
Abstract
Chronic lymphocytic leukaemia (CLL) is a molecularly heterogeneous disease as revealed by recent genomic studies. Among genetic lesions that are recurrent in CLL, few clinically validated prognostic markers, such as TP53 mutations and 17p deletion, are available for the use in clinical practice to guide treatment decisions. Recently, several novel molecular markers have been identified in CLL. Though these mutations have not yet gained the qualification of predictive factors for treatment tailoring, they have shown to be promising to refine the prognostic stratification of patients. The introduction of targeted drugs is changing the genetics of CLL, and has disclosed the acquisition of previously unexpected drug resistant mutations in signalling pathway genes. Ultra-deep next generation sequencing has allowed to reach deep levels of resolution of the genetic portrait of CLL providing a precise definition of its subclonal genetic architecture. This approach has shown that small subclones harbouring drug resistant mutations anticipate the development of a chemorefractory phenotype. Here we review the recent advances in the definition of the genomic landscape of CLL and the ongoing research to characterise the clinical implications of old and new molecular lesions in the setting of both conventional chemo-immunotherapy and targeted drugs.
Similar content being viewed by others

Main
Chronic lymphocytic leukaemia (CLL) is an indolent B-cell malignancy characterised by the accumulation of mature CD5+/CD19+/CD23+ lymphocytes with weak surface expression of a monoclonal immunoglobulin (Müller-Hermelink et al, 2008). The clinical course of CLL ranges from a very indolent condition, with a nearly normal life expectancy, to rapidly progressive leading to early death. Asymptomatic patients are managed with watch and wait until the development of symptoms that result from cytopenias, adenopathy, or splenomegaly, as outlined by the 2008 International Workshop on CLL (Hallek et al, 2008).
An extensive array of effective options are available when treatment is required, though the disease still remains incurable. Combination chemo-immunotherapy with fludarabine, cyclophosphamide and rituximab (FCR) is the most effective regimen for young and fit patients, as it yields excellent long-term results. As CLL often affects elderly individuals, more tolerable therapeutic approaches have been successfully applied, such as chemo-immunotherapy combining chlorambucil with an anti-CD20 antibody (rituximab, obinutuzumab, ofatumumab), and bendamustine-based regimens (Ghia and Hallek, 2014). Most recently, therapies targeting the B-cell-receptor (BCR) signalling pathway, such as ibrutinib and idelalisib, have shown high-response rates and tolerability (Byrd et al, 2014).
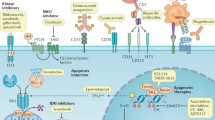
CLL pathogenesis might be viewed as a cooperation between microenvironmental mechanisms and tumour genetics. The chronic lymphocytic leukaemia (CLL) genome carries ∼2000 molecular lesions, including ∼20 non-synonymous mutations and ∼5 gross structural abnormalities Puente et al, 2015). At variance with other indolent B-cell lymphoproliferative disorders, where one predominant gene is molecularly altered in virtually all cases, CLL has a heterogeneous genetic profile and no unifying lesions have been so far identified. Recurrent balanced translocations, which commonly occur in many types of mature B-cell tumours, are extremely rare in CLL, being limited to the t(14;18) translocation involving the BCL2 genes in ∼2% of unselected cases (Cavazzini et al, 2008). Few molecular alterations recur at a frequency >5% in CLL, whereas a large number of biologically and clinically uncharacterised genes are mutated at lower frequencies (Landau et al, 2015; Puente et al, 2015). The most recurrent molecular lesions of CLL point to the deregulation of cellular programs of clinical importance, namely: (i) apoptosis; (ii) DNA damage response; and (iii) cell signalling (Figure 1). Though the pathogenic implications of CLL genetic lesions has not been fully characterised in most instances, their usefulness as prognostic biomarkers (i.e. biomarkers that provide information on the likely outcome of CLL), or predictive biomarkers (i.e. biomarkers that provide information on the likely benefit from a specific CLL treatment) has been investigated to a certain extent.

Significantly mutated genes and pathways in CLL. Cellular programs that are affected by the most recurrent molecular lesions are represented. Boxes show the genes that are recurrently mutated in each cellular programme and the clinical implications of gene mutations. Arrows indicate the positive or negative prognostic impact of the genetic lesions.
Apoptotic Checkpoint Deregulation
Deletion 13q14 is the most frequent genetic lesion of CLL occurring in 50–60% of cases. The minimal deleted region on 13q14 contains the miR15A and miR16A microRNAs (Calin et al, 2002). In normal cells, miR15A and miR16A inhibit the expression of multiple genes, including BCL2, the cyclins CCND1 and CCND3, and cyclin-dependent kinase 6 (CDK6) (Klein et al, 2010). Deletion of miR15A and miR16A abrogates this inhibitory effect, favours the constitutive survival and cycling of tumour B-cells, and causes CLL in mouse models. In a relevant fraction (∼25%) of CLL patients, deletion of 13q14 occurs in the absence of any concomitant driver genetic lesion. Patients harbouring solely 13q14 deletion have an excellent clinical outcome with a progression rate of <1% per year and an expected survival only slightly lower than that of the general population (Rossi et al, 2013). BCL2 is one of the genes that are upregulated in CLL as a consequence of miR15A/miR16A deletion. Consistent with the central contribution of BCL2 activation in the pathogenesis of CLL, selective inhibition of BCL2 through the BH3 mimetic venetoclax results into high-response rates in relapsed or refractory patients, including those harbouring high-risk genetic abnormalities (Roberts et al, 2016).
DNA Damage Response Deregulation
TP53 codes for a central regulator of the DNA-damage response pathway and, when functional, triggers CLL cell apoptosis in response to chemotherapy. TP53 may be disrupted in CLL by deletions, mutations or a combination of both. Mutations represent the most frequent form of TP53 inactivation in CLL and are frequently (∼70% of the cases) accompanied by the loss of the second allele through 17p13 deletion (Rossi et al, 2009). The frequency of mutations lacking 17p13 deletion varies among different studies depending on patient cohort and the methodology used, but in general it represents ∼30% of all TP53 defects, whereas sole 17p13 deletion with the absence of TP53 mutation are less frequent (∼10% of all TP53 defects). The high proportion of TP53 mutations in the absence of 17p13 deletion may be in part attributed to the presence of two TP53 mutations on individual alleles in CLL cells. In some patients, neutral loss of heterozygosity (uniparental disomy) of the TP53 locus copy number was described, which results in duplication of the mutant allele. The direct clinical implication of these molecular observations is that, in order to perform a comprehensive evaluation of the TP53 gene status in CLL, it is recommended to assess both the presence of chromosome 17p13 deletion by FISH, which is the more sensitive approach for deletion assessment, and of TP53 mutations by gene sequencing (Pospisilova et al, 2012). At diagnosis, the incidence of TP53 abnormalities has been reported to be 4–8%. As disease progresses, the incidence rises to 10–12% at the time of first line treatment, 40% in fludarabine-refractory CLL, and 50–60% in Richter syndrome.
The clinical importance of TP53 abnormalities in CLL is tightly linked to the poor prognosis marked by this genetic lesion and its close association with chemorefractoriness, as documented by a number of observational studies and prospective trials led in both the chemotherapy and immunochemotherapy era. Among newly diagnosed CLL, patients harbouring TP53 abnormalities have the worst outcome, with an estimated median overall survival (OS) of 3–5 years (∼30% of cases are alive at 10 years, accounting for a ∼70% reduction of the expected survival compared to the general population) (Rossi et al, 2013). However, in situations without treatment indication, TP53 abnormalities should not be routinely tested, as they might turn a ‘watch and wait’ strategy into a ‘watch and worry’ situation for the patient without any immediate therapeutic consequences. Indeed, it is important to stress that there is a small subgroup of patients with 17p13 deletion (and mostly mutated IGHV genes) who may exhibit stable disease for years without treatment indications.
The outcome of patients with TP53 abnormalities and need for treatment is poor if treated with chemo/chemoimmuno-therapy, as they will very rarely achieve complete response. Such poor response to chemo+/−immunotherapy translates into an estimated progression free survival of 1 year or less and an OS in the range of 2–3 years from the time of front-line treatment (Zenz et al, 2010; Stilgenbauer et al, 2014). The Bruton tyrosine kinase (BTK) inhibitor ibrutinib as a single agent or combined to rituximab induces a response rate >80%, and a progression free survival >80% after 2 years of follow-up in untreated patients with TP53 abnormalities (Farooqui et al, 2015). Similar proportions of response and progression free survival rates are observed with the PI3Kδ inhibitor idelalisib combined to rituximab (Furman et al, 2014). Therefore, it is recommended that patients with TP53 deletion/mutation are treated with novel inhibitors (ibrutinib, idelalisib and rituximab) (Eichhorst et al, 2015). Although these results appear significantly better than every previous historical control in CLL with TP53 abnormalities, ibrutinib and idelalisib-rituximab per se do not promise long-lasting remissions, especially in the setting of relapsed disease. Indeed, TP53 defects predispose to the development of complex karyotype, which in turn marks genetic instability and favors the accumulation of resistance mutations. Consistently, complex karyotype is a powerful predictor of outcomes for ibrutinib-treated patients (Thompson et al, 2015). Therefore, allogeneic stem cell transplantation should still be offered and discussed in patients with CLL harbouring TP53 defects, a complex karyotype, sufficient physical fitness and an available donor (Dreger et al, 2014).
Given their value as predictive biomarkers of resistance to chemo+/−immunotherapy, current guidelines recommend to test 17p13 deletion and TP53 mutations in CLL patients requiring therapy (Hallek et al, 2008; Pospisilova et al, 2012; Eichhorst et al, 2015; Zelenetz et al, 2015). Because leukaemic clones may evolve, FISH for 17p13 deletion and TP53 mutation analyses should be repeated at each disease progression requiring treatment. Sanger sequencing is the currently recommended approach for TP53 mutation analysis. However, due to its limited sensitivity, conventional Sanger sequencing misclassifies as wild type those CLL cases harbouring TP53 mutations of low-clonal abundance (<10% of the alleles). Such small TP53 mutated subclones occur in a significant fraction of CLL, have the same unfavourable prognostic impact as clonal TP53 defects, and anticipate the development of a chemorefractory phenotype among CLL patients requiring treatment (Landau et al, 2013; Rossi et al, 2014; Landau et al, 2015). Thanks to its high sensitivity (down to 1–0.1%), deep next generation sequencing is capable of detecting these minor, but clinically relevant, TP53 mutated subclones. Therefore, deep next generation sequencing should be considered as a useful tool for a comprehensive assessment of TP53 disruption in CLL.
The ATM gene encodes a nuclear serine/threonine kinase whose activity is induced by chromosomal double-strand breaks that arise endogenously or after exposure to DNA-damaging agents, including chemotherapeutic drugs. ATM protects the integrity of the genome by regulating the cell-cycle arrest at G1/S and G2/M to prevent processing of damaged DNA, and by activating the DNA-repair pathways or, alternatively, inducing apoptosis if the DNA damage cannot be repaired. As for TP53, the ATM gene is inactivated in CLL by both deletion and/or somatic mutations, which result in impaired DNA damage responses. Deletion of 11q22-23 always includes ATM and occurs in <10% newly diagnosed CLL, whereas its prevalence rises to ∼20% at the time of first treatment. Deletion 11q22-23 co-occurs with ATM mutations in 30–40% of patients. ATM mutations generally consist in a mixture of missense substitutions distributed across the ATM coding sequence, with no clear hotspots, and have been observed in ∼10–15% of newly diagnosed patients and in ∼15% of progressive CLL requiring first treatment. By combining mutations and deletions, genetic lesions of ATM occur in ∼20% of diagnostic samples of CLL and in ∼35% cases requiring first treatment (Skowronska et al, 2012).
From a clinical perspective, the presence of ATM deletion at the time of CLL presentation identifies a group of patients with intermediate-risk disease (∼40% of cases are alive at 10 years, accounting for a ∼50% reduction of the expected survival compared to the general population) (Rossi et al, 2013). Among CLL requiring treatment, the presence of 11q22-23 deletion alone or combined to ATM mutations associates with poor response to chemotherapy. The addition of rituximab to chemotherapy significantly improved the outcome of CLL patients harbouring ATM lesions. However, even among CLL treated with FCR, 11q22-23 deletion still remains an adverse factor that identifies a group of patients with intermediate-risk disease that are projected to progress in a relatively short time (Stilgenbauer et al, 2014).
SF3B1 is a core component of the mRNA splicing machinery. SF3B1 mutations occur with a prevalence that ranges from 7 to 10% of unselected CLL, are enriched in cases harbouring unmutated IGHV genes, and tend to co-occur with ATM deletion or mutation. SF3B1 mutations in CLL are generally represented by missense nucleotide changes that recurrently target hotspots (codons 662, 666, 700, 704, 742), with a single amino-acid substitution (K700E) accounting for ∼50% of all SF3B1 mutations (Quesada et al, 2011; Rossi et al, 2011; Wang et al, 2011).
Though the role of SF3B1 mutations in CLL pathogenesis still remain largely unclear, the recent notion that the spliceosome participates in the ATM-induced DNA damage response and the observation that SF3B1 mutations are associated with a defective response to DNA damage, suggest that SF3B1 mutations may have a similar role as ATM abnormalities in deregulating the DNA damage programme (Te Raa et al, 2015). Consistently, similar to ATM defects, also the presence of SF3B1 mutations at the time of CLL presentation identifies a group of patients with intermediate-risk disease (40% of cases are alive at 10 years, accounting for a ∼50% reduction of the expected survival compared to the general population) (Rossi et al, 2013). Among CLL requiring treatment, SF3B1 mutations can potentially help refining prognostication of treatment relapse, though they do not represent a predictive biomarker for treatment tailoring. Indeed, the SF3B1 status does not impact on the chance of achieving responses to chemo+/−immunotherapy, though patients harbouring SF3B1 mutations show a shorter PFS than SF3B1 wild type cases (Stilgenbauer et al, 2014).
Signalling Pathway Deregulation
At variance with other B-cell tumours, genes encoding for components of the BCR signalling machinery are not usually targeted by somatic mutations in unselected CLL. The introduction of targeted drugs inhibiting the BCR signalling is changing the genetics of the disease, and has disclosed the acquisition of previously unexpected drug resistant mutations in the BCR pathway genes, including mutations affecting the BTK binding site of ibrutinib or gain-of-function mutations in PLCG2 (Woyach et al, 2014). Ibrutinib resistant mutations of the BCR pathway are not detectable before ibrutinib exposure, thus indicating that they are biologically irrelevant in the absence of selective pressures imposed by the drug (Famà et al, 2014). From a clinical standpoint, BTK and PLCG2 mutation analysis may be of help in the assessment of patients that progress under ibrutinib therapy.
The load of somatic hypermutation of the rearranged immunoglobulin heavy-variable genes (IGHV) genes, which is associated with antigen binding capacity and functions of the BCR, is one of the most important prognostic biomarkers in CLL. At the time of presentation, CLL cases carrying mutated IGHV genes (∼60% of patients) experience an indolent course compared with patients carrying unmutated IGHV genes (Hamblin et al, 1999). At the time of treatment requirement, CLL patients harbouring mutated IGHV genes, but lacking both TP53 and ATM defects (∼30% of cases), have a high chance of achieving a durable remission after FCR (Rossi et al, 2015). Therefore, FCR is currently the best option in physically fit patients with progressive CLL whose disease has a low-risk molecular profile (mutated IGHV genes without TP53 and ATM defects). Because the IGHV mutation status is a powerful biomarker for the prediction of duration of response after FCR, its assessment is recommended by the current guidelines in patients requiring therapy (Eichhorst et al, 2015).
The NOTCH receptor genes encode a family of heterodimeric transmembrane proteins (NOTCH1 to NOTCH4) that function as ligand-activated transcription factors. When the NOTCH receptors interact with their ligands through the extracellular subunit, two consecutive proteolytic cleavages of the NOTCH proteins are initiated and lead to pathway activation. Upon activation, the cleaved intracellular portion of the NOTCH receptors (ICN) translocates into the nucleus where it recruits a transcriptional complex that modifies the expression of a number of target genes, including MYC and NF-κB signalling components. The most prominent mechanism of NOTCH signal suppression is operated through its PEST domain of the ICN, which is recognised by the FBXW7 ubiquitin protein ligase and directed towards proteasomal degradation.
NOTCH1 mutations characterise ∼15% of unselected CLL and are represented by frameshift or non-sense events clustering within exon 34, including the highly recurrent c.7544_7545delCT deletion (∼80% of all mutations), as well as by non-coding mutations affecting the 3′ UTR region of NOTCH1, which cause aberrant splicing events resulting in a deletion of the last 158 coding bases of exon 34 (Puente et al, 2015). NOTCH1 mutations in CLL are selected to disrupt the PEST domain of the protein, resulting in NOTCH1 impaired degradation, stabilization of the active ICN, and deregulated NOTCH signalling. NOTCH1 is preferentially targeted in specific biological groups of CLL. In fact, NOTCH1 mutations are significantly more common in CLL with unmutated IGHV genes, and are enriched in CLL harbouring +12 (Puente et al, 2015).
The clinical implication of NOTCH1 mutations affecting exon 34 has been clarified to a certain extent. Conversely, though mutations in the 3′ UTR of the gene seem to behave similarly to exon 34 mutations, their role as prognostic biomarker needs further validation. At the time of CLL presentation, the presence of NOTCH1 mutations in exon 34 identifies a group of patients with intermediate-risk disease (∼40% of cases are alive at 10 years, accounting for a ∼50% reduction of the expected survival compared to the general population) and those in whom CLL is more likely to transform into RS (cumulative incidence of transformation at 10 years of ∼50%) (Rossi et al, 2012, 2013). Among CLL requiring treatment, cases harbouring NOTCH1 mutations in exon 34 seem not to benefit from the addition of an anti-CD20 monoclonal antibody to chemotherapy. Indeed, among CLL harbouring NOTCH1 mutations, treatment with FCR does not result into the expected increase in minimal residual disease response nor into an improvement in PFS or OS compared to treatment with the sole FC (Stilgenbauer et al, 2014). NOTCH1 mutations in exon 34 associate with low CD20 levels in CLL and are responsible for a dysregulation of HDAC-mediated epigenetic repression of CD20 expression, which may explain the lower sensitivity to anti-CD20 treatment of CLL harbouring NOTCH1 mutations (Pozzo et al, 2015).
In CLL, NF-κB signalling is generally upregulated through specific interactions between protective microenvironmental niches and CLL cells. At least in a fraction of cases, CLL gain active NF-κB signalling by mutating NF-κB genes. The non-canonical NF-κB pathway is engaged by CD40 and BAFF receptors. Upon receptor binding, the TRAF3/MAP3K14-TRAF2/BIRC3 negative regulatory complex of non-canonical NF-κB signalling is disrupted, allowing the cytoplasmic release and stabilization of MAP3K14, the central activating kinase of non-canonical NF-κB signalling. The stabilised MAP3K14 activates the IKKα kinase, which in turns directly phosphorylates NF-κB/p100, inducing partial proteolysis of p100 to p52 by the proteasome. The p52 protein dimerises with RelB to translocate into the nucleus, where it regulates gene transcription.
The Baculoviral IAP repeat containing 3 (BIRC3) gene, which cooperates in the TRAF3/MAP3K14-TRAF2/BIRC3 negative regulatory complex of non-canonical NF-κB signalling, is mutated in ∼2% of unselected CLL (Rossi et al, 2013). At the biochemical level, BIRC3 mutations cause the truncation of the C-terminal RING domain of the BIRC3 protein, whose E3 ubiquitin ligase activity is essential for switching off MAP3K14 through proteosomal degradation, thus leading to constitutive non-canonical NF-κB activation. From a clinical standpoint, BIRC3 mutations identify a genetic subgroup of cases characterised by poor risk disease (Rossi et al, 2013).
NF-κB comprises a small family of transcription factors, including the NF-κB/Rel members RelA, RelB, c-Rel, NF-κB1, and NF-κB2. These proteins are kept inactive by cytoplasmic association with the IκB inhibitory proteins. The NF-κB inhibitor epsilon (NFKBIE) belongs to the IκB inhibitory protein family and counteracts NF-κB activation via cytoplasmic retention of the Rel proteins. The NFKBIE gene is affected by a recurrent 4 bp deletion in ∼5% unselected CLL. NFKBIE mutation results in protein truncation, reduced inhibitory interaction with the Rel transcription factor, and enhanced NF-κB activation (Mansouri et al, 2015). Though the precise clinical implication of NFKBIE mutations remains to be clarified, their enrichment among CLL presenting in advanced stage suggests that they might be involved in disease progression.
In B-cells, Toll-like receptors are central to the BCR-independent response to antigens by sensing a variety of pathogen-associated molecular patterns derived from bacteria, viruses, and fungi. Adaptor proteins, including the myeloid differentiation factor 88 (MYD88), are essential for initiating Toll-like receptors signalling. MYD88 has a modular structure with a death domain (DD) at the N terminus, and a Toll-IL-1 receptor (TIR) domain at the C terminus. The TIR domain of MYD88 is crucial for signal transduction as it mediates contacts with the intracellular TIR domains of the TLRs upon signalling activation. The DD domain allows oligomerization of the active MYD88 and its interaction with the respective DD of the serine-threonine kinases IRAK1-4, thus resulting in a multimeric complex. This complex propagates the signal and leads to activation of a series of cascades and transcription factors, such as NF-κB, AP-1 and STAT3. Most MYD88 mutations in CLL are represented by the L265P missense substitution, which affects the evolutionarily conserved beta-beta loop of the TIR domain of MYD88, suggesting that it has been selected to change the structure of MYD88 and to allow spontaneous homodimerization and recruitment of IRAK1 and IRAK4. Consistently, in B-cell tumours, mutant MYD88 results in uncontrolled formation of the MYD88/IRAK complex, which translates into the recruitment of TRAF6, constitutive phosphorylation of TAK1 and, ultimately, the elevation of NF-κB activity and cytokine secretion. MYD88 gene mutations occur in ∼3% of unselected CLL, whereas they are enriched in a specific clinical subgroup of patients characterised by young age at presentation, mutated IGHV genes and expected survival similar to that of the age–sex matched normal population (Martínez-Trillos et al, 2014).
Conclusion
The use of genetic prognostic markers is not recommended in patients that are not in need of treatment. In the era of personalised medicine, the challenges for the treatment of patients with CLL will involve correctly matching therapies to the unique genetic composition of each individual tumour. Predictive biomarkers that are recommended by the guidelines and that should be routinely applied in the clinical practice for treatment tailoring are: (i) IGHV mutations, that mark durable remission after FCR; and (ii) TP53 defects, that mark resistance to imunochemotherapy and represent an indication to targeted agents (i.e. ibrutinib or idelalisib plus rituximab). NOTCH1 mutation status, that associates with resistance to anti-CD20 monoclonal antibody, as well as complex karyotype, BTK and PLCG2 mutations, that associate with resistance to ibrutinib, might represent novel predictive biomarkers. However, their value needs to be confirmed before they can be used in clinical routine.
References
Byrd JC, Jones JJ, Woyach JA, Johnson AJ, Flynn JM (2014) Entering the era of targeted therapy for chronic lymphocytic leukemia: impact on the practicing clinician. J Clin Oncol 32: 3039–3047.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM (2002) Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 99: 15524–15529.
Cavazzini F, Hernandez JA, Gozzetti A, Russo Rossi A, De Angeli C, Tiseo R, Bardi A, Tammiso E, Crupi R, Lenoci MP, Forconi F, Lauria F, Marasca R, Maffei R, Torelli G, Gonzalez M, Martin-Jimenez P, Maria Hernandez J, Rigolin GM, Cuneo A (2008) Chromosome 14q32 translocations involving the immunoglobulin heavy chain locus in chronic lymphocytic leukaemia identify a disease subset with poor prognosis. Br J Haematol 142: 529–537.
Dreger P, Schetelig J, Andersen N, Corradini P, van Gelder M, Gribben J, Kimby E, Michallet M, Moreno C, Stilgenbauer S, Montserrat E European Research Initiative on CLL (ERIC) and the European Society for Blood and Marrow Transplantation (EBMT) (2014) Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood 124: 3841–3849.
Eichhorst B, Robak T, Montserrat E, Ghia P, Hillmen P, Hallek M, Buske C ESMO Guidelines Committee (2015) Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol Suppl 5: v78–v84.
Famà R, Bomben R, Rasi S, Dal BoM, Ciardullo C, Monti S, Rossi F, D’Agaro T, Zucchetto A, Gattei V, Gaidano G, Rossi D (2014) Ibrutinib-naïve chronic lymphocytic leukemialacks Bruton tyrosine kinase mutations associated with treatment resistance. Blood 124: 3831–3833.
Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, Herman SE, Tian X, Marti G, Soto S, Hughes TE, Jones J, Lipsky A, Pittaluga S, Stetler-Stevenson M, Yuan C, Lee YS, Pedersen LB, Geisler CH, Calvo KR, Arthur DC, Maric I, Childs R, Young NS, Wiestner A (2015) Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemiawith TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol 16: 169–176.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM (2014) Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 370: 997–1007.
Ghia P, Hallek M (2014) Management of chronic lymphocytic leukemia. Haematologica 99: 965–972.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ International Workshop on Chronic Lymphocytic Leukemia (2008) Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111: 5446–5456.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK (1999) Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94: 1848–1854.
Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R (2010) The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40.
Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, Vartanov A, Fernandes SM, Saksena G, Cibulskis K, Tesar B, Gabriel S, Hacohen N, Meyerson M, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ (2013) Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–726.
Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter SL, Cibulskis K, Mertens D, Sougnez CL, Rosenberg M, Hess JM, Edelmann J, Kless S, Kneba M, Ritgen M, Fink A, Fischer K, Gabriel S, Lander ES, Nowak MA, Döhner H, Hallek M, Neuberg D, Getz G, Stilgenbauer S, Wu CJ (2015) Mutations driving CLL and their evolution in progression and relapse. Nature 526: 525–530.
Mansouri L, Sutton LA, Ljungström V, Bondza S, Arngården L, Bhoi S, Larsson J, Cortese D, Kalushkova A, Plevova K, Young E, Gunnarsson R, Falk-Sörqvist E, Lönn P, Muggen AF, Yan XJ, Sander B, Enblad G, Smedby KE, Juliusson G, Belessi C, Rung J, Chiorazzi N, Strefford JC, Langerak AW, Pospisilova S, Davi F, Hellström M, Jernberg-Wiklund H, Ghia P, Söderberg O, Stamatopoulos K, Nilsson M, Rosenquist R (2015) Functional loss of IκBɛ leads to NF-κB deregulation in aggressive chronic lymphocytic leukemia. J Exp Med 212: 833–843.
Martínez-Trillos A, Pinyol M, Navarro A, Aymerich M, Jares P, Juan M, Rozman M, Colomer D, Delgado J, Giné E, González-Díaz M, Hernández-Rivas JM, Colado E, Rayón C, Payer AR, Terol MJ, Navarro B, Quesada V, Puente XS, Rozman C, López-Otín C, Campo E, López-Guillermo A, Villamor N (2014) Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood 123: 3790–3796.
Müller-Hermelink HK, Montserrat E, Catovsky D, Campo E, Harris NL, Stein H (2008) Chronic lymphocytic leukemia/small lymphocytic lymphoma. In World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, Swerdlow SH, Campo E, Harris NL (eds). 180–182. IARC: Lyon, France.
Pospisilova S, Gonzalez D, Malcikova J, Trbusek M, Rossi D, Kater AP, Cymbalista F, Eichhorst B, Hallek M, Döhner H, Hillmen P, van Oers M, Gribben J, Ghia P, Montserrat E, Stilgenbauer S, Zenz T European Research Initiative on CLL (ERIC) (2012) ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 26: 1458–1461.
Pozzo F, Bittolo T, Arruga F, Bulian P, Macor P, Tissino E, Gizdic B, Rossi FM, Bomben R, Zucchetto A, Benedetti D, Degan M, D’Arena G, Chiarenza A, Zaja F, Pozzato G, Rossi D, Gaidano G, Del Poeta G, Deaglio S, Gattei V, Dal Bo M (2015) NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia 30 (1): 182–189.
Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, Munar M, Rubio-Pérez C, Jares P, Aymerich M, Baumann T, Beekman R, Belver L, Carrio A, Castellano G, Clot G, Colado E, Colomer D, Costa D, Delgado J, Enjuanes A, Estivill X, Ferrando AA, Gelpí JL, González B, González S, González M, Gut M, Hernández-Rivas JM, López-Guerra M, Martín-García D, Navarro A, Nicolás P, Orozco M, Payer ÁR, Pinyol M, Pisano DG, Puente DA, Queirós AC, Quesada V, Romeo-Casabona CM, Royo C, Royo R, Rozman M, Russiñol N, Salaverría I, Stamatopoulos K, Stunnenberg HG, Tamborero D, Terol MJ, Valencia A, López-Bigas N, Torrents D, Gut I, López-Guillermo A, López-Otín C, Campo E (2015) Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 526 (7574): 519–524.
Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Giné E, Hernández JM, González-Díaz M, Puente DA, Velasco G, Freije JM, Tubío JM, Royo R, Gelpí JL, Orozco M, Pisano DG, Zamora J, Vázquez M, Valencia A, Himmelbauer H, Bayés M, Heath S, Gut M, Gut I, Estivill X, López-Guillermo A, Puente XS, Campo E, López-Otín C (2011) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47–52.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF (2016) Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 374: 311–322.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, De Paoli L, Spina V, Gattei V, Capello D, Forconi F, Lauria F, Gaidano G (2009) The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res 15: 995–1004.
Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, Marasca R, Laurenti L, Bruscaggin A, Cerri M, Monti S, Cresta S, Famà R, De Paoli L, Bulian P, Gattei V, Guarini A, Deaglio S, Capello D, Rabadan R, Pasqualucci L, Dalla-Favera R, Foà R, Gaidano G (2012) Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 119: 521–529.
Rossi D, Terzi-di-Bergamo L, De Paoli L, Cerri M, Ghilardi G, Chiarenza A, Bulian P, Visco C, Mauro FR, Morabito F, Cortelezzi A, Zaja F, Forconi F, Laurenti L, Del Giudice I, Gentile M, Vincelli I, Motta M, Coscia M, Rigolin GM, Tedeschi A, Neri A, Marasca R, Perbellini O, Moreno C, Del Poeta G, Massaia M, Zinzani PL, Montillo M, Cuneo A, Gattei V, Foà R, Gaidano G (2015) Molecular prediction of durable remission after first line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 126 (16): 1921–1924.
Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, Fangazio M, Vaisitti T, Monti S, Chiaretti S, Guarini A, Del Giudice I, Cerri M, Cresta S, Deambrogi C, Gargiulo E, Gattei V, Forconi F, Bertoni F, Deaglio S, Rabadan R, Pasqualucci L, Foà R, Dalla-Favera R, Gaidano G (2011) Mutations of the SF3B1splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118: 6904–6908.
Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R, Rasi S, Monti S, Deambrogi C, De Paoli L, Wang J, Gattei V, Guarini A, Foà R, Rabadan R, Gaidano G (2014) Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 123: 2139–2147.
Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, Deambrogi C, Khiabanian H, Serra R, Bertoni F, Forconi F, Laurenti L, Marasca R, Dal-Bo M, Rossi FM, Bulian P, Nomdedeu J, Del Poeta G, Gattei V, Pasqualucci L, Rabadan R, Foà R, Dalla-Favera R, Gaidano G (2013) Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 121: 1403–1412.
Skowronska A, Parker A, Ahmed G, Oldreive C, Davis Z, Richards S, Dyer M, Matutes E, Gonzalez D, Taylor AM, Moss P, Thomas P, Oscier D, Stankovic T (2012) Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol 30: 4524–4532.
Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Döhner K, Bühler A, Böttcher S, Ritgen M, Kneba M, Winkler D, Tausch E, Hoth P, Edelmann J, Mertens D, Bullinger L, Bergmann M, Kless S, Mack S, Jäger U, Patten N, Wu L, Wenger MK, Fingerle-Rowson G, Lichter P, Cazzola M, Wendtner CM, Fink AM, Fischer K, Busch R, Hallek M, Döhner H (2014) Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 123: 3247–3254.
Te Raa GD, Derks IA, Navrkalova V, Skowronska A, Moerland PD, van Laar J, Oldreive C, Monsuur H, Trbusek M, Malcikova J, Lodén M, Geisler CH, Hüllein J, Jethwa A, Zenz T, Pospisilova S, Stankovic T, van Oers MH, Kater AP, Eldering E (2015) The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia 29: 1133–1142.
Thompson PA, O’Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, Burger JA, Estrov Z, Jain N, Kantarjian HM, Keating MJ (2015) Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 121: 3612–3621.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, Zhang W, Vartanov AR, Fernandes SM, Goldstein NR, Folco EG, Cibulskis K, Tesar B, Sievers QL, Shefler E, Gabriel S, Hacohen N, Reed R, Meyerson M, Golub TR, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506.
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ, Byrd JC (2014) Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 370: 2286–2294.
Zelenetz AD, Gordon LI, Wierda WG, Abramson JS, Advani RH, Andreadis CB, Bartlett N, Byrd JC, Czuczman MS, Fayad LE, Fisher RI, Glenn MJ, Habermann TM, Harris NL, Hoppe RT, Horwitz SM, Kelsey CR, Kim YH, Krivacic S, LaCasce AS, Nademanee A, Porcu P, Press O, Rabinovitch R, Reddy N, Reid E, Saad AA, Sokol L, Swinnen LJ, Tsien C, Vose JM, Wilson L, Yahalom J, Zafar N, Dwyer M, Sundar H (2015) Chronic lymphocytic leukemia/small lymphocytic lymphoma, version 1.2015. J Natl Compr Canc Netw 13: 326–362.
Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, Bühler A, Edelmann J, Bergmann M, Hopfinger G, Hensel M, Hallek M, Döhner H, Stilgenbauer S (2010) TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 28: 4473–4479.
Acknowledgements
The work by the authors described in this review was supported by Fondazione Cariplo, Grant No. 2012-0689; Special Program Molecular Clinical Oncology 5 × 1000 No. 10007 and My First AIRC Grant No. 13470, Associazione Italiana per la Ricerca sul Cancro Foundation Milan, Italy; Progetto Ricerca Finalizzata RF-2010-2307262 and RF-2011-02349712, Ministero della Salute, Rome, Italy; Futuro in Ricerca 2012 Grant No. RBFR12D1CB, Ministero dell’Istruzione, dell’Università e della Ricerca, Rome, Italy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Rossi, D., Gaidano, G. The clinical implications of gene mutations in chronic lymphocytic leukaemia. Br J Cancer 114, 849–854 (2016). https://doi.org/10.1038/bjc.2016.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.78
Keywords
This article is cited by
-
Front-line fludarabine-cyclophosphamide-rituximab (FCR) in 110 patients with chronic lymphocytic leukaemia (CLL): real-life experience with long-term outcomes, toxicities and responses to second-line therapies
International Journal of Hematology (2023)
-
Clonal evolution in chronic lymphocytic leukemia is associated with an unmutated IGHV status and frequently leads to a combination of loss of TP53 and TP53 mutation
Molecular Biology Reports (2022)
-
Multivariate transcriptome analysis identifies networks and key drivers of chronic lymphocytic leukemia relapse risk and patient survival
BMC Medical Genomics (2021)
-
Novel Therapies in Chronic Lymphocytic Leukemia: A Rapidly Changing Landscape
Current Treatment Options in Oncology (2020)
-
Chronic Lymphocytic Leukaemia in 2020: the Future Has Arrived
Current Oncology Reports (2020)
