic benefit in patients with mCRC (Taniguchi et al, 2015; Van Cutsem et al, 2015; Yoshino et al, 2015) However, the response rate (RR) to anti-EGFR monotherapy remains low, at ∼20% in later line, in patients with mCRC having wild-type RAS, indicating additional biomarkers beyond expanded RAS are needed (Peeters et al, 2013).
BRAF is a serine-threonine kinase, located downstream of EGFR in the Ras/Raf/mitogen-activated protein kinase (MAPK) pathway (Mercer and Pritchard, 2003; Roskoski, 2010). The hotspot of BRAF mutations in CRC is substitution from valine to glutamic acid at codon 600 (V600E), located in exon 15, leading to 130- to 700-fold increased BRAF kinase activity compared with that of wild-type BRAF; these mutations are reported in ∼5–12% of cases (Davies et al, 2002; Andreadi et al, 2012). In addition, BRAFV600E mutations are more frequently observed in tumours in the right-sided colon than in tumours in the left-sided colon and rectum, and are prognostic biomarkers in CRC and could be potential predictive biomarker for anti-EGFR antibody treatment in pretreated mCRC (Kawazoe et al, 2015).
Recent clinical studies have shown that the primary location of the tumour may be associated with the therapeutic effects of anti-EGFR antibody treatment. Tumours in the right-sided colon showed worse outcomes than those in the left-sided colon and rectum in patients with mCRC with wild-type RAS, suggesting that genetic alterations other than BRAF V600E could be responsible for the poor prognosis of right-sided tumours (von Einem et al, 2014; Brulé et al, 2015).
In contrast, few reports have described mCRC with BRAF mutations other than BRAFV600E (BRAFnon-V600E mutations), for which the incidence ranges from 1.6% to 5.1% (Shen et al, 2013; Ciardiello et al, 2014; Cremolini et al, 2015). BRAFnon-V600E mutations can be classified on the basis of kinase activity as either high activity, intermediate activity or impaired activity (130- to 700-fold; high activity mutants, 1.3- to 64-fold; intermediate activity mutants and 30–80%, respectively) (Wan et al, 2004). Furthermore, BRAF mutation with impaired kinase activity also enhances MAPK kinase (MEK) phosphorylation by heterodimerising with wild-type CRAF (Haling et al, 2014). However, little is known regarding the clinicopathological features and anti-EGFR antibody sensitivity of BRAFnon-V600E-mutated mCRC.
Here we reported the clinicopathological features of BRAFnon-V600E-mutated mCRC and the clinical significance of these mutations with regard to the therapeutic effects of anti-EGFR antibody treatment in pretreated mCRC.
Materials and methods
Study design
The Biomarker Research for Anti-EGFR Monoclonal Antibodies by Comprehensive Cancer Genomics (BREAC) study was a multicentre, translational research study aiming to investigate novel predictive biomarkers of anti-EGFR antibody treatment in patients with mCRC harbouring wild-type or unknown KRAS exon 2 (details in Supplementary Protocol). We had the following study design; patients were divided into two independent cohorts named ‘exploratory’ and ‘inference’ cohorts according to the duration of anti-EGFR antibody treatment. The exploratory cohort included subjects who were considered as ‘super-responders’ or ‘super-nonresponders’ among the entire mCRC cohort (403 patients) who received cetuximab including treatment as salvage line between September 2008 and May 2010 at seven major institutions in Japan. We put a strong assumption that associations between relatively minor gene mutations and patient prognosis become more remarkable in the ‘super-responders’ plus ‘nonresponders’ cohort than associations observed in the entire cohort, leading to a power increase in statistical tests (Supplementary Figure S1). The possible mutations founded in the exploratory cohort were then evaluated by targeted resequencing of the patients in the inference cohort who were treated by anti-EGFR antibody during the different period from the exploratory cohort.
Study conduct
In the inference cohort, patients with mCRC were consecutively enroled between June 2010 and November 2011 from seven institutions to validate the associations of candidate biomarkers identified in the exploratory cohort with the efficacy of anti-EGFR antibody treatment in pretreated mCRC harbouring wild-type or unknown KRAS exon 2. The details of selection criteria for the inference cohort are described in the Supplementary Appendix.
This study was approved by the Institutional Review Board of each participating centre. Written informed consent was obtained from patients who were alive when initiating this study. For deceased patients and their relatives at that time, we disclosed the study design on the website of each centre and allowed the relatives to approve or deny inclusion in the study. This study was conducted in accordance with the Ethical Guidelines for the human genome and genetic analysis research of the Ministry of Education, Culture, Sports, Science and Technology, Ministry of Health, Labour and Welfare and Ministry of Economy, Trade and Industry.
Collection of clinical and pathological data
An electronic data capture system (Viedoc; PCG Solutions, Uppsala, Sweden) was used for registration of patients and collection of clinical and pathological data by the Office of Translational Research, Exploratory Oncology Research and Clinical Trial Center (EPOC), National Cancer Center, Chiba, Japan.
Patient characteristics including age, sex, site of primary lesion, histology, site of metastases, prior treatments, clinical outcome of anti-EGFR antibody treatment, subsequent treatment, and severe adverse events related to anti-EGFR antibody treatment, were collected. Sites of primary lesions were divided into right-sided colon, left-sided colon, and rectum. Right-sided tumours were defined as those arising anywhere from the caecum to the transverse colon, and left-sided tumours were defined as those arising anywhere from the splenic flexure to the rectosigmoid junction.
Primary investigators were blinded to cancer genome alterations analysed in the study; investigators evaluated the antitumour effect according to Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1 (Eisenhauer et al, 2009) and confirmed the safety of the treatment based on the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 (National Cancer Institute, 2009).
Targeted capture resequencing
Archived FFPE tissue specimens collected before administration of anti-EGFR antibody were used for target resequencing. Candidate biomarkers identified from the exploratory cohort were validated by target resequencing, which covered the full length of all the candidate genes, including KRAS, NRAS, and BRAF. The details of preparation of clinical samples, DNA extraction, identification of single nucleotide variants (SNVs) and insertion-deletion mutations (INDELs) and target resequencing are described in the Supplementary Appendix.
BRAF activity assay
To clarify the activity of newly identified BRAF mutations, we assessed the phosphorylation status of downstream molecules of EGFR by western blotting using HEK293 cells transfected with the BRAF mutant vector (Supplementary Appendix).
Statistical analysis
The efficacy endpoints were progression-free survival (PFS), defined as the duration from the initiation of anti-EGFR antibody treatment to disease progression or death from any cause; overall survival (OS), defined as the duration from the initiation of anti-EGFR antibody treatment to death from any cause; RR, defined as the proportion of patients who had a complete or partial response with anti-EGFR antibody treatment; and disease control rate (DCR), defined as the proportion of patients who had a complete or partial response or stable disease. For PFS and OS, survival curves according to each mutational status were estimated by the Kaplan–Meier method and were compared using log-rank test.
Univariate and multivariate Cox regression analyses were performed to evaluate the prognostic impact of any RAS/BRAFV600E/BRAFnon-V600E mutant (herein referred to as RAS/BRAF mutant) vs wild-type. Covariates in the regression analyses included RAS/BRAF (mutant vs wild-type), age, gender, ECOG PS, histology, primary site, primary tumour resection, adjuvant chemotherapy, metastasis (synchronous vs metachronous), combined use of irinotecan, and prior oxaliplatin treatment. Considering the limited number of death events, backward elimination procedure, setting the removal criteria as a P-value of <0.20, was performed; four covariates (gender, ECOG PS, primary site, and combined use of irinotecan) were forcibly retained as potential confounding factors.
All statistical analyses were performed using SAS Release 9.3 (SAS Institute, Inc., Cary, NC, USA). All P values were obtained from two-sided statistical tests with a significance level of 0.05.
Results
Summary of the exploratory cohort
In the exploratory cohort, 92 patients with mCRC, comprising 57 super-responders and 35 nonresponders to anti-EGFR antibody treatment (90 KRAS exon 2 wild type and 2 unknown), were selected (Supplementary Figure S2). FFPE clinical samples of both cancerous and noncancerous areas were subjected to whole exon sequencing. Briefly, the exomes were captured using the SureSelect Human All Exon V4+UTRs Kit (Agilent Technologies) and sequenced using a HiSeq 2000 system (Illumina) to generate 100 bppaired-end data. The average base coverage of the targeted regions in the tumour and normal samples was 162.5-fold (range: 10.2–389.7) and 166.2-fold (range: 2.7–377.4), respectively. We identified 182.7±97.1 (range: 37.0–509.0) (5.7±3.0 per Mb, range: 1.2–15.9) somatic SNVs and 8.1±4.2 (range: 1.0–23.0) (0.3±0.1 per Mb, range: 0.0–0.7) somatic INDELs in the tumour tissues. Thirty-one candidate biomarker genes, including KRAS/NRAS/BRAF, in which mutations significantly deviated from either super-responders or nonresponders, were selected for further analysis with the inference cohort. Detailed data of the exploratory cohort is described elsewhere. Here we focused on the association of expanded RAS and BRAF mutation with efficacy endpoints in the inference cohort.
Genomic alternations and patient characteristics according to RAS/BRAF status in the inference cohort
A total of 184 patients were selected in the inference cohort. Target resequencing of the candidate biomarker genes, including KRAS/NRAS and BRAF, was successful in 156 patients, while 28 clinical samples were not analysed due to insufficient FFPE samples (n=6) and sequencing failure (n=22). The average base coverage of the targeted regions in the tumour and normal samples was 671.9-fold (range: 66.8–1735.0) and 731.6-fold (range: 70.4–1699.5), respectively. We identified 1.4±1.3 (range: 0.0–8.0) variants in the 31 candidate genes from tumour tissues. Additionally, six patients were excluded due to ineligibility (n=5) and acquisition of the specimen after anti-EGFR antibody treatment (n=1). Accordingly, 150 patients were included in the biomarker analysis population (Supplementary Figure S3). Baseline patient characteristics and clinical outcomes were similar between the whole population (N=184) and biomarker analysis population (N=150; data not shown).
KRAS, NRAS, and BRAF mutations were detected in 29 (19.3%), 11 (7.3%), and 16 (10.7%) patients, respectively. RAS and BRAF mutations were identified in a mutually exclusive manner. Nine of 16 BRAF mutations (6.0%) were BRAFV600E mutations, and seven were BRAFnon-V600E mutations (4.7%) located in the kinase domain as one G469A (high activated subtype in exon 11) with co-mutation of MAP2K1, one L485F (intermediate subtype in exon 12), one Q524L, one L525R (intermediate subtypes in exon 13), two D594G (impaired subtype in exon 15) and one V600R (high subtype in exon 15) with co-mutations of MSP2 and PPFIA2 (Table 1). Q524L and L525R were newly identified mutations that were not registered in either the Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov) or the Catalogue of Somatic Mutation in Cancer (COSIMIC; http://cancer.sanger.ac.uk/cosmic) databases.
Baseline patient characteristics, based on the RAS and BRAF mutational status, are shown in Table 2. Both BRAFV600E and BRAFnon-V600E mutant tumours were more commonly associated with the right-sided colon (44.4% and 57.1%, respectively) than the RAS/BRAF wild-type and RAS mutant tumours (13.8% and 27.5%, respectively). BRAFnon-V600E mutant tumours tended to have more lymph node metastases (71.4%) than with other mutational subtypes, RAS and BRAFV600E mutations (27.5% and 11.1%, respectively).
RRs to anti-EGFR antibody treatment according to RAS/BRAF mutation status in the inference cohort
The RR was 20.7% in all patients. The RRs in patients with RAS, BRAFV600E, and BRAFnon-V600E mutations were lower in comparison with patients harbouring wild-type RAS/BRAF (2.5%, 0%, and 0% vs 31.9%, respectively). In addition, the proportion of SD more than 6 months in patients with BRAFnon-V600E mutant was 14.3%, which was similar to that in patients with RAS or BRAFV600E mutations rather than wild-type RAS/BRAF (Table 3).
Survival and safety analysis according to RAS and BRAF status in the inference cohort
The median follow-up time was 12.1 months as of the cutoff date of December 24, 2014. The median PFS and OS of all patients were 4.0 months (95% confidence interval (CI), 3.4–4.8 months) and 12.4 months (95% CI, 9.8–14.0), respectively.
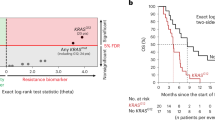
The median PFS of patients with BRAFnon-V600E mutations was 2.4 months (95% CI, 2.1–4.0), similar to that in patients with RAS or BRAFV600E mutations (2.1 months, 95% CI, 1.9–2.6 and 1.6 months, 95% CI, 1.1–3.4, respectively) but significantly worse than that in patients with wild-type RAS/BRAF (5.9 months, 95% CI, 4.9–7.7, P<0.0001; Table 3, Figure 1).

Merged survival curves for anti-EGFR antibody treatment. Kaplan–Meier curves for (A) progression-free survival (PFS) and (B) overall survival (OS) from the initiation of anti-EGFR antibody treatment in patients with pretreated mCRC according to mutational status. Wild-type RAS/BRAF was defined as all wild-type sequences with RAS, BRAFV600E and BRAFnon-V600E. A total of 150 patients in the inference cohort were classified according to RAS/BRAF WT (n=94); RAS MT (n=40); BRAFV600E MT (n=9); and BRAFnon-V600E MT (n=7). For comparison, Kaplan–Meier curves of RAS WT (n=110; yellow solid) and RAS/BRAFV600E WT (n=101; blue solid) were added. MT, mutant; WT, wild-type.
The median OS of patients with BRAFnon-V600E mutations was 8.1 months (95% CI, 5.3–16.9), similar to that in patients with RAS or BRAFV600E mutations (6.3 months, 95% CI, 4.6–8.4 and 4.6 months, 95% CI, 1.3–21.2, respectively) but worse than that in patients with wild-type RAS/BRAF (14.5 months, 95% CI, 12.6–16.2; Table 3, Figure 1).
Univariate and multivariate analyses for PFS and OS
Univariate and multivariate analyses for PFS and OS are shown in Table 4. Mutation subtype with RAS/BRAF was a strong negative prognostic factor for both PFS (HR, 3.49; 95% CI, 2.43–5.00) and OS (HR, 2.14; 95% CI, 1.51–3.04) in univariate analyses. Similarly, the RAS/BRAF subtype was also a strong negative prognostic factor for both PFS (HR, 5.43; 95% CI, 3.45–8.55) and OS (HR, 3.37; 95% CI, 2.20–5.16) in multivariate analyses.
BRAF activity assays for the newly identified mutations Q524L and L525R
To evaluate the kinase activity of newly identified BRAF mutants, mutant- and wild-type BRAF-expressing vectors were transiently transfected into EGFR-expressing HEK293 cells. The transfection efficiency was more than 70%, as assessed by EGFR-expressing control plasmid vector transfection (data not shown). Western blot analysis showed that extracellular signal-regulated kinase (ERK) phosphorylation in cells with BRAFV600E overexpression was significantly increased compared with that in cells with wild-type BRAF overexpression (Figure 2). BRAFL525R induced increased ERK phosphorylation to a level similar to that induced by BRAFV600E. However, BRAFQ524L activity was similar to that of wild-type BRAF.

BRAF activity assay. Phosphorylation status of ERK was assessed by western blotting. HEK293 cells were transiently transfected with wild-type, V600E, Q524L, or L525R BRAF and were then treated with 0.5 or 5 μg/ml cetuximab.
In the control vector-transfected cells, cetuximab reduced ERK phosphorylation in a concentration-dependent manner. Additionally, cetuximab reduced ERK phosphorylation level, which was enhanced by wild-type BRAF or BRAFQ524L expression. On the other hand, ERK phosphorylation enhanced by BRAFV600E and BRAFL525R was not affected by cetuximab, suggesting that cells with BRAFV600E or BRAFL525R mutants were resistant to cetuximab-induced inhibition of EGFR.
Discussion
To the best of our knowledge, this is the first report for the clinical significance of BRAFnon-V600E mutations focusing on the therapeutic effects of anti-EGFR monoclonal antibodies in patients with pretreated mCRC.
Few studies have reported the clinicopathological features of BRAFnon-V600E mutations because BRAFV600E-mutant tumours are most frequently observed in CRC (Shen et al, 2013; Ciardiello et al, 2014). According to studies in Western countries, the incidence of BRAFnon-V600E in mCRC was reported to range from 1.6% to 2.7% (Ciardiello et al, 2014; Cremolini et al, 2015). In contrast, the incidence of BRAFnon-V600E in 676 Chinese patients with mCRC was reported to be 5.1%, consistent with that in our cohort (4.7%) (Shen et al, 2013). On the other hand, the racial differences in terms of the incidence of BRAF V600E mutations were reported from the analysis of large-scale adjuvant trial in US, suggesting the incidence appeared to be lower in Asians than in blacks or whites (Yoon et al, 2015). Instead, the incidence of BRAFnon-V600E mutations might be higher in Asian than that in Caucasian patients.
Two meta-analyses suggested that primary tumours in the right-sided colon showed worse prognoses than those in the left-sided colon and rectum in patients treated with anti-EGFR antibodies (Arnold et al, 2017; Holch et al, 2017). In addition, integrated analysis of two randomised panitumumab studies showed that they had consistent results, even when the BRAFV600E mutations were excluded (Boeckx et al, 2017). In our series, a similar tendency was observed clinical outcomes; specifically, patients with wild-type RAS mCRC with primary tumours in the right-sided colon had poorer prognoses compared with those having primary tumours in the left-sided colon and rectum, although the difference was not significant. However, if limited to wild-type RAS/BRAF tumours, there were no clear differences in OS among sites of primary lesions (Supplementary Figure 4). Thus, the unresponsiveness of primary tumours in the right-sided colon to anti-EGFR antibodies in later line might be partially explained by underlying BRAFnon-V600E-mutated tumours.
Subtypes of BRAF mutations in the kinase domain can be classified into high, intermediate, and impaired activity subtypes based on their kinase activity (Wan et al, 2004). The BRAFV600E mutation belongs to the high activity subtype, whereas the BRAFG469A, BRAFL485F, and BRAFV600R mutations observed in this study belong to the intermediate subtype and the BRAFD594G mutation belongs to the impaired subtype. Moreover, the BRAFL525R mutant observed in this study had enhanced kinase activity, and the enhanced downstream signal of BRAFL525R may contribute to primary resistance to cetuximab, consistent with the lack of response to anti-EGFR antibody treatment in our series. In contrast, the newly identified BRAFQ524L mutant identified in this study had intermediate kinase activity and did not induce resistance to cetuximab in an in vitro cell model. However, such in vitro experiments in non-CRC epithelial cell lines may not fully predict the clinical outcome.
One possible explanation for the similar behaviours of BRAFnon-V600E and BRAFV600E-mutated tumours in terms of unresponsiveness to anti-EGFR antibody treatment may be the incomplete blockade of the MEK pathway by modestly upregulated kinase activity of BRAF and/or by additional signalling through wild-type CRAF (Wan et al, 2004). Therefore, it is necessary to establish patient-derived xenograft models harbouring BRAFnon-V600E mutations to clarify the mechanisms of primary resistance to anti-EGFR antibody treatment in BRAFnon-V600E-mutated mCRC.
Regarding BRAFD594G, classified as an impaired subtype in the study, Cremolini et al reported 10 cases with BRAF mutations in codons 594 or 596, the number of which was similarly small showing a favourable prognosis from first line (Cremolini et al, 2015). More recently, it was reported that OS was significantly longer for 101 patients with BRAFnon-V600E mutations than for the control group of 99 patients with BRAFV600E mutations (60.7 vs 11.4 months) (Jones et al, 2017); however, our analysis focused on pretreated population. In addition, according to the European Consortium, De Roock et al reported that two patients harbouring BRAFD594G-mutated mCRC achieved a partial response to cetuximab monotherapy (De Roock et al, 2010). In contrast, the two patients with BRAFD594G-mutated tumours in our study did not achieve objective response to anti-EGFR antibody treatment. Considering small patient’s number of each reports as well as heterogeneous population, it is difficult to conclude predictive impact of BRAFnon-V600E mutation. The overall data in this study supported that BRAFnon-V600E mutations were prognostic, as a similar magnitude to the presence of BRAFV600E and RAS mutations in later line, and the outcome appeared similar to patients with RAS mutations as well, who do not benefit from anti-EGFR therapy. The present study has some limitations. It was a retrospective study with a small number of subgroups of BRAFnon-V600E mutations, using archived FFPE samples. In addition, it is difficult to conduct further analyses by subdividing the group into ‘non-V600E kinase activity’ and ‘non-V600E non- kinase activity’ subgroups, due to the small number of BRAFnon-V600E mutations. The emergence of secondary RAS and BRAF gene mutations in ctDNA was recently reported after treatment with anti-EGFR antibody (Bettegowda et al, 2014). However, in the case of patients receiving systemic chemotherapy without anti-EGFR antibody, secondary gene alterations are rare (Kawamoto et al, 2012); therefore, we believed that adding the archived FFPE samples before anti-EGFR antibody administration would be reasonable. Further investigation in large-scale data set from such as randomised control trials is necessary to clarify the significance of anti-EGFR antibody treatment for each BRAFnon-V600E mutational variants as the role of the predictive value.
In conclusion, although identified BRAFnon-V600E mutations were rare and unestablished molecular subtype in mCRC, overall clinical outcomes of BRAFnon-V600E mutations in the kinase domain, similar to those of RAS- and BRAFV600E-mutant tumours, appeared to be significantly worse than those in wild-type RAS/BRAF tumours. Certain BRAFnon-V600E mutations might contribute to a lesser benefit of anti-EGFR monoclonal antibody treatment. This novel knowledge provides an intriguing background to investigate new target approaches in patients with BRAFnon-V600E mutations and represents substantial progression toward more precision medicine.
References
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26: 1626–1634.
Andreadi C, Noble C, Patel B, Jin H, Aguilar Hernandez MM, Balmanno K, Cook SJ, Pritchard C (2012) Regulation of MEK/ERK pathway output by subcellular localization of B-Raf. Biochem Soc Trans 40: 67–72.
Arnold D, Lueza B, Douillard JY, Peeters M, Lenz HJ, Venook A, Heinemann V, Van Cutsem E, Pignon JP, Tabernero J, Cervantes A, Ciardiello F (2017) Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomised trials. Ann Oncol 28: 1713–1729.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih lM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr (2014) Detection of circulating tumour DNA in early- and late-stage human malignancies. Sci Transl Med 6 (224): 224ra24.
Boeckx N, Koukakis R, Op de Beeck K, Rolfo C, Van Camp G, Siena S, Tabernero J, Douillard JY, André T, Peeters M (2017) Primary tumour sidedness has an impact on prognosis and treatment outcome in metastatic colorectal cancer: results from two randomized first-line panitumumab studies. Ann Oncol 28 (8): 1862–1868.
Brulé SY, Jonker DJ, Karapetis CS, O'Callaghan CJ, Moore MJ, Wong R, Tebbutt NC, Underhill C, Yip D, Zalcberg JR, Tu D, Goodwin RA (2015) Location of colon cancer (right-sided vs left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur J Cancer 51: 1405–1414.
Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, Giuliani F, Barone C, Cartenì G, Rachiglio AM, Montesarchio V, Tonini G, Rizzi D, Cinieri S, Bordonaro R, Febbraro A, De Vita F, Orditura M, Fenizia F, Lambiase M, Rinaldi A, Tatangelo F, Botti G, Colucci G (2014) Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol 25: 1756–1761.
Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, Sartore-Bianchi A, Fontanini G, Milione M, Lauricella C, Siena S, Falcone A, de Braud F, Loupakis F, Pietrantonio F (2015) BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol 26: 2092–2097.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417: 949–954.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753–762.
Douillard J-Y, Oliner KS, Siena S, Abernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369: 1023–1034.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45 (2): 228–247.
Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S (2014) Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling cancer. Cell 26: 402–413.
Holch JW, Ricard I, Stintzing S, Modest DP, Heinemann V (2017) The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta-analysis of first-line clinical trials. Eur J Cancer 70: 87–98.
Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, Kasi PM, Voss JS, Leal AD, Sun J, Ross J, Ali SM, Hubbard JM, Kipp BR, McWilliams RR, Kopetz S, Wolff RA, Grothey A (2017) Non-V600 BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol 35 (23): 2624–2630.
Kawamoto Y, Tsuchihara K, Yoshino T, Ogasawara N, Kojima M, Takahashi M, Ochiai A, Bando H, Fuse N, Tahara M, Doi T, Esumi H, Komatsu Y, Ohtsu A (2012) KRAS mutations in primary tumours and post-FOLFOX metastatic lesions in cases of colorectal cancer. Br J Cancer 107 (2): 340–344.
Kawazoe A, Shitara K, Fukuoka S, Kuboki Y, Bando H, Okamoto W, Kojima T, Fuse N, Yamanaka T, Doi T, Ohtsu A, Yoshino T (2015) A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer 15: 258.
Mercer KE, Pritchard CA (2003) Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta 1653: 25–40.
National Cancer Institute (2009) NIH Publication No. 09-5410. Available at http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek J, Reese D, Patterson SD (2013) Massively parallel tumour multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 19: 1902–1912.
Roskoski R (2010) RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Commun 399: 313–317.
Shen Y, Wang J, Han X, Yang H, Wang S, Lin D, Shi Y (2013) Effectors of epidermal growth factor receptor pathway: the genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS ONE 8: e81628.
Taniguchi H, Yamazaki K, Yoshino T, Muro K, Yatabe Y, Watanabe T, Ebi H, Ochiai A, Baba E, Tsuchihara K Japanese Society of Medical Oncology (2015) Japanese Society of Medical Oncology Clinical Guidelines: RAS (KRAS/NRAS) mutation testing in colorectal cancer patients. Cancer Sci 106: 324–327.
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360: 1408–1417.
Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, Ciardiello F (2015) Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 33: 692–700.
von Einem JC, Heinemann V, von Weikersthal LF, Vehling-Kaiser U, Stauch M, Hass HG, Decker T, Klein S, Held S, Jung A, Kirchner T, Haas M, Holch J, Michl M, Aubele P, Boeck S, Schulz C, Giessen C, Stintzing S, Modest DP (2014) Left-sided primary tumours are associated with favorable prognosis in patients with KRAS codon 12/13 wild-type metastatic colorectal cancer treated with cetuximab plus chemotherapy: an analysis of the AIO KRK-0104 trial. J Cancer Res Clin Oncol 140: 1607–1614.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R Cancer Genome Project (2004) Mechanism of activation of the Raf-MEK signaling pathway by oncogenic mutations of B-Raf. Cell 116: 856–867.
Yoon HH, Shi Q, Alberts SR, Goldberg RM, Thibodeau SN, Sargent DJ, Sinicrope FA Alliance for Clinical Trials in Oncology (2015) Racial differences in BRAF/KRAS mutation rates and survival in stage iii colon cancer patients. J Natl Cancer Inst 107 (10): djv186.
Yoshino T, Muro K, Yamaguchi K, Nishina T, Denda T, Kudo T, Okamoto W, Taniguchi H, Akagi K, Kajiwara T, Hironaka S, Satoh T (2015) Clinical validation of a multiplex kit for RAS mutations in colorectal sancer: results of the RASKET (RAS KEy Testing) prospective, multicenter study. EBioMedicine 2: 317–323.
Acknowledgements
We would like to express special thanks to all participating patients and their families; all investigators, including the participating radiologists Takeshi Aramaki and Toshihiro Iguchi; and Izumi Miki, Akiko Nakayama, and Yoriko Kato at the Office of Translational Research, Exploratory Oncology Research and Clinical Trial Center, National Cancer Center, Chiba, Japan. The BREAC study was supported by the Adaptable and Seamless Technology Transfer Program (ASTEP) through target-driven R&D, Practical Application Development by SME Start-up, Japan Science and Technology Agency (JST), National Cancer Center Research and Development Fund (23-A-2), G & G Science Co., Ltd., and Medical & Biological Laboratories Co., Ltd.
Author contributions
Conception and design: TY, KT, SF, YS, KA, HE, MS, NN, MM, YK, YA, and AO. Provision of study materials or patients: ES, KY, TY, KM, TN, KY, SY KS, and HB. Collection and assembly of data: TY and SN. Genome sequencing: SM, CN, and KT. Cell-based experiments: KM and CN. Data analysis and interpretation: All authors. Manuscript writing: TY, KT, and ES. Final approval of manuscript: All authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Presentations: American Society of Clinical Oncology Annual Meeting, Chicago, May 2015 and Gastrointestinal Cancers Symposium, San Francisco, January 2015.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shinozaki, E., Yoshino, T., Yamazaki, K. et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br J Cancer 117, 1450–1458 (2017). https://doi.org/10.1038/bjc.2017.308
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.308
Keywords
This article is cited by
-
Regulation of MEK inhibitor selumetinib sensitivity by AKT phosphorylation in the novel BRAF L525R mutant
International Journal of Clinical Oncology (2023)
-
Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies
Journal of Experimental & Clinical Cancer Research (2021)
-
Biomarker-guided therapy for colorectal cancer: strength in complexity
Nature Reviews Clinical Oncology (2020)
-
Clinicopathological and Molecular Study of Peritoneal Carcinomatosis Associated with Non-Small Cell Lung Carcinoma
Pathology & Oncology Research (2020)
-
Long term response on Regorafenib in non-V600E BRAF mutated colon cancer: a case report
BMC Cancer (2019)
