
Abstract
Background:
KW-2478 is a novel non-ansamycin Hsp90 inhibitor with modest single-agent activity in relapsed/refractory myeloma but which shows synergistic antimyeloma activity with bortezomib (BTZ) in preclinical studies. This study determined the safety, preliminary clinical activity, and pharmacokinetics of KW-2478, an Hsp90 inhibitor, in combination with BTZ in patients with relapsed/refractory multiple myeloma (MM).
Methods:
Phase I dose escalation determined the recommended phase II dose (RP2D) of KW-2478 plus BTZ, which was then used during phase II.
Results:
The maximum tolerated dose was not reached during phase I and the RP2D was KW-2478 175 mg m−2 plus BTZ 1.3 mg m−2 on days 1, 4, 8, and 11 every 3 weeks. In the efficacy evaluable phase I/II population treated at the RP2D (n=79), the objective response rate was 39.2% (95% confidence interval: 28.4–50.9%), clinical benefit rate 51.9% (40.4–63.3%), median progression-free survival 6.7 (5.9-not reached (NR)) months, and median duration of response 5.5 (4.9-NR) months. In the phase I/II safety population (n=95), the most frequently observed treatment-related grade 3/4 adverse events were diarrhoea, fatigue, and neutropenia (each in 7.4% of patients), and nausea and thrombocytopenia (each in 5.3%).
Conclusions:
KW-2478 plus BTZ was well tolerated with no apparent overlapping toxicity in patients with relapsed/refractory MM. The antimyeloma activity of KW-2478 in combination with BTZ as scheduled in this trial appeared relatively modest; however, the good tolerability of the combination would support further exploration of alternate dosing schedules and combinations.
Similar content being viewed by others

Main
It has been estimated that multiple myeloma (MM) accounted for 30 330 new cancer cases and 12 650 deaths in 2016 in the United States (Siegel et al, 2016). In the past decade, advances in therapy, including high-dose chemotherapy, autologous stem cell transplantation, and introduction of immunomodulatory drugs such as thalidomide or lenalidomide and proteasome inhibitors such as bortezomib (BTZ) into treatment regimens have improved clinical outcomes (Kumar et al, 2014). However, nearly all patients eventually relapse or become refractory following first- or second-line treatment (Badros et al, 2009). These patients with relapsed/refractory MM present a therapeutic challenge because of their poor clinical outcome (Sinha et al, 2012). There is therefore a need for novel agents that act in a mechanistically different manner to proteasome inhibitors or thalidomide derivatives, which can be used sequentially or in combination with currently available agents for patients with relapsed/refractory MM.
One approach has been the development of agents that target heat shock protein 90 (Hsp90), a chaperone protein involved in the stabilisation of certain cellular proteins involved in tumour growth. Heat shock protein 90 is uniformly expressed in different human MM cell lines (Mitsiades et al, 2006; Sharp and Workman, 2006). Various Hsp90 inhibitors have shown antitumour activity in clinical trials, particularly when combined with other anticancer agents (Lu et al, 2012; Whitesell et al, 2012; Soga et al, 2013).
KW-2478 is a potent, intravenous (i.v.), non-ansamycin, non-purine Hsp90 inhibitor that exhibits antitumour activity in preclinical in vitro and in vivo MM models (Nakashima et al, 2010). This antitumour activity in MM cells is retained in the presence of bone marrow stromal cells (Juliger et al, 2008), suggesting that KW-2478 overcomes the protective effect of the bone marrow microenvironment. The combination of KW-2478 and BTZ showed greater antitumour activity than either agent alone in preclinical MM models (Ishii et al, 2012). In a phase I single-agent study, KW-2478 was well tolerated, the maximum tolerated dose (MTD) was not reached at doses up to 176 mg m−2 on days 1–5 every 2 weeks, and preliminary signs of clinical activity were seen (Yong et al, 2016). The purpose of this phase I/II trial was to determine the safety, preliminary clinical activity, and pharmacokinetics of KW-2478 in combination with BTZ in patients with relapsed/refractory MM.
Patients and methods
The overall design was an open-label, multicentre, two-part trial (phases I and II). It was carried out in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice, the ethical principles of the Declaration of Helsinki, and all applicable local regulatory requirements. The trial was conducted at 26 institutions (13 in the United Kingdom, 9 in the United States, and 4 in The Philippines) and approved by the Institutional Review Board of each participating institution. Written informed consent was obtained from each patient prior to recruitment. The trial was registered at ClinicalTrials.gov (NCT01063907).
Patients
Adults (⩾18 years) with a confirmed diagnosis of MM by International Myeloma Working Group (IMWG) criteria (Durie et al, 2006; Kyle and Rajkumar, 2009), who had relapsed or failed to respond at least one prior MM regimens with an Eastern Cooperative Oncology Group (ECOG) performance status ⩽2 and a life expectancy ⩾3 months were eligible for the trial. Patients had to have disease that could be evaluated by serum or urinary M protein (⩾1 g dl−1 or ⩾200 mg 24 h−1, respectively), or serum-free light chains (⩾10 mg dl−1 with abnormal κ/λ ratio (normal range, 0.26–1.65)) in the absence of measurable M protein in serum or urine. Patients could not have progressed while receiving BTZ or another proteasome inhibitor alone or in combination, and >60 days must have elapsed since that treatment. Prior therapy had to be completed >4 weeks (>6 weeks for nitrosoureas) previously and monoclonal antibody therapy >6 weeks prior to entry. A stable dose of bisphosphonate treatment for >3 months prior to entry was permitted.
Patients had to have adequate haematological (absolute neutrophil count ⩾1 × 109 l−1, haemoglobin ⩾9 g dl−1, and platelets ⩾75 × 109 l−1), hepatic (total bilirubin <1.5 times the institutional upper limit of normal (ULN) excluding Gilbert’s syndrome, and aspartate aminotransferase and alanine aminotransferase <2.5 times ULN), and renal function (serum creatinine ⩽2 mg dl−1 or calculated creatinine clearance >30 ml min−1 1.73 m−2 if serum creatinine >2 mg dl−1). Excluding these specifications, patients had to have resolution of any significant toxicity from prior anticancer therapy to grade ⩽1 according to Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0, 2009. Women of child-bearing potential and men with female partners of child-bearing potential had to agree to use a highly effective method of birth control (i.e., condom or diaphragm plus spermicide, some intrauterine devices, hormonal devices, hormonal contraceptive, true sexual abstinence, or male vasectomy) during study drug treatment and until 3 months after last BTZ administration.
Patient exclusion criteria were significant uncontrolled intercurrent illness, intracranial or epidural disease, history of macular degeneration or blindness in one eye, or any other ocular diagnosis where the benefit-risk ratio would favour exclusion from the trial; non-secretory or biclonal MM; known hypersensitivity to boron or mannitol; prior treatment with any Hsp90 inhibitor; grade >1 sensory and/or motor neuropathy; known HIV infection or AIDS-related illness, known hepatitis B or C or other active liver disease, and active herpes zoster infection (those with a prior history of herpes zoster infection were permitted entry into the trial if treated with prophylactic acyclovir unless contraindicated); pregnancy or breast-feeding; use of immunosuppressive therapy other than corticosteroids at a dose equivalent to dexamethasone ⩽2.5 mg d−1; use of medication known to commonly cause QTc interval prolongation; Fridericia-correct QTc ⩾480 ms on screening; inability or unwillingness to receive blood or platelet transfusion that might be indicated to manage haematological complication; major surgery <6 weeks prior to screening; receipt of allogeneic stem cell transplantation; other malignant conditions unless disease-free for >5 years, except for adequately treated basal or squamous cell cutaneous carcinoma or cervical carcinoma in situ; clinically significant findings on screening slit-lamp retinal examination (phase I only) or any change in visual acuity or other ocular symptoms at screening or prior to the first dose (phases I and II); and requirement to use potent cytochrome P450 isoenzyme 3A4 (CYP3A4) or CYP2C19 inhibitors or inducers.
Study design
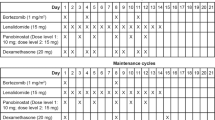
The primary objective of the phase I study was to determine the recommended phase II dose (RP2D), safety, and preliminary evidence of efficacy of KW-2478 plus BTZ in patients with relapsed/refractory MM. A secondary objective was to characterise the pharmacokinetics of the combination. The design was a standard 3+3 study of KW-2478 (130 or 175 mg m−2) plus BTZ (1 or 1.3 mg m−2) on days 1, 4, 8, and 11 of a 21-day cycle utilising four dose-escalation cohorts (cohorts 1–4) (see Table 1). Three patients had to complete one treatment cycle without a dose-limiting toxicity (DLT) in a cohort before progression to the next cohort. If patients discontinued prior to completing one cycle of therapy, they were replaced. Two dose de-escalation cohorts were planned but were not eventually required. The RP2D was defined as either the highest dose allowed by the dose-escalation schema or the MTD if DLTs were encountered. After two cycles of treatment, dose escalation of KW-2478 and BTZ was allowed provided the safety of the higher dose had been established; eventually, patients were permitted to continue to participate in the study at up to the RP2D if they met criteria for further treatment, but remained in phase I as a separate population.
Dose-limiting toxicity was defined by any of the following events occurring during the first cycle of treatment that was considered possibly, probably, or definitely related to KW-2478 or KW-2478 in combination with BTZ: any non-haematologic grade ⩾3 toxicity (with the exception of nausea, vomiting, or diarrhoea that can be medically controlled or reduced to grade ⩽2) or grade 4 haematologic toxicity based on CTCAE version 4.0 criteria (with the exception of neutropenia lasting ⩽5 days). If grade ⩾3 nausea, vomiting, and/or diarrhoea could be reduced to grade ⩽1 within 24 h and did not exceed grade 2 in subsequent treatments with prophylaxis, it was not considered a DLT.
The following rationale was used for dose selection. The maximum approved dose of BTZ is 1.3 mg m−2 administered on days 1, 4, 8, and 11 of a 21-day cycle, although it can also be administered as 0.7 or 1 mg m−2 doses (Velcade (bortezomib) for Injection, 2009). The proposed starting dose of BTZ 1 mg m−2 was chosen to allow treatment at a potentially effective dose but maintain a margin of safety in combination with KW-2478 with an intention to escalate to the approved dose. The starting dose of single-agent KW-2478, 130 mg m−2, is lower than the dose previously administered in a phase I study in patients with advanced, relapsed/refractory B-cell malignancies (predominantly with MM) with the intent to escalate to a dose of 176 mg m−2. These doses of KW-2478 would also result in a degree of exposure lower than that seen in the prior phase I study, where a higher dose intensity was employed.
The primary objective of the phase II study was to determine the safety, tolerability, and overall response rate (ORR) for KW-2478 plus BTZ at the RP2D in patients with relapsed/refractory MM. Secondary objectives were to determine progression-free survival (PFS) and duration of response with the combination. Patients who did not complete two full cycles of treatment (at least three of the four full doses of KW-2478 and BTZ for each cycle) for reasons other than disease progression were replaced. A Simon 2-stage design was used for the phase II portion of the trial (Simon, 1989). The trial was designed so that if 11 responses were observed in the first 27 evaluable patients, an additional 50 evaluable patients would be enroled with a target of more than 33 responses, corresponding to a response rate of 35%. All patients, including those in the phase I portion of the study, who were evaluable for response at the RP2D were considered in the final response analysis.
Treatment
KW-2478 and BTZ were administered on days 1, 4, 8, and 11 of a 21-day cycle. Bortezomib was administered according to the manufacturer’s instructions as an i.v. bolus dose over 3–5 s (Velcade (bortezomib) for Injection, 2009). KW-2478 (Kyowa Hakko Kirin Co. Ltd., Tokyo, Japan) was supplied as a sterile solution, which was diluted in 0.9% saline prior to infusion. KW-2478 was administered by i.v. infusion over 1 h on days 1, 4, 8, and 11 of a 21-day cycle 30 min after completion of BTZ dose administration. BP and pulse rate were monitored during infusion and patients were closely observed for infusion-related reaction. Treatment with KW-2478 plus BTZ was up to 8 cycles (24 weeks) in the absence of disease progression, drug intolerance or unacceptable toxicity, consent withdrawal, or non-compliance. Bortezomib could be continued past the initial eight cycles in responding patients if it was considered in the best interest of the patient by the investigator and was approved by the prescribing information for the relevant country. KW-2478 could be continued for up to 1 year for patients who had a response or stable disease (SD) but in whom BTZ had to be discontinued. For phase I patients after the first cycle and for all phase II patients, doses of KW-2478 were adjusted (decreased and/or delayed) in case of specified toxicities according to specific guidelines in the study protocol, while doses of BTZ were adjusted in accordance with approved prescribing information applicable to the country where the patient was being treated.
Patients were not allowed to receive any other therapy for MM during the trial, apart from steroids equivalent to dexamethasone <2.5 mg d−1 for the first four treatment cycles. After this, therapeutic doses of dexamethasone were permitted in patients with SD. Addition of bisphosphonates during the trial was not permitted, but patients already on bisphosphonates at the time of trial entry were permitted to continue therapy. Haematopoietic growth factors were not permitted during the 2 weeks prior to and during the first cycle of treatment for patients in phase I, but were allowed in all other patients and cycles.
Assessments
Demographic data and medical history were determined during screening. Vital signs, weight, ECOG performance status, assessment of vision, peripheral neuropathy and adverse events (AEs), haematology, and clinical chemistry were determined during screening, on days 1, 4, 8, 11, and 16–18 of each cycle as well as at the end of treatment and 30-day follow-up. Physical examination was performed during screening, on days 1, 4, 8, 11, and 16–18 of the first cycle, and on day 1 of subsequent cycles, as at the end of treatment and 30-day follow-up. Coagulation profile and urinalysis were determined during screening and at the end of the study. Twelve-lead ECG was performed during screening, on days 1, 4, 8, and 11 of the first cycle, on day 11 of each subsequent cycle, and at the end of the study. Bence–Jones protein was determined at screening and was repeated on days 16–18 of each cycle and at the end of the study in those who were positive at screening. Serum β2-microglobulin was determined during screening, on day 4 of the first cycle, on day 1 of subsequent cycles, and at the end of the study. Skeletal survey and bone marrow aspiration/biopsy were performed during screening and as required to confirm complete response.
Efficacy was determined using the IMWG criteria (Durie et al, 2006) with the addition of minimal response (MR) as per the American Society of Hematology-Food and Drug Administration panel (Anderson et al, 2008). Patients failing to achieve stringent complete response (sCR), complete response (CR), very good partial response (VGPR), partial response (PR), or MR were considered to have SD until they progressed, or were censored without achieving a response or showing progressive (PD). Patients were evaluated every 3 weeks for response, PD, and relapse according to the IMWG criteria (Durie et al, 2006), although only objective evidence of PD was considered (CRAB features were excluded). Additional efficacy end points included duration of response, clinical benefit rate (CBR) (MR or better response), and PFS. Duration of response was measured from the time of CR/PR/MR, whichever was first recorded, until PD or death (as specified in the protocol). PFS was the time from the first day of treatment until PD or death.
AEs were classified and graded according to CTCAE version 4.0 (Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0, 2009). Treatment-related AEs were those classified as possibly, probably, or definitely related to the individual study drugs by the investigator.
The safety population included patients who had received at least one dose of study medication. The efficacy evaluable population included all patients who had completed ⩾2 cycles of treatment, who had baseline data, and had at least one on-study assessment of response. The calculation of efficacy parameters was performed after 8 cycles of therapy, with patients who withdrew prior to cycle 8 because of PD being included using their best response.
Pharmacokinetics
Blood samples were taken on days 1 and 11 for pharmacokinetic analysis in phase I. Predose samples were taken immediately prior to BTZ administration for the determination of both KW-2478 and BTZ. Samples were taken at the following nominal times after the start of KW-2478 infusion for determination of KW-2478: 1 (end of infusion), 1.08, 1.25, 1.5, 2, 4, and 7 h on days 1 and 11, and 25 h on day 1.
Plasma KW-2478 concentrations were determined at a central laboratory (Quintiles AB Bioanalytical Laboratory, Uppsala, Sweden) using a validated method that employed solid-phase extraction followed by quantitation using a liquid chromatography-mass spectrometry/mass spectrometry method.
Pharmacokinetic parameters were determined using non-compartmental methods using WinNonlin v.5.2 (Pharsight Co., Mountain View, CA, USA). The following variables were calculated using this model: Cmax (maximum serum concentration); Tmax (time to Cmax); AUC0–∞ and AUC0–t (area under the serum concentration−time curve extrapolated to infinity and to the last measurable time point, respectively); CL (total plasma clearance); Vz (volume of distribution based on the elimination phase); t1/2 (elimination half-life); and R (accumulation ratio calculated as day 11 AUC0–7 h/day 1 AUC0–7 h).
Statistics
Overall response rate was estimated with 95% Clopper–Pearson confidence intervals (CIs). The overall significance for the Simon two-stage design was 0.05 with a power of 80% assuming a reference ORR of 35% and a target ORR of 50%. The sample size was computed using PASS 2008 software (NCSS, LLC, Kaysville, UT, USA). Duration of response and PFS were estimated using the Kaplan–Meier method. Efficacy was determined among evaluable patients from the phase II population plus those from phase I who were treated at the RP2D. Exploratory post hoc determination of efficacy was determined for BTZ-naive and BTZ-pretreated as well as lenalidomide-naive and lenalidomide-pretreated subpopulations.
Results
Patients
The study was conducted between 26 May 2010 and 30 November 2013. Ninety-five patients were enroled: 15 in phase I and 80 in phase II. The baseline demographic and clinical characteristics of the patients are summarised in Table 2. All patients had either relapsed (71.6%) or relapsed/refractory (28.4%) MM. The majority had IgG (61.1%) or IgA (24.2%) isotype disease; ECOG performance status was 0 (56.8%) or 1 (34.7%), with a few classed as 2 (7.4%). The patients had received 1 (34.7%), 2 (24.2%), or ⩾3 (41.1%) prior lines of MM therapy. All 95 patients in phases I and II were included in the safety population and 87 patients in the efficacy evaluable population. Eighteen patients had previously received both bortezomib and lenalidomide. Twenty-four patients had received prior lenalidomide therapy. The number of prior therapies were comparable between the lenalidomide-naive and lenalidomide-pretreated subgroups. However, the number of prior therapies in the bortezomib-naive subgroup were less compared to the bortezomib-pretreated subgroup.
Phase I determination of RP2D
One patient experienced a DLT (grade 3 vasovagal reaction), which occurred in cohort 4 (the maximum tested dose combination) (Table 1). The MTD was not therefore reached and the RP2D was KW-2478 175 mg m−2 plus BTZ 1.3 mg m−2 on days 1, 4, 8, and 11 every 3 weeks, which was used in phase II.
Safety
The median (range) number of treatment cycles received was 4.0 (1–13) during phase I, 5.5 (1–17) during phase II, and 5.0 (1–17) overall. Treatment-emergent AEs are summarised in Table 3. The most common all-grade treatment-related AEs were diarrhoea (58.9%), nausea (49.5%), fatigue (43.2%), and vomiting (36.8%). The most common grade 3/4 treatment-related AEs were diarrhoea (7.4%), fatigue (7.4%), neutropenia (7.4%), nausea (5.3%), and thrombocytopenia (5.3%). No patient developed any significant abnormality on routine ophthalmological examination or fundus autofluorescence imaging. Seven of 95 patients (7.4%) developed blurred vision considered at least possibly related to treatment and all were grade 1/2. One patient with grade 2 blurred vision possibly related to treatment was considered serious and was withdrawn from treatment, which was followed by full recovery.
There were two deaths during the study but neither was related to treatment (MM and pneumonia, respectively). Eighteen patients experienced 28 treatment-related serious AEs (lung infection n=7; vomiting and diarrhoea, each n=3; and abdominal pain, anaemia, blurred vision, dehydration, dyspnoea, haematuria, hyponatremia, nausea, neurogenic bladder, pancreatitis, peripheral neuropathy, presyncope, rash, inappropriate antidiuretic hormone secretion, and transient ischaemic attack, each n=1). Ten patients were discontinued because of 11 treatment-related AEs (peripheral neuropathy n=2; and abdominal pain, blurred vision, constipation, dehydration, diarrhoea, ventricular extrasystoles, macular degeneration, rash, and reduced visual acuity, each n=1). The two patients withdrawn because of peripheral neuropathy were grade 2 and 3, respectively. Grade 4 treatment-related AEs were infrequent, with three patients (3.2%) experiencing neutropenia and one patient (1.1%) hyponatremia.
There were no clinically meaningful treatment-related changes in haematologic and biochemistry values, vital signs, body weight, and ECG results.
Efficacy
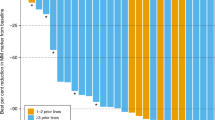
Results for the efficacy evaluable populations in phase I, phase II, phase I/II, and phase I/II at the RP2D are summarised in Table 4. We focus on results for the phase I/II population treated at the RP2D (KW-2478 175 mg m−2 plus BTZ 1.3 mg m−2 on days 1, 4, 8, and 11 every 3 weeks) (n=79), as it formed the largest group treated at a fixed dose. Overall response rate (sCR+CR+VGPR+PR) was 39.2% (95% CI: 28.4–50.9%) with the following responses: CR 3.8%, VGPR 12.7%, and PR 22.8%. Median PFS was 6.8 (95% CI: 5.9-not reached (NR)) months and median duration of response was 5.6 (95% CI: 4.9-NR) months. The CBR (sCR+CR+VGPR+PR+MR) was 51.9% (95% CI: 40.4–63.3%). We subsequently performed subgroup analyses in the phase I/II population treated at the RP2D depending on prior bortezomib or lenalidomide pretreatment (Table 5). Subgroup analysis in BTZ-naive patients treated at the RP2D (n=43) revealed an ORR of 44.2% with the following responses: CR 7.0%, VGPR 11.6%, and PR 25.6%. Clinical benefit rate was 53.5% (95% CI: 37.7–68.8%). The percentage of DCR was 95.3. Corresponding results among BTZ-pretreated patients treated at the RP2D (n=36) were ORR 33.3% (95% CI: 18.6–51.0%) with the following responses: VGPR 13.9% and PR 19.4%. Clinical benefit rate was 50.0% (95% CI: 32.9–67.1%). The percentage of DCR was 88.9. Subgroup analysis in lenalidomide-naive patients treated at the RP2D (n=55) revealed an ORR of 45.5% (95% CI: 32.0–59.4%) with the following responses: CR 5.5%, VGPF 12.7%, and PR 27.3%. Clinical benefit rate was 61.8% (95% CI: 47.7–74.6%). The percentage of DCR was 94.4. Corresponding results among lenalidomide-pretreated patients treated at the RP2D (n=24) were ORR 25.0% (95% CI: 9.8–46.7%) with VGPR 12.5% and PR 12.5%. Clinical benefit rate was 29.2% (95% CI: 12.6–51.1%). The percentage of DCR was 87.5.
Pharmacokinetics
Mean plasma concentration vs time curves for KW-2478 on days 1 and 11 during cycle 1 of cohorts 1–4 in phase I are shown in Supplementary Figure S1 (available online). Mean pharmacokinetic parameters for KW-2478 are summarised in Supplementary Table S1 (available online). No formal statistical analysis was undertaken. Mean t1/2 for KW-2478 ranged from 5.27 to 5.79 h on day 1 and from 1.77 to 2.02 h on day 11, and did not appear related to dose. The shorter t1/2 on day 11 was related to the shorter plasma sampling interval on day 11 compared to day 1 (7 h vs 25 h, respectively). Mean CL of KW-2478 was higher at the 175 mg m−2 compared to the 130 mg m−2 dose (32.2–62.0 l h−1 vs 25.9–33.3 l h−1). Systemic exposure (Cmax and AUC values) increased in a dose-related manner. Accumulation did not occur. There were no notable differences in KW-2478 pharmacokinetic parameters comparing cohorts 1 and 2 or comparing cohorts 3 and 4, that is, comparing the effect of co-administered BTZ 1 and 1.3 mg m−2, indicating that the increase in BTZ dose did not affect KW-2478 pharmacokinetics.
Discussion
The aim of this trial was to determine the safety, preliminary clinical activity, and pharmacokinetics of KW-2478 plus BTZ in patients with relapsed/refractory MM. The MTD was not reached during the phase I part of the trial and the RP2D was the maximum tested dose level of KW-2478 175 mg m−2 plus BTZ 1.3 mg m−2 on days 1, 4, 8, and 11 every 3 weeks. This RP2D for the combination of KW-2478 plus BTZ demonstrated clinical activity in patients with relapsed/refractory MM. Overall response rate in the efficacy evaluable population in phase I/II treated at the RP2D (n=79) was 39.2% and CBR was 51.9%. Median PFS was 6.8 months and median duration of response was 5.6 months. The study was originally designed in 2009 based on a reference ORR of 35% for BTZ alone and a target ORR of 50% for the KW-2478 plus BTZ combination. The ORR of 35% for BTZ alone was based on the results of the SUMMIT trial (Richardson et al, 2006). The ORR of 39.2% in the efficacy evaluable population was within the lower 95% CI for the target ORR for the KW-2478 plus BTZ combination.
The safety signals seen in the present study were expected, although the relative contribution of BTZ compared to KW-2478 to particular AEs was difficult to assess. Overall, KW-2478 plus BTZ was well tolerated when administered at the doses and schedule studied. There appeared to be no overlapping toxicities between KW-2478 and BTZ and, in general, the profile of treatment-related grade 3/4 AEs appeared characteristic for that seen with single-agent BTZ therapy in patients with relapsed/refractory MM (Velcade (bortezomib) for Injection, 2009). The most frequent individual grade 3/4 AEs with single-agent BTZ therapy in large-scale phase III clinical trials of patients with relapsed/refractory MM were neutropenia (14.5%) and thrombocytopenia (15.4–29.3%) (Richardson et al, 2005; Orlowski et al, 2007): this compares with 7.4% and 5.3%, respectively, in our study. The combination of KW-2478 with BTZ did not therefore exacerbate the frequency of severe and/or life-threatening neutropenia and thrombocytopenia.
Ocular toxicity has been a concern with respect to the first class of ansamycin Hsp90 inhibitors (Zhou et al, 2013). KW-2478 is a second-generation non-ansamycin Hsp90 inhibitor. No patient developed any significant abnormality on routine ophthalmological examination or fundus autofluorescence imaging. KW-2478 did not raise any ocular safety concerns.
Robust historical data concerning the efficacy of single-agent BTZ in the treatment of relapsed and/or refractory MM have been available from large-scale phase III clinical trials (Richardson et al, 2005; Orlowski et al, 2007). Single-agent BTZ was reported to have an ORR of ∼40% and a median PFS of ∼6 months. These results are similar to those found for the combination of KW-2478 plus BTZ in our current cohort (ORR 39.2%, median PFS 6.7 months). However, none of the patients recruited in the two phase III studies of single-agent BTZ had received prior BTZ therapy, whereas a considerable proportion of the patients (∼40%) in our study had received prior BTZ therapy. Since prior BTZ therapy may compromise response to subsequent BTZ therapy (Mateos et al, 2010), a more valid comparison of efficacy would involve the BTZ-naive subpopulation from our study. BTZ-naive patients in our study had an ORR of 44.2% compared to ∼40% for single-agent BTZ in previous phase III clinical trials. There was therefore insufficient evidence that KW-2478 added to the efficacy of BTZ alone.
The subset analyses of this study revealed two important points. First, the KW-2478 plus BTZ combination regimen showed an ORR of 44.2% for BTZ-naive patients, which is higher than that for BTZ-pretreated patients (33.3%). Also, the KW-2478 plus BTZ combination showed somewhat better responses (CR 7%+VGPR 11.6%) in BTZ-naive patients compared to BTZ-pretreated patients (VGPR 13.9%). These data are consistent with in vitro preclinical studies that showed synergism between KW-2478 and BTZ in BTZ-naive myeloma cell lines (Ishii et al, 2012). It is noteworthy that KW-2478 did not enhance the toxicity of BTZ in the combination regimen. Second, the KW-2478 plus BTZ combination showed an ORR of 45.5% for lenalidomide-naive patients, which appeared higher than that for lenalidomide-pretreated patients (25.0%). This trend is reflected in somewhat better responses in lenalidomide-naive patients (CR 5.5%, VGPR 12.7%, and PR 27.3%) than for lenalidomide-pretreated patients (CR 0%, VGPR 12.5%, and PR 12.5%). This was an unexpected finding from the preclinical standpoint. However, recent evidence that lenalidomide binds to cereblon thus inducing the selective degradation of Ikaros proteins suggests that there might be an overlap for the mechanisms of action for Hsp90 inhibitors and lenalidomide (Lu et al, 2012, 2014). Further studies are needed to clarify this point.
The pharmacokinetics of KW-2478 in combination with BTZ did not show any notable differences compared to those found after single-agent KW-2478 administration (data on file, Kyowa Kirin Pharmaceutical Development, Inc.).
Other Hsp90 inhibitors (tanespimycin and retaspimycin) have shown activity and were well tolerated as single-agent therapy in patients with relapsed/refractory MM in phase I clinical trials (Richardson et al, 2010; Siegel et al, 2011). Tanespimycin plus BTZ showed modest activity in patients with relapsed/refractory MM in a phase II clinical trial (Richardson et al, 2011): among 67 evaluable patients, tanespimycin plus BTZ resulted in an ORR of 14.9% (95% CI: 7.4–25.7%). The efficacy results with KW-2478 plus BTZ in our study appear more favourable, although the study of tanespimycin plus BTZ exclusively recruited who had received ⩾2 prior MM therapies.
The antimyeloma activity of KW-2478 in this phase II study might have been improved by increasing drug exposure. The MTD was not reached using KW-2478 176 mg m−2 on days 1, 4, 8, and 11 every 3 weeks. In the previous phase I study (Yong et al, 2016), KW-2478 175 mg m−2 was administered as a single agent on days 1–5 every 2 weeks. Alternative dosing schedules and the inclusion of dexamethasone may serve to optimise this combination of drugs for antimyeloma therapy.
In conclusion, this phase I/II trial demonstrated that the combination of KW-2478 (175 mg m−2) plus BTZ (1.3 mg m−2) on days 1, 4, 8, and 11 every 3 weeks was well tolerated albeit with modest antimyeloma activity in patients with relapsed/refractory MM. Post hoc subgroup analysis suggests that combination with lenalidomide might be an alternative approach for KW-2478. The good tolerability of this combination would support further exploration of alternate dosing schedules, perhaps in combination with dexamethasone, and indeed combinations of KW-2478 with other antimyeloma agents.
References
Anderson KC, Kyle RA, Rajkumar SV, Stewart AK, Weber D, Richardson P (2008) Clinically relevant end points and new drug approvals for myeloma. Leukemia 22 (22): 231–239.
Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zwiebel J, Wright JJ, Espinoza-Delgado I, Baer MR, Holleran JL, Egorin MJ, Grant S (2009) Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res 15 (16): 5250–5257.
Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 (2009) US Department of Health and Human Services. Available at: http://www.hrc.govt.nz/sites/default/files/CTCAE%20manual%20-%20DMCC.pdf (accessed 25 August 2015).
Durie BGM, Harousseau JL, Miguel JS, Bladé J, Barlogie B, Anderson K, Gertz M, Dimopoulos M, Westin J, Sonneveld P, Ludwig H, Gahrton G, Beksac M, Crowley J, Belch A, Boccadaro M, Cavo M, Turesson I, Joshua D, Vesole D, Kyle R, Alexanian R, Tricot G, Attal M, Merlini G, Powles R, Richardson P, Shimizu K, Tosi P, Morgan G, Rajkumar SV International Myeloma Working Group (2006) International uniform response criteria for multiple myeloma. Leukemia 20 (9): 1467–1473.
Ishii T, Seiki T, Nakashima T, Juliger S, Maharaj L, Soga S, Akinaga S, Cavenagh J, Joel S, Shiotsu Y (2012) Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J 2 (4): e68.
Juliger S, Nakashima T, Maharaj L, Ishii T, Nakagawa H, Kanda Y, Oakervee H, Cavenagh J, Akinaga S, Shiotsu Y, Joel SP (2008) A novel heat shock protein (Hsp) 90 inhibitor KW-2478 shows activity in B-cell malignancies in vitro and in vivo. Blood 112 (11): abstract 1625.
Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, Kapoor P, Dingli D, Hayman SR, Leung N, Lust J, McCurdy A, Russell SJ, Zeldenrust SR, Kyle RA, Rajkumar SV (2014) Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 28 (5): 1122–1128.
Kyle RA, Rajkumar SV (2009) Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 23 (1): 3–9.
Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin Jr WG (2014) The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343 (6168): 305–309.
Lu X, Xiao L, Wang L, Ruden DM (2012) Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem Pharmacol 83 (8): 995–1004.
Mateos MV, Richardson PG, Schlad R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Esseltine DL, Liu K, Cakana A, van de Velde H, San Miguel JF (2010) Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in patients with previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol 28 (13): 2259–2266.
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R, Munshi NC, Richardson PG, Hideshima T, Chauhan D, Gu X, Bailey C, Joseph M, Libermann TA, Rosen NS, Anderson KC (2006) Antimyeloma activity of heat shock protein-90 inhibition. Blood 107 (3): 1092–1100.
Nakashima T, Ishii T, Tagaya H, Seike T, Nakagawa H, Kanda Y, Akinaga S, Soga S, Shiotsu Y (2010) New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin Cancer Res 16 (10): 2792–2802.
Orlowski RZ, Nagler A, Sonneveld P, Bladé J, Hajek R, Spencer A, San Miguel J, Robak T, Dmoszynska A, Horvath N, Spicka I, Sutherland HJ, Suvorov AN, Zhuang SH, Parekh T, Xiu L, Yuan Z, Rackoff W, Harousseau JL (2007) Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 25 (25): 3892–3901.
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin DH, Rajkumar SV, Srkalovic G, Alsina M, Anderson KC (2006) Extended follow-up of a phase II trial in relapsed, refractory multiple myeloma: final time-to-event results from the SUMMIT trial. Cancer 106 (6): 1316–1319.
Richardson PG, Chanan-Khan AA, Lonial S, Krishnan AY, Carroll MP, Alsina M, Albitar M, Berman D, Messina M, Anderson KC (2011) Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol 153 (6): 729–740.
Richardson PG, Channan-Khan AA, Alsina M, Albitar M, Berman D, Messina M, Mitsiades CS, Anderson KC (2010) Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase I dose-escalation study. Br J Haematol 150 (4): 438–445.
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352 (24): 2487–2498.
Sharp S, Workman P (2006) Inhibitors of the HSP90 molecular chaperone: current status. Adv Cancer Res 95: 323–348.
Siegel D, Jagannath S, Vesole DH, Borello I, Mazumder A, Mitsiades C, Goddard J, Dunbar J, Normant E, Adams J, Grayzel D, Anderson KC, Richardson P (2011) A phase I study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 52 (12): 2308–2315.
Siegel R, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66 (1): 7–30.
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10 (1): 1–10.
Sinha S, Lacy M, Mikhael J, Hayman S, Buadi F, Detweiler-Short K, Dispenzieri A, Gertz M, Dingli D, Rajkumar SV, Kumar SK (2012) Response to salvage therapies and outcome in patients with multiple myeloma relapsing after pomalidomide therapy. Leukemia 26 (4): 839–841.
Soga S, Akinaga S, Shiotsu Y (2013) Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr Pharm Des 19 (3): 366–376.
Velcade (bortezomib) for Injection (2009) Highlights of prescribing information. Update on 30 December 2009. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021602s019s020lbl.pdf (accessed 25 August 2015).
Whitesell L, Santagata S, Lin NU (2012) Inhibiting HSP90 to treat cancer: a strategy in evolution. Curr Mol Med 12 (9): 1108–1124.
Yong K, Cavet J, Johnson P, Morgan G, Williams C, Nakashima D, Akinaga S, Oakerveee H, Cavenagh J (2016) Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br J Cancer 114 (1): 7–13.
Zhou D, Liu Y, Ye J, Ying W, Ogawa LS, Inoue T, Tatsuta N, Wada Y, Koya K, Huang Q, Bates RC, Sonderfan AJ (2013) A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol Appl Pharmacol 273 (2): 401–409.
Acknowledgements
Kyowa Kirin Pharmaceutical Development, Inc. (Princeton, NJ, USA) (KKD) funded the study. Prior to the start of the study, KKD agreed that the authors had full rights to submit the manuscript for publication, KKD approval of the manuscript was not required, and publication of the manuscript was not contingent upon the approval of KKD. The authors acknowledge Peter Todd, PhD (Tajut Ltd., Kaiapoi, New Zealand) for editorial assistance in the preparation of this manuscript, for which he received financial support from KKD. We also thank Takahiro Ito, Li Liu, PhD, and Rocco Ballerini, PhD (KKD) for statistical support, and the study investigators for their invaluable contribution.
Author contributions
All authors contributed equally to data analysis and interpretation, figure creation, and writing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MK is an employee of Kyowa Kirin Pharmaceutical Development, Inc. (NJ, USA). NS, DN, and SA are employees of Kyowa Hakko Kirin Co. Ltd. KY receives support from the NIHR Biomedical Research Centre (Guy’s and St Thomas’ NHS Foundation Trust and King’s College, London, UK). The remaining authors declare no competing financial interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Cavenagh, J., Oakervee, H., Baetiong-Caguioa, P. et al. A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br J Cancer 117, 1295–1302 (2017). https://doi.org/10.1038/bjc.2017.302
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.302
