
Abstract
There is now a resurgent interest in the role of mitochondria in cancer. Long considered controversial or outright unimportant, mitochondrial biology is now increasingly recognised as an important tumour driver. The underlying mechanisms remain to be fully elucidated. But recent studies have uncovered a complex landscape where reprogramming of mitochondrial homoeostasis, including organelle dynamics, metabolic output, apoptosis control and redox status converge to promote tumour adaptation to an unfavourable microenvironment and inject new traits of aggressive disease. In particular, mechanisms of subcellular mitochondrial trafficking have unexpectedly emerged as central regulators of metastatic competence in disparate tumours. Some of these pathways are druggable, opening fresh therapeutic opportunities for advanced and disseminated disease.
Similar content being viewed by others

Main
Among the uniqueness of cancer is a pervasive reprogramming of cellular metabolism, which shifts from oxidative bioenergetics in mitochondria to a process of glycolysis even in the presence of oxygen. As one of the hallmarks of cancer, this ‘Warburg effect’ (Vander Heiden et al, 2009), produces biomass for malignant expansion, introduces new cancer traits and reprograms accessory cells in the microenvironment (Vander Heiden et al, 2009). At least in certain tumours, a glycolysis signature is a strong indicator of worse patient outcome. Against this backdrop, it is not surprising that mitochondrial oxidative metabolism has been viewed as unimportant in cancer, or even dubbed as a ‘tumour suppressor’, at least in certain malignancies.
Recent studies, however, have changed that perspective. In fact, work from several groups have now uncovered a far more complex landscape, where mitochondrial homoeostasis in general and, more specifically, oxidative phosphorylation critically contribute to tumour fitness, promote adaptation to an ever-changing and mostly unfavourable microenvironment, and inject new traits of aggressive disease. For instance, oxidative phosphorylation still accounts for a large portion of ATP generated in tumours (Moreno-Sanchez et al, 2014), supports malignant growth in patients (Sellers et al, 2015) and enables key cancer traits of ‘stemness’ (Janiszewska et al, 2012), drug resistance (Roesch et al, 2013) and metastatic competence (LeBleu et al, 2014).
The present mini-review is not intended to revisit the broad array of mitochondrial functions implicated in cancer. Excellent contributions on this topic have recently appeared in the literature (Vyas et al, 2016; Zong et al, 2016). Instead, we will focus on an emerging role of subcellular mitochondrial trafficking as an unexpected requirement of tumour cell motility, invasion and metastasis. This process and its underlying mechanisms reflect mitochondrial reprogramming to microenvironment stress, and may provide new therapeutic targets in advanced disease (Caino et al, 2013).
Subcellular mitochondria trafficking as a novel requirement of tumour cell motility and metastasis
Most cancer death are due to metastatic disease: the successful dissemination of tumour cells to distant organs. Available therapeutic options in these settings are scarce, producing only palliative, short-lived clinical responses, if at all. Considerable progress has been made in our mechanistic understanding of the metastatic process. However, a basic question of how tumours that utilise an inefficient glycolytic metabolism (Vander Heiden et al, 2009) accomplish among the most energy-intensive processes of membrane-cytoskeletal dynamics, chemotaxis and invasion across basement membrane(s) (Roussos et al, 2011) has remained unanswered.
Recent studies have provided a unique perspective to this question. Exposure of tumour cells to small molecule inhibitors of the phosphatidylinositol-3-kinase (PI3K), a key ‘cancer’ gene and therapeutic target in humans (Fruman and Rommel, 2014), was found to upregulate multiple signalling pathways of cell motility, invasion and metastasis (Caino et al, 2015). These results were confirmed experimentally, as PI3K inhibitors dramatically stimulated membrane-cytoskeletal dynamics, tumour chemotaxis and invasion across basement membranes or in 3D spheroids (Caino et al, 2015). Such a paradoxical response to molecular therapy, where treated cells actually acquire more malignant traits, is not without precedent. For instance, angiogenesis inhibitors (Ebos et al, 2009) or anti-Hsp90 therapy (Yano et al, 2008) have also been associated with increased metastatic propensity in tumour models, in vivo. What was unique in the tumour response to PI3K inhibitors was a dynamic behaviour of mitochondria, which migrated from their polarised and perinuclear localisation to the cortical cytoskeleton of treated cells (Caino et al, 2015), accumulating in physical proximity of focal adhesion complexes, which are effectors of cell motility (Roussos et al, 2011). Functionally, these cortical mitochondria supported membrane lamellipodia dynamics, actin cytoskeleton remodelling and phosphorylation of cell motility kinases, resulting in increased tumour cell motility and invasion (Caino et al, 2015). This pathway was not an oddity of PI3K therapy: chemotactic stimuli produced the same response, repositioning mitochondria from their perinuclear localisation to the cell periphery to support invasion and metastasis in mouse models (Rivadeneira et al, 2015). One requirement of the pathway was that mitochondria had to be energetically active for successful subcellular trafficking. Tumour cells with poisoned mitochondria or where oxidative phosphorylation was pharmacologically inhibited failed to reposition mitochondria to the cortical cytoskeleton, and had lost the ability to migrate and invade across basement membranes (Caino et al, 2015; Rivadeneira et al, 2015). Independent studies validated the model of mitochondrial trafficking as a requisite of tumour cell movements (Jung et al, 2016), and linked mitochondrial infiltration of polarised lamellipodia protrusions to increased ‘regional’ oxidative metabolism to fuel tumour cell movements (Cunniff et al, 2016).
The concept that mitochondria are highly motile organelles is not new. In fact, we know that mitochondria actively travel along the microtubule network in neurons and accumulate at sites of high energy demands, such as synapses and growth cones (Birsa et al, 2013). We also know that this pathway relies on an elaborate mitochondrial-cytoskeletal machinery of cellular motors, adapter proteins and GTPases that controls both anterograde (from nuclei to periphery) and retrograde (from periphery to nuclei) mitochondrial movements (Sheng, 2014). Although originally considered ‘neuronal specific’, some aspects of this pathway are clearly at work in other cell types. Consistent with recent findings (Caino et al, 2015; Rivadeneira et al, 2015; Jung et al, 2016), mitochondria have been shown to redistribute along the actin and tubulin networks in migrating lymphocytes (Campello et al, 2006), as well as tumour cells (Mills et al, 2016), and localise to membrane protrusions implicated in directional cell movements in both cell types (Desai et al, 2013; Morlino et al, 2014). Some of the signalling requirements of this pathway have also come into better focus. For instance, heightened activity of the energy sensor, AMPK has been associated with increased mitochondrial infiltration of leading edge lamellipodia, resulting in localised increase in mitochondrial mass and ‘regional’ ATP production (Cunniff et al, 2016). So, is a neuronal machinery of mitochondrial trafficking exploited in cancer to control metastatic competence, one of the most deleterious traits of progressive disease?
Exploitation of a neuronal network of mitochondrial trafficking for metastasis
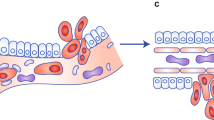
To answer this question and identify novel regulators of mitochondrial regulated tumour cell invasion, a genome-wide short hairpin RNA screen was recently carried out. Unexpectedly, one of the top hits in the screen was syntaphilin (SNPH) (Caino et al, 2016), a molecule that halts mitochondrial movements in neurons at subcellular sites of high energy demands (Sheng, 2014). Although originally characterised as ‘neuronal specific’, SNPH was in fact expressed in multiple non-neuronal tissues, including cancer (Caino et al, 2016). Functionally, silencing of SNPH in tumour cells was sufficient, in the absence of other stimuli, to cause exaggerated mitochondrial repositioning to the cortical cytoskeleton (Figure 1), increased focal adhesion complex dynamics and heightened tumour cell motility and invasion (Caino et al, 2016). As in neurons, this was due to the ability of SNPH to inhibit mitochondrial movements in tumour cells, decreasing the time that organelles spend in motion, or processivity, compared to stationary mitochondria. These findings had a clear disease-relevance. Analysis of genomic databases as well as primary patient cohorts showed that SNPH became downregulated or even lost during disease progression, correlating with worse patient outcome (Caino et al, 2016). Conversely, re-introduction of SNPH in invasive tumour cells was sufficient to reduce metastatic dissemination in a mouse model, in vivo. So, does this mean that other effectors of ‘neuronal’ mitochondrial trafficking are equally exploited in cancer (Sheng, 2014)? And does SNPH represent a novel class of ‘metastasis suppressors’, shutting off the energy requirements of invasive tumours? More work is required to reach these conclusions. Key questions of what controls the extraneuronal expression of SNPH, how is endogenous SNPH downregulated in advanced tumours, and whether other ‘positive’ regulators of ‘neuronal’ mitochondrial trafficking become upregulated in cancer remain to be experimentally demonstrated. However, it is intriguing that atypical mitochondrial GTPases, Miro-1 or -2, which support mitochondrial trafficking in neurons (Birsa et al, 2013) become prominently upregulated in disparate cancers (Caino et al, 2016), and have been implicated in mitochondrial trafficking (Mills et al, 2016) and directional cell movements in tumour cells (Desai et al, 2013).

Mitochondrial trafficking to the cortical cytoskeleton. Prostate adenocarcinoma PC3 cells were transfected with control non-targeting siRNA or SNPH-directed siRNA and analysed for mitochondrial repositioning to the cortical cytoskeleton by confocal microscopy. A 3D isosurface rendering of mitochondria was obtained by staining fixed cells with an antibody to mitochondria (green), followed by Alexa Fluor 488-conjugated secondary antibody. DNA (blue) was stained with DAPI.
Is it only about ‘regional’ ATP production?
Although bioenergetics is critical for cellular functions, mitochondria do more than producing ATP, and it is possible that these additional functions may also contribute to tumour chemotaxis. For instance, changes in SNPH levels in tumour cells have been associated with increased cycles of mitochondrial fusion and fission, i.e., dynamics (Caino et al, 2016). A key regulator of organelle shape and size, recent work has suggested that mitochondrial dynamics is broadly exploited for a host of tumour traits, such as proliferation, invasion, redox balance and ‘stemness’ (Senft and Ronai, 2016). In addition, changes in mitochondrial dynamics have been linked to tumour cell migration and invasion (Zhao et al, 2013; Ferreira-da-Silva et al, 2015). In turn, mitochondrial dynamics couples to mitophagy, a process of organelle quality control that clears damaged or energetically impaired mitochondria (Youle and Narendra, 2011). The role of mitophagy in cancer remains poorly understood, but recent findings suggest an important tumour suppressor function, involving both Parkin-dependent and -independent mechanisms (Bernardini et al, 2017), and there is initial evidence that mitophagy defects may favour disease progression (Chourasia et al, 2015). Whether mitochondrial dynamics (Senft and Ronai, 2016) and/or mitophagy (Youle and Narendra, 2011) contribute to organelle trafficking and tumour cell motility remain to be established. However, it is intriguing that deletion of SNPH significantly increases cycles of both mitochondrial fusion and fission in tumour cells, and that changes in organelle size influence the velocity of mitochondrial repositioning to the cortical cytoskeleton (Caino et al, 2016).
Mitochondrial trafficking as part of tumour plasticity
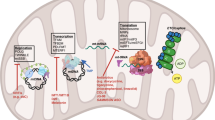
A straightforward interpretation of the data briefly summarised above is that mitochondrial repositioning to the cortical cytoskeleton provides an efficient, ‘regional’ energy source to fuel membrane-cytoskeletal dynamics and lamellipodia formation (Cunniff et al, 2016) that are essential for cell movements (Roussos et al, 2011). Per this model, it may not be the ‘how much’ ATP is generated, but rather the ‘when’ and ‘where’ that are important to support tumour cell invasion, and, therefore, metastasis. But what is the biological context for this response? The fact that molecular therapy stimulates mitochondrial trafficking and cell invasion (Caino et al, 2015) suggests that ‘stress’ conditions in the microenvironment may function as central drivers of this pathway (Gillies et al, 2012). This is consistent with other data that mitochondrial metabolism was required to sustain tumour cell invasion and metastasis under restrictive conditions of the microenvironment, such as glucose or amino acid depletion (Caino et al, 2013). Taken together, in an as yet fanciful scenario, it could be hypothesised that mitochondrial trafficking to the cortical cytoskeleton and the ensuing heightened cell motility constitute an adaptive response to ‘stress’, enabling tumour cells to ‘escape’ from an unfavourable microenvironment (Caino et al, 2015), no longer able to support their expansion (Gillies et al, 2012). In this context, there is evidence that mitochondrial reprogramming contributes to multiple aspects of tumour adaptation to environmental ‘stress’ stimuli (Figure 2). Published work from several groups suggests that this involves flexible titration of an anti-apoptotic threshold, control of ‘retrograde’ gene expression signalling, modulation of multiple metabolic pathways and redox status and sustained tumour cell proliferation even in noxious conditions (Vyas et al, 2016; Zong et al, 2016; Figure 2). These mechanisms help tumours cope with a rapidly evolving and often unfavourable microenvironment, while conferring new adaptive traits, such as drug resistance and metastatic competence that are key for disease progression in the clinic.

Mitochondrial reprogramming contributes to tumour plasticity. Stress stimuli of the tumour microenvironment modulate mitochondrial functions in apoptosis inhibition, cell motility and invasion, proliferation, ‘retrograde’ gene expression, metabolic reprogramming and organelle dynamics, including subcellular trafficking. In turn, these mitochondrial functions expand tumour diversity, buffer environmental stress stimuli and introduce new traits of aggressive disease, including drug resistance and metastatic competence.
Translational implications – mitochondria-directed cancer therapy?
The impressive technological advances of the past decade have changed the paradigm in cancer therapy and ushered the concept (and hope) of personalised, or precision medicine: that if we sequence the genome of every patient, we will successfully match genetic alterations to specific inhibitor(s) and bring about durable clinical responses, even cures (Carneiro et al, 2016). Unfortunately, the reality in the clinic proved different, as all molecularly targeted agents so far produce short-lived responses, quickly supplanted by the emergence of drug-resistant and metastatic disease (Tannock and Hickman, 2016). The data reviewed above point to mitochondrial reprogramming (Vyas et al, 2016; Zong et al, 2016) in response to environmental ‘stress’ (Gillies et al, 2012) as a novel, key determinant of disease progression (Figure 2). We now know that some of these pathways are druggable, with manageable or minimal toxicity for normal tissues (Fulda et al, 2010). Differently from the premise of personalised medicine, targeting mitochondrial reprogramming is expected to simultaneously disable multiple, fundamental mechanisms of disease progression irrespective of driver mutation(s) (Figure 2). This approach may be applicable across a broad spectrum of genetically heterogeneous tumours, and has the potential to bypass the powerful mechanisms of tumour selection that are at work to confer drug resistance in face of single oncogene targeting. Clearly, much work remains to be done, and it is not a foregone conclusion that disabling an entire organelle system, that is, mitochondria, may be sufficiently tolerated in humans. On the other hand, initial proof-of-concept studies have suggested that pharmacological disruption of mitochondrial protein folding quality control (Chae et al, 2013; Cole et al, 2015) or inhibition of oxidative metabolism (Rivadeneira et al, 2015) is feasible in different tumour models, causes irreversible collapse of multiple mitochondrial functions and delivers promising anticancer activity with manageable toxicity, in vivo.
References
Bernardini JP, Lazarou M, Dewson G (2017) Parkin and mitophagy in cancer. Oncogene 36 (10): 1315–1327.
Birsa N, Norkett R, Higgs N, Lopez-Domenech G, Kittler JT (2013) Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem Soc Trans 41 (6): 1525–1531.
Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O’Connell M, Jernigan D, Fatatis A, Languino LR, Bosari S, Altieri DC (2013) Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest 123 (7): 2907–2920.
Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, Webster MR, Weeraratna AT, Bosari S, Languino LR, Altieri DC (2015) PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA 112 (28): 8638–8643.
Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, Hayden JE, Vaira V, Morotti A, Ferrero S, Bosari S, Gabrilovich DI, Languino LR, Cohen AR, Altieri DC (2016) A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun 7: 13730.
Campello S, Lacalle RA, Bettella M, Manes S, Scorrano L, Viola A (2006) Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med 203 (13): 2879–2886.
Carneiro BA, Costa R, Taxter T, Chandra S, Chae YK, Cristofanilli M, Giles FJ (2016) Is personalized medicine here? Oncology (Williston Park) 30 (4): 293–303, 307.
Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, Powers JF, Tischler AS, Pacak K, Fliedner S, Michalek RD, Karoly ED, Wallace DC, Languino LR, Speicher DW, Altieri DC (2013) Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun 4: 2139.
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16 (9): 1145–1163.
Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, Restall I, Wang X, Jeyaraju DV, Sukhai MA, Prabha S, Bashir S, Ramakrishnan A, Leung E, Qia YH, Zhang N, Combes KR, Ketela T, Lin F, Houry WA, Aman A, Al-Awar R, Zheng W, Wienholds E, Xu CJ, Dick J, Wang JC, Moffat J, Minden MD, Eaves CJ, Bader GD, Hao Z, Kornblau SM, Raught B, Schimmer AD (2015) Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27 (6): 864–876.
Cunniff B, McKenzie AJ, Heintz NH, Howe AK (2016) AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27 (17): 2662–2674.
Desai SP, Bhatia SN, Toner M, Irimia D (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 104 (9): 2077–2088.
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15 (3): 232–239.
Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S (2015) Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One 10 (3): e0122308.
Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13 (2): 140–156.
Fulda S, Galluzzi L, Kroemer G (2010) Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 9 (6): 447–464.
Gillies RJ, Verduzco D, Gatenby RA (2012) Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 12 (7): 487–493.
Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I (2012) Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev 26 (17): 1926–1944.
Jung JU, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, Sitcheran R (2016) NIK/MAP3K14 regulates mitochondrial dynamics and trafficking to promote cell invasion. Curr Biol 26 (24): 3288–3302.
LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R (2014) PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16 (10): 992–1003.
Mills KM, Brocardo MG, Henderson BR (2016) APC binds the Miro/Milton motor complex to stimulate transport of mitochondria to the plasma membrane. Mol Biol Cell 27 (3): 466–482.
Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ, Rodriguez-Enriquez S (2014) Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol 50: 10–23.
Morlino G, Barreiro O, Baixauli F, Robles-Valero J, Gonzalez-Granado JM, Villa-Bellosta R, Cuenca J, Sanchez-Sorzano CO, Veiga E, Martin-Cofreces NB, Sanchez-Madrid F (2014) Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Mol Cell Biol 34 (8): 1412–1426.
Rivadeneira DB, Caino MC, Seo JH, Angelin A, Wallace DC, Languino LR, Altieri DC (2015) Survivin promotes oxidative phosphorylation, subcellular mitochondrial repositioning, and tumor cell invasion. Sci Signal 8 (389): ra80.
Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23 (6): 811–825.
Roussos ET, Condeelis JS, Patsialou A (2011) Chemotaxis in cancer. Nat Rev Cancer 11 (8): 573–587.
Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, Lane AN, Fan TW (2015) Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125 (2): 687–698.
Senft D, Ronai ZA (2016) Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol 39: 43–52.
Sheng ZH (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol 204 (7): 1087–1098.
Tannock IF, Hickman JA (2016) Limits to personalized cancer medicine. N Engl J Med 375 (13): 1289–1294.
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324 (5930): 1029–1033.
Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and cancer. Cell 166 (3): 555–566.
Yano A, Tsutsumi S, Soga S, Lee MJ, Trepel J, Osada H, Neckers L (2008) Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostate carcinoma cells in bone. Proc Natl Acad Sci USA 105 (40): 15541–15546.
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12 (1): 9–14.
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32 (40): 4814–4824.
Zong WX, Rabinowitz JD, White E (2016) Mitochondria and cancer. Mol Cell 61 (5): 667–676.
Acknowledgements
I thank all members of the Altieri laboratory, who over the years have steadily and assiduously contributed to this line of research. Dr Cecilia M Caino prepared the 3D isosurface rendering of mitochondrial trafficking in response to SNPH knockdown shown in Figure 1. This work was supported by National Institutes of Health (NIH) grants P01 CA140043, R01 CA78810 and CA190027, the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No. W81XWH-13-1-0193, and a Challenge Award from the Prostate Cancer Foundation (PCF). Support for Core Facilities utilised in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Altieri, D. Mitochondria on the move: emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br J Cancer 117, 301–305 (2017). https://doi.org/10.1038/bjc.2017.201
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.201
Keywords
This article is cited by
-
Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management
Journal of Hematology & Oncology (2022)
-
PGC-1α induced mitochondrial biogenesis in stromal cells underpins mitochondrial transfer to melanoma
British Journal of Cancer (2022)
-
Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion
Nature Communications (2020)
-
Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells
Oncogene (2019)
-
Depletion of mitochondrial protease OMA1 alters proliferative properties and promotes metastatic growth of breast cancer cells
Scientific Reports (2019)
