
Abstract
Background:
Germline mutations within DNA-repair genes are implicated in susceptibility to multiple forms of cancer. For prostate cancer (PrCa), rare mutations in BRCA2 and BRCA1 give rise to moderately elevated risk, whereas two of ∼100 common, low-penetrance PrCa susceptibility variants identified so far by genome-wide association studies implicate RAD51B and RAD23B.
Methods:
Genotype data from the iCOGS array were imputed to the 1000 genomes phase 3 reference panel for 21 780 PrCa cases and 21 727 controls from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium. We subsequently performed single variant, gene and pathway-level analyses using 81 303 SNPs within 20 Kb of a panel of 179 DNA-repair genes.
Results:
Single SNP analyses identified only the previously reported association with RAD51B. Gene-level analyses using the SKAT-C test from the SNP-set (Sequence) Kernel Association Test (SKAT) identified a significant association with PrCa for MSH5. Pathway-level analyses suggested a possible role for the translesion synthesis pathway in PrCa risk and Homologous recombination/Fanconi Anaemia pathway for PrCa aggressiveness, even though after adjustment for multiple testing these did not remain significant.
Conclusions:
MSH5 is a novel candidate gene warranting additional follow-up as a prospective PrCa-risk locus. MSH5 has previously been reported as a pleiotropic susceptibility locus for lung, colorectal and serous ovarian cancers.
Similar content being viewed by others

Main
Prostate Cancer (PrCa) is the most frequently diagnosed cancer among men in developed countries and despite high survival rates also one of the highest for mortality (Cancer Research UK, 2014; Quaresma et al, 2015). However, as the majority of prostate neoplasms develop extremely slowly, many do not require clinical intervention, which coupled with the low specificity of the prostate-specific antigen test for clinically relevant forms of the disease could potentially lead to considerable over-diagnosis and overtreatment of patients for relatively modest reductions in mortality (Ilic et al, 2013). In conjunction with the establishment of improved biomarkers for lethal PrCa, the identification of individuals at greater risk of developing prostate tumours that require clinical intervention would also help inform more targeted and appropriate application of treatment. The heritability of PrCa is believed to be the highest of all the common forms of cancer (Hjelmborg et al, 2014). This is consistent with observations from genome-wide association studies (GWAS), which have to date identified >100 low-penetrance susceptibility variants for PrCa, two of which implicate the DNA-repair genes RAD51B and RAD23B (Xu et al, 2012; Al Olama et al, 2014; Eeles et al, 2014; Amin Al Olama et al, 2015). In addition, rare germline mutations in a small number of genes have been reported, with varying degrees of evidence, as potentially conferring greater risks of PrCa, including the DNA-repair genes ATM, BRCA1, BRCA2, BRIP1, CHEK2 and NBN (Dong et al, 2003; Kote-Jarai et al, 2009, 2011; Leongamornlert et al, 2012, 2014; Robinson et al, 2015). Recently, increasing evidence has demonstrated that these germline DNA-repair gene mutation carriers are at increased likelihood of experiencing advanced disease, metastatic spread and poorer survival outcome; yet these mutations also hold promise as potentially clinically actionable and responsive to targeted treatments (Castro et al, 2013; Cybulski et al, 2013; Leongamornlert et al, 2014; Robinson et al, 2015). In spite of these discoveries, the majority of the excess familial risk of PrCa still remains to be explained (Attard et al, 2015), with the contribution of DNA-repair gene variants identified to date making them attractive candidates for further investigation. In this study, using data from the iCOGS project imputed to the 1000 Genomes Phase 3 reference panel, we have analysed a large panel of DNA-repair gene variants for 21 780 PrCa cases and 21 727 controls of European ancestry from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) Consortium (Eeles et al, 2013). Analyses were performed at single variant, gene and pathway levels to maximise the power to detect putative associations with lower frequency variants or those with modest effect sizes.
Materials and methods
Samples
Samples for the iCOGS study were drawn from 25 studies participating in the PRACTICAL Consortium. The majority of studies were population-based or hospital-based case–control studies, or nested case–control studies; some studies selected samples by age or oversampled for cases with a family history of prostate cancer. Further information regarding the samples from the PRACTICAL Consortium included on the iCOGS array may be found within the original publication (Eeles et al, 2013). Analyses for DNA-repair gene variants were restricted to samples of European ancestry. In total, genotype data for 21 780 PrCa cases and 21 727 matched controls were available after quality control.
Genotyping and imputation
Genotyping was performed as part of the iCOGS project. This utilised a custom genotyping array designed in collaboration between the PRACTICAL, BCAC (Breast Cancer Association Consortium), OCAC (Ovarian Cancer Association Consortium) and CIMBA (Consortium of Investigators of Modifiers of BRCA1/2) consortia. Detailed information about the design, genotyping and quality control procedures for iCOGS can be found within the original publication (Eeles et al, 2013). In total 211 155 SNPs were genotyped on the iCOGS array, of which 3510 were situated within our defined DNA-repair gene regions. Imputation of the iCOGS PRACTICAL data was performed based on sequence data for 2504 samples from the 1000 Genomes phase 3 reference panel (IMPUTE2 haplotype panel, October 2014 release; https://mathgen.stats.ox.ac.uk/impute/1000GP%20Phase%203%20haplotypes%206%20October%202014.html) using SHAPEIT (v2 r778) and IMPUTE v2.3.1 in 588 chunks with a median size of 5 Mb (Howie et al, 2009; Delaneau et al, 2013). Imputed data for variants with INFO scores ⩾0.3 and MAF >0.001 were included in these analyses, which retained a total of 81 303 variants within the studied DNA-repair gene regions.
Gene/region selection
We identified a total of 179 genes with a core function in DNA-damage repair from the literature that intersected imputed iCOGS genotype data. We annotated DNA-repair genes to a single primary DNA-repair pathway according to previous curations (Wood et al, 2005; Kang et al, 2012). The genes analysed in this study represent the pathways Homologous recombination/Fanconi Anaemia signalling network (HR/FA), base excision repair (BER), non-homologous end joining (NHEJ), mismatch repair (MMR), nucleotide excision repair (NER), translesion synthesis (TLS), ATM signalling (ATM), RECQ helicase family (RECQ), cross-link repair (XLR), and other miscellaneous DNA-repair genes with functions including endonuclease/exonuclease activity and modification of chromatin structure (Other). Gene coordinates were assigned according to GENCODE release 19 (GRCh37.p13), with a 20-kb flank added to define the study region for each gene, in order to focus primarily on capturing gene and promoter centric variation over that within regulatory elements, which can be located at variable and potentially relatively large distances from the gene itself. Variants were annotated using wANNOVAR to facilitate designation as coding, intronic, UTR, splice and intergenic (Wang et al, 2010; Chang and Wang, 2012). A full list of the DNA-repair genes analysed in this study, their pathway annotations, region coordinates and the number of typed and imputed variants available is included in Supplementary Table 1.
Statistical analyses
Analyses were adjusted for study groups and the first eight principal components. For single-SNP analyses the genome-wide significance threshold was employed (P<5 × 10−8), whereas for gene and pathway level tests the Bonferroni correction was used to determine multiple testing adjusted significance thresholds (gene P<2.7 × 10−4, pathway P<5.56 × 10−3).
All analyses were carried out using R. For single-SNP analyses, per allele odds ratios were estimated using logistic regression. SKAT tests were performed using the SKAT package for R (http://CRAN.R-project.org/package=SKAT). We used the SKAT-O and SKAT-C tests for optimal analyses of the combined effect of multiple rare variants and common and rare variants, respectively (Wu et al, 2011; Lee et al, 2012; Ionita-Laza et al, 2013). Tests were conducted using default parameters and a common/rare cutoff threshold of MAF=0.01 for the SKAT-C test. StepAIC and SKAT leave one out were used to further interrogate the significant SKAT signal at the MSH5 gene for the individual variants that best described the signal.
Analyses for low-grade vs high-grade PrCa were carried out based on two clinical criteria. For stringent comparison of non-aggressive and aggressive PrCa, we defined NCCN stage 1 patients as non-aggressive PrCa and individuals with metastatic disease (M+) or nodal spread (N+) as aggressive (395 NCCN1 vs 1391 M+/N+), whereas to enhance the sample panel available for this analysis we also compared patients with Gleason Stage (GS) ⩽6 against those with GS ⩾8 disease (9626 GS ⩽6 vs 2776 GS ⩾8).
Results
Using genotype data from the iCOGS study imputed to the 1000 genomes phase 3 reference panel we analysed 81 303 SNPs within a 20-kb flanking region of 179 genes with a core function in DNA-damage repair (Supplementary Table 1). Rare and uncommon variants represented a substantial proportion of the data set, with 29 503 variants of MAF ⩽1%, 16 689 with MAF 1–5% and 35 111 with MAF >5% (Supplementary Figure 1a). Variants were categorised as SNPs, insertions and deletions, annotated using wANNOVAR (Wang et al, 2010; Chang and Wang, 2012), and classified into five categories; coding, UTR, splice, intronic and intergenic. Variants available for this analysis were predominantly situated within non-coding (intronic or intergenic) regions, with 3943 variants annotated as coding, splice or UTR in total, whereas most were single-base substitutions, with 3914 insertions and 5576 deletions, respectively. All of the insertion and deletion variants were imputed, with the vast majority located within non-coding regions (Supplementary Figure 1b–d, Supplementary Table 2). All analyses were adjusted for study population and the first eight principal components. For single-variant level analyses the genome-wide significance threshold (P<5 × 10−8) was used to determine significantly associated variants, whereas for gene and pathway level analyses the significance threshold was defined according to the Bonferroni correction (gene P<2.7 × 10−4, pathway P<5.56 × 10−3).
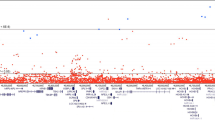
Single-variant analysis for association of DNA-repair gene variants with PrCa identified only the previously reported association with RAD51B at Chr14q24 (rs371311594, P=1.29 × 10−10). Several other gene loci showed suggestive association peaks; however, no other variants were within one order of magnitude of genome-wide significance (Figure 1, Supplementary Table 3).

Single SNP case–control Manhattan Plot. In total, 81 303 SNPs from 179 DNA-repair genes were analysed for association with PrCa. Only the previously reported association within the RAD51B gene was identified, with suggestive, non-significant association peaks observed at a small number of other loci.
We observed evidence for modest inflation within our association data (λ=1.105); nonetheless, departure from the null was apparent towards the extremity of the P-value distribution and this persisted to a more modest extent even after the RAD51B region was excluded (Supplementary Figure 2). We subsequently performed gene level association tests, in an attempt to ascertain whether additional putative PrCa-risk signals might be present among the genes within which no individual variant achieved significance after adjustment for multiple testing, arising through a cumulative effect of several low MAF or low penetrance variants. We performed two gene-level association tests using the SKAT; SKAT-C, which is optimised for combined testing of rare and common variants and SKAT-O, which attempts to maximise power for rare variant testing (Lee et al, 2012; Ionita-Laza et al, 2013). Gene-level analysis identified a novel significant association with the MSH5 gene using the SKAT-C test (Chr6p21; P=1.68 × 10−4) (Figure 2, Supplementary Table 4). We used stepAIC and leave one out for SKAT to further interrogate the MSH5 data for the individual variants that best explain the signal. This test selected three variants at the MSH5 locus, rs61036903 (known as 6 : 31713892 within the reference panel) intronic within the gene and two variants 10-kb downstream within an adjacent gene VWA7, rs805825 and rs185333600. These were all among the top-ranking variants in the single-SNP analysis (rs61036903: MAF=0.14, OR 0.92, P=8.06 × 10−5; rs805825: MAF=0.45, OR 0.94, P=4.05 × 10−5; rs185333600: MAF=0.003, OR 1.57, P=6.83 × 10−4).

Case–control Manhattan Plots for the 179 DNA-repair genes analysed by SKAT. (A) A significant association was observed for the MSH5 gene using the SKAT-C test that examines the combined effect of common and rare variants. (B) No significant association was detected for any gene under the SKAT-O test that primarily focuses on rare variant association testing.
We subsequently examined the iCOGS data set at the pathway level under the SKAT test to supplement the gene-level analyses. We again used the Bonferroni correction to define the significance threshold (pathway P<5.56 × 10−3). No pathway achieved significance at this threshold, with suggestive associations under the SKAT-O test observed with the translesion synthesis pathway (P=6.18 × 10−3) and mismatch-repair pathway (P=0.056).
Variants within the coding sequence of DNA-repair genes could be more likely to influence PrCa risk than those in non-coding regions. We therefore performed an additional SKAT test to assess whether the coding DNA-repair gene variants available for this study, when collapsed as a single entity, could stratify case and control status. We observed a significant association when using the SKAT-C test (P=0.003), which suggests that variants that affect the coding sequence of genes participating in DNA-repair processes contribute to PrCa risk. We attempted to further elaborate upon this finding by analysing coding variation within each pathway separately. Despite relatively modest numbers of coding variants available within each pathway, we continued to observe suggestive associations under the SKAT-O test for the translesion synthesis pathway (P=0.026) and mismatch-repair pathway (P=0.055), in addition to the HR/FA pathway under the SKAT-C test (P=0.011).
To complement the tests designed to identify potential PrCa susceptibility variants and genes, we also performed case–case analyses to investigate whether individual or cumulative germline DNA-repair gene and pathway variants in the iCOGS imputed data set correlated with phenotypic characteristics of more aggressive PrCa. This analysis was limited by lack of complete phenotypic data for all patients within the iCOGS sample set and low numbers of samples within individual phenotypic subgroups; therefore, we utilised two separate criteria to define aggressive and non-aggressive disease. For a stringent comparison of non-aggressive and aggressive PrCa, we analysed NCCN stage 1 patients against individuals with metastatic disease (M+) or nodal spread (N+) (395 NCCN1 vs 1391 M+/N+), whereas to maximise the number of samples available we also compared patients with GS ⩽6 disease against those with GS ⩾8 (9626 GS ⩽6 vs 2776 GS ⩾8). No significant associations with aggressive PrCa were identified at either the variant or gene levels for either of the phenotypic criteria tested. (Supplementary Figure 3, Supplementary Table 5). When we examined PrCa aggressiveness at the pathway level, we observed associations at P<0.05 for the HR/FA pathway under both tests for the GS ⩽6 vs GS ⩾8 phenotype cohort (SKAT-C P=0.011, SKAT-O P=0.040). This pathway was also the highest ranked for the NCCN1 vs M+/N+ phenotype cohort under the SKAT-C test (P=0.052). When these analyses were restricted to only coding variants, an association at P<0.05 remained for the HR/FA pathway for the NCCN1 vs M+/N+ cohort and the SKAT-O test (P=0.021). These suggestive associations were not however significant after adjustment for multiple testing (Supplementary Table 5).
Discussion
DNA-repair genes have a crucial role in the correction of damage to the genome of a cell and therefore their impairment can lead to carcinogenesis. Although these detrimental genetic alterations frequently originate within somatic cells during an individual’s lifetime, a number of rare, hereditary mutations within specific DNA-repair genes have been identified that confer substantially increased risks to the individual of PrCa and other cancers. GWAS have also previously identified common, low-penetrance variants in close proximity to the DNA-repair genes RAD51B and RAD23B that contribute to PrCa susceptibility (Xu et al, 2012; Eeles et al, 2013; Amin Al Olama et al, 2015). However, even relatively well-powered genetic association studies may have been limited in their ability to reliably interrogate variants with lower MAFs or associations with modest effect sizes; therefore, additional-risk variants that confer their functional effect though DNA-repair genes may remain to be discovered. We have recently imputed PrCa data from the iCOGS study to the 1000 Genomes phase 3 reference panel, thereby enhancing the capability to interrogate this data set for untyped variants within tagged regions. In particular, a far greater number of lower MAF and insertion and deletion variants were available for analysis, although these are predominantly situated in non-coding regions. Imputation performance of lower MAF variants is improved by larger reference panel size and ethnic diversity and higher marker density on the genotyping array; however, rare variants still regularly remain challenging to impute without an additional reference panel enriched for specific low-frequency variants of known interest, and may also be more sensitive to differences in the imputation approach employed (Hoffmann and Witte, 2015). Our relatively large sample size provided good power to detect associations with PrCa for rare variants with greater effect sizes (e.g., for a variant at our 0.1% MAF cutoff with OR 2.5, we had 78% power) as well as common, low-penetrance variants (e.g., for a variant with OR 1.1 and a MAF of 20%, power was 86%). We were however limited with respect to the detection of variants with the combination of both modest allele frequency and effect size.
We have examined all variants in the imputed iCOGS data set situated within 20-kb of a panel of 179 DNA-repair genes for association with PrCa or more aggressive phenotypic presentation. No novel risk variants were identified in our single-SNP analysis, with only the previously reported signal at RAD51B on Chr14q24 genome-wide significant (Figure 1, Supplementary Table 3). Our analysis did not detect the previously reported signal at the RAD23B locus on Chr9q31, which was originally identified in the Chinese population and recently also confirmed in Europeans with the most significantly associated variant rs1771718 and the signal also an eQTL for RAD23B in normal prostate tissue in the TCGA data set (Xu et al, 2012; Amin Al Olama et al, 2015). rs1771718 is located ∼57 kb downstream of RAD23B, which is the closest neighbouring gene but located in a distinct recombination block from these risk variants. As no variant among the 509 within the gene centric region that we interrogated in this study showed substantial evidence for association (P⩾2.94 × 10−3), it appears likely that risk at this locus is modulated through a nearby regulatory element controlling expression of the gene as opposed to intragenic causal functional variants (Supplementary Figure 4).
We conducted two gene-level analyses in an attempt to identify whether there may be additional signals among the several loci that demonstrated suggestive but non-significant association peaks in our single-SNP analysis, but for which no individual variant had achieved significance. SKAT-C tests for the combined effects of common and rare variants, whereas SKAT-O adaptively combines the burden test and SKAT test in an attempt to maximise power for rare variant association testing (Lee et al, 2012; Ionita-Laza et al, 2013). We identified a significant PrCa-risk association after adjustment for multiple testing at the MSH5 gene at Chr6p21 using the SKAT-C test, implying that multiple common, or a combination of common and rare variants within this gene may contribute to PrCa risk. Although caution must be taken with respect to this finding until replicated and deconstructed, this evidence implicates MSH5 as a prospective PrCa susceptibility locus that warrants additional follow-up. MSH5 had previously been reported as a plausible candidate gene for the lung cancer-risk locus at Chr6p21.33, for which the most strongly associated variant rs3117582 is intronic in BAT3; however, is highly correlated to rs3131379 in intron 10 of MSH5 (Wang et al, 2008; Kazma et al, 2012). A recent study examining cancer pleiotropy among DNA-repair and DNA-damage signalling pathway variants has also reported a highly significant association with lung cancer for rs3115672, a synonymous variant within MSH5, in addition to weaker associations with colon and serous ovarian cancers (pleiotropic OR 1.18, 95% CI 1.12-1.24, P=2.53 × 10−8) (Scarbrough et al, 2016). This variant was however non-significant for PrCa within their study of 14 160 PrCa cases and 12 724 controls (OR 0.96, P=0.21). Within our larger study (of which 2614 cases and 2679 controls overlapped with those of Scarbrough et al), in the single-SNP analysis, rs3115672 remained non-significant after adjustment for multiple testing (OR 0.94, 95% CI 0.90–0.98, P=5.69 × 10−3). However, a number of other variants among the 312 within the MSH5 gene in our analysis were more strongly associated, the top individual variant of which was rs9281573 (OR 0.94, P=4.01 × 10−5). StepAIC combined with SKAT leave one out selected two common and one rare variant as best explaining the SKAT-C association, all of which were among the top variants in the single-SNP analysis. This implies that a combination of common and rare variants could potentially underpin this signal.
We annotated these three variants for evidence of functionality using HaploReg v4.1 (Ward and Kellis, 2016); this annotation included chromatin state data for cell lines derived from multiple tissue types provided by the Roadmap Epigenomics Consortium (Roadmap Epigenomics et al, 2015); however, no data for prostate tissue were available. rs61036903, which is intronic to MSH5, showed limited direct evidence for functionality itself. Both of the variants situated around the MSH5 promoter region, within VWA7, showed strong evidence for being located within enhancer elements that are active across a wide range of tissue types. In addition, expression data from the GTEx Consortium indicates that rs805825 is an eQTL for a number of genes from the MHC region (HLA-DRB1, HLA-DRB5, LY6G5C, DDAH2, LY6G6C, HSPA1B and C4B) (GTEx Consortium, 2015). These genes are clustered closely centromeric and telomeric of MSH5 and VWA7 within a gene dense locus; however, no eQTL with MSH5 or VWA7 was observed for this variant.
Although the MSH5 gene is routinely classified as a member of the MMR pathway along with all other homologues of MutS (Wood et al, 2005; Ji et al, 2012; Scarbrough et al, 2016), functional evidence to date provides limited support for a role in MMR for MSH5 itself. Instead, this gene has been implicated primarily in the processes of meiotic recombination, maintenance of chromosome integrity and DNA double-strand break repair (Clark et al, 2013; Wu et al, 2013). RNA-seq data from GTEx Analysis Release V6 for 2712 total samples across 51 normal human tissues (including 106 prostate tissue samples) demonstrates that MSH5 is expressed at broadly similar levels across a wide range of tissue types, including prostate (GTEx Consortium, 2015; accessed via. http://www.gtexportal.org/home/gene/MSH5). Data from TCGA further support this expression profile across a range of normal tissues and also indicates that MSH5 is consistently overexpressed for almost all tumour types in comparison with their respective normal tissues. For TCGA prostate tissue, a median RSEM (log2) value of 8.08 was observed across 498 tumour samples compared with 6.85 from 52 normal samples (http://cancergenome.nih.gov/; accessed via. http://firebrowse.org/viewGene.html?gene=msh5).
Taken together, these information demonstrate that although the MSH5 gene represents a strong biological candidate for the PrCa-risk association that we have observed, additional functional follow-up studies will be required to dissect the precise functional variants, genes, regulatory elements or processes that underpin this signal.
It is worth noting that the gene level analyses in this study did not identify significant associations with any genes previously implicated in PrCa susceptibility. This was irrespective of whether the known risk mechanisms are believed to operate through multiple common, low-penetrance variants (e.g., RAD51B; SKAT-O P=0.05, SKAT-C P=2.76 × 10−3) or rare coding variants (e.g., BRCA2; SKAT-O P=0.46, SKAT-C P=0.15). In the case of BRCA2 and other genes in which rare, moderate penetrance, protein truncating PrCa susceptibility variants had previously been identified, this is likely to reflect the fact that even using the latest 1000 Genomes reference panel, rare variants expected to confer greater phenotypic consequences may remain absent from the reference panel and consequently unimputable. This is consistent with the poor representation of coding insertion and deletion variants within our data set and would have rendered us underpowered to detect the effects of this class of variation in our analysis. Our observations do however imply that any additional contribution from common, lower penetrance variation at these genes may be minimal. This includes the rs11571833 nonsense polymorphism in the terminal exon of BRCA2, which is a reported lung cancer susceptibility variant, but was not associated with PrCa in this study (OR 1.03, 95% CI 0.89–1.19, P=0.74) (Wang et al, 2014). It is perhaps more surprising that RAD51B did not achieve significance under the SKAT-C test, which considers the potential contribution towards association of both common and rare variants within a region, given that three independent associations have previously been identified at this locus (Amin Al Olama et al, 2015). However, a suggestive association was observed under this test, which may be an indication that the cumulative effect size of the independent low-penetrance-risk variants within this region were insufficient to be conclusively disambiguated through this methodology.
Our pathway-level analysis identified suggestive but non-significant associations for two pathways under the SKAT-O test; translesion synthesis and mismatch repair. Although this study did not therefore provide sufficient evidence to implicate genes within these pathways in PrCa susceptibility, given the inherently conservative nature of the Bonferroni correction with respect to type II error and the relatively low proportion of coding variants within our data set, these observations may still justify further evaluation. In particular, as these suggestive associations were observed under the SKAT-O test that maximises power for rare variant association analyses and were not abrogated when the analyses were restricted only to coding variants, if substantiated, these nascent observations could be underpinned by direct effects of rare variants on the protein structure and function. Consequently, sequencing studies designed to comprehensively analyse the entire coding sequence of genes within the translesion synthesis and mismatch repair pathways could potentially yield further insight towards the mechanisms of susceptibility to developing PrCa. It is also worth noting that somatic mutations in translesion synthesis pathway genes, in particular the POLK gene, have been observed in prostate tumours previously (Makridakis and Reichardt, 2012; Yadav et al, 2015), whereas a rare germline nonsynonymous variant in the POLI gene has also been reported to predispose towards the occurrence of the TMPRSS2-ERG fusion in PrCa patients (Luedeke et al, 2009).
Increasing evidence suggests that moderate-penetrance germline mutations within DNA-repair genes also correlate with a more aggressive phenotypic presentation of PrCa and poorer prognosis (Castro et al, 2013; Cybulski et al, 2013; Leongamornlert et al, 2014; Robinson et al, 2015). This could in turn signify that DNA-repair gene variants might exist that do not confer greater risk of developing PrCa per se, yet do modify the likelihood of developing more aggressive disease in individuals that develop PrCa owing to other risk factors or exposures. We therefore also performed case–case analyses to further explore this hypothesis using two distinct phenotypic criteria. No significant or suggestive associations with aggressive disease were identified at the individual variant or gene levels under either definition; however, suggestive non-significant associations with the HR/FA pathway were observed. These analyses were, however, limited by relatively low sample numbers within each comparison group, which would have reduced our power to detect associations, particularly for rare and uncommon variants. We cannot therefore exclude the existence of additional DNA-repair gene variants that promote increased PrCa aggressiveness rather than risk of the disease itself; however, our data would suggest that any that exist are more likely to be rare than common.
Overall, this study represents the most comprehensive interrogation of the role of DNA-repair gene variants in PrCa susceptibility that we are aware of to date. We confirmed the presence of low-penetrance susceptibility loci situated at the RAD51B locus and found evidence to implicate a novel gene, MSH5, in PrCa susceptibility. We also share preliminary observations that rare germline variation in genes within the translesion synthesis pathway, in particular variants within the coding sequence, could be worthy of further investigation as candidates for PrCa risk.
The main limitations of our study relate to the challenges in imputing rare, potentially pathogenic variants to array genotype data from population-based reference panels and in performing association tests on low-frequency variants in a large multi-population study while controlling for population stratification. Therefore, additional sequencing studies would still be warranted to further explore the contribution of rare DNA-repair gene variants to PrCa risk. In addition, incomplete availability of phenotypic data and the fact that the iCOGS study did not specifically select individuals with low- or high-grade disease may have reduced our ability to examine any potential influence of these variants on PrCa aggressiveness. Future studies, whether array or sequencing based, that specifically select patients from these cohorts for inclusion would facilitate investigation of this aspect; which might in turn help to enhance stratification of patients that require altered clinical management pathways.
References
Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, Benlloch S, Hazelett DJ, Wang Z, Saunders E, Leongamornlert D, Lindstrom S, Jugurnauth-Little S, Dadaev T, Tymrakiewicz M, Stram DO, Rand K, Wan P, Stram A, Sheng X, Pooler LC, Park K, Xia L, Tyrer J, Kolonel LN, Le Marchand L, Hoover RN, Machiela MJ, Yeager M, Burdette L, Chung CC, Hutchinson A, Yu K, Goh C, Ahmed M, Govindasami K, Guy M, Tammela TL, Auvinen A, Wahlfors T, Schleutker J, Visakorpi T, Leinonen KA, Xu J, Aly M, Donovan J, Travis RC, Key TJ, Siddiq A, Canzian F, Khaw KT, Takahashi A, Kubo M, Pharoah P, Pashayan N, Weischer M, Nordestgaard BG, Nielsen SF, Klarskov P, Roder MA, Iversen P, Thibodeau SN, McDonnell SK, Schaid DJ, Stanford JL, Kolb S, Holt S, Knudsen B, Coll AH, Gapstur SM, Diver WR, Stevens VL, Maier C, Luedeke M, Herkommer K, Rinckleb AE, Strom SS, Pettaway C, Yeboah ED, Tettey Y, Biritwum RB, Adjei AA, Tay E, Truelove A, Niwa S, Chokkalingam AP, Cannon-Albright L, Cybulski C, Wokolorczyk D, Kluzniak W, Park J, Sellers T, Lin HY, Isaacs WB, Partin AW, Brenner H, Dieffenbach AK, Stegmaier C, Chen C, Giovannucci EL, Ma J, Stampfer M, Penney KL, Mucci L, John EM, Ingles SA, Kittles RA, Murphy AB, Pandha H, Michael A, Kierzek AM, Blot W, Signorello LB, Zheng W, Albanes D, Virtamo J, Weinstein S, Nemesure B, Carpten J, Leske C, Wu SY, Hennis A, Kibel AS, Rybicki BA, Neslund-Dudas C, Hsing AW, Chu L, Goodman PJ, Klein EA, Zheng SL, Batra J, Clements J, Spurdle A, Teixeira MR, Paulo P, Maia S, Slavov C, Kaneva R, Mitev V, Witte JS, Casey G, Gillanders EM, Seminara D, Riboli E, Hamdy FC, Coetzee GA, Li Q, Freedman ML, Hunter DJ, Muir K, Gronberg H, Neal DE, Southey M, Giles GG, Severi G Breast, Prostate Cancer Cohort C, Consortium P, Consortium C, Consortium G-OE Cook MB, Nakagawa H, Wiklund F, Kraft P, Chanock SJ, Henderson BE, Easton DF, Eeles RA, Haiman CA ( 2014 ) A meta-analysis of 87 040 individuals identifies 23 new susceptibility loci for prostate cancer . Nat Genet 46 (10) : 1103 – 1109 .
Amin Al Olama A, Dadaev T, Hazelett DJ, Li Q, Leongamornlert D, Saunders EJ, Stephens S, Cieza-Borrella C, Whitmore I, Benlloch Garcia S, Giles GG, Southey MC, Fitzgerald L, Gronberg H, Wiklund F, Aly M, Henderson BE, Schumacher F, Haiman CA, Schleutker J, Wahlfors T, Tammela TL, Nordestgaard BG, Key TJ, Travis RC, Neal DE, Donovan JL, Hamdy FC, Pharoah P, Pashayan N, Khaw KT, Stanford JL, Thibodeau SN, McDonnell SK, Schaid DJ, Maier C, Vogel W, Luedeke M, Herkommer K, Kibel AS, Cybulski C, Wokolorczyk D, Kluzniak W, Cannon-Albright L, Brenner H, Butterbach K, Arndt V, Park JY, Sellers T, Lin HY, Slavov C, Kaneva R, Mitev V, Batra J, Clements JA, Spurdle A, Teixeira MR, Paulo P, Maia S, Pandha H, Michael A, Kierzek A, Govindasami K, Guy M, Lophatonanon A, Muir K, Vinuela A, Brown AA Consortium P, Initiative C-CG-E, Australian Prostate Cancer B, Collaborators UKGPCS, Collaborators UKPS Freedman M, Conti DV, Easton D, Coetzee GA, Eeles RA, Kote-Jarai Z ( 2015 ) Multiple novel prostate cancer susceptibility signals identified by fine-mapping of known risk loci among Europeans . Hum Mol Genet 24 (19) : 5589 – 5602 .
Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, de Bono JS ( 2015 ) Prostate cancer . Lancet 387 (10013) : 70 – 82 .
Cancer Research UK ( 2014 ) Cancer Statistics Report: Cancer Incidence and Mortality in the UK, September 2014 .
Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, Sawyer E, Wilkinson R, Ardern-Jones A, Ellis S, Frost D, Peock S, Evans DG, Tischkowitz M, Cole T, Davidson R, Eccles D, Brewer C, Douglas F, Porteous ME, Donaldson A, Dorkins H, Izatt L, Cook J, Hodgson S, Kennedy MJ, Side LE, Eason J, Murray A, Antoniou AC, Easton DF, Kote-Jarai Z, Eeles R ( 2013 ) Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer . J Clin Oncol 31 (14) : 1748 – 1757 .
Chang X, Wang K ( 2012 ) wANNOVAR: annotating genetic variants for personal genomes via the web . J Med Genet 49 (7) : 433 – 436 .
Clark N, Wu X, Her C ( 2013 ) MutS homologues hMSH4 and hMSH5: genetic variations, functions, and implications in human diseases . Curr Genomics 14 (2) : 81 – 90 .
Cybulski C, Wokolorczyk D, Kluzniak W, Jakubowska A, Gorski B, Gronwald J, Huzarski T, Kashyap A, Byrski T, Debniak T, Golab A, Gliniewicz B, Sikorski A, Switala J, Borkowski T, Borkowski A, Antczak A, Wojnar L, Przybyla J, Sosnowski M, Malkiewicz B, Zdrojowy R, Sikorska-Radek P, Matych J, Wilkosz J, Rozanski W, Kis J, Bar K, Bryniarski P, Paradysz A, Jersak K, Niemirowicz J, Slupski P, Jarzemski P, Skrzypczyk M, Dobruch J, Domagala P, Narod SA, Lubinski J Polish Hereditary Prostate Cancer C ( 2013 ) An inherited NBN mutation is associated with poor prognosis prostate cancer . Br J Cancer 108 (2) : 461 – 468 .
Delaneau O, Zagury JF, Marchini J ( 2013 ) Improved whole-chromosome phasing for disease and population genetic studies . Nat Methods 10 (1) : 5 – 6 .
Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W ( 2003 ) Mutations in CHEK2 associated with prostate cancer risk . Am J Hum Genet 72 (2) : 270 – 280 .
Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, Easton D, Kote-Jarai Z ( 2014 ) The genetic epidemiology of prostate cancer and its clinical implications . Nat Rev Urol 11 (1) : 18 – 31 .
Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth-Little S, Dadaev T, Neal DE, Hamdy FC, Donovan JL, Muir K, Giles GG, Severi G, Wiklund F, Gronberg H, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Lindstrom S, Kraft P, Hunter DJ, Gapstur S, Chanock SJ, Berndt SI, Albanes D, Andriole G, Schleutker J, Weischer M, Canzian F, Riboli E, Key TJ, Travis RC, Campa D, Ingles SA, John EM, Hayes RB, Pharoah PD, Pashayan N, Khaw KT, Stanford JL, Ostrander EA, Signorello LB, Thibodeau SN, Schaid D, Maier C, Vogel W, Kibel AS, Cybulski C, Lubinski J, Cannon-Albright L, Brenner H, Park JY, Kaneva R, Batra J, Spurdle AB, Clements JA, Teixeira MR, Dicks E, Lee A, Dunning AM, Baynes C, Conroy D, Maranian MJ, Ahmed S, Govindasami K, Guy M, Wilkinson RA, Sawyer EJ, Morgan A, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As NJ, Woodhouse CJ, Thompson A, Dudderidge T, Ogden C, Cooper CS, Lophatananon A, Cox A, Southey MC, Hopper JL, English DR, Aly M, Adolfsson J, Xu J, Zheng SL, Yeager M, Kaaks R, Diver WR, Gaudet MM, Stern MC, Corral R, Joshi AD, Shahabi A, Wahlfors T, Tammela TL, Auvinen A, Virtamo J, Klarskov P, Nordestgaard BG, Roder MA, Nielsen SF, Bojesen SE, Siddiq A, Fitzgerald LM, Kolb S, Kwon EM, Karyadi DM, Blot WJ, Zheng W, Cai Q, McDonnell SK, Rinckleb AE, Drake B, Colditz G, Wokolorczyk D, Stephenson RA, Teerlink C, Muller H, Rothenbacher D, Sellers TA, Lin HY, Slavov C, Mitev V, Lose F, Srinivasan S, Maia S, Paulo P, Lange E, Cooney KA, Antoniou AC, Vincent D, Bacot F, Tessier DC Initiative CO-CRUG-E, Australian Prostate Cancer BOncology UKGPCSCBAoUSSo, Collaborators UKPS, Consortium P Kote-Jarai Z, Easton DF ( 2013 ) Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array . Nat Genet 45 (4) : 385 – 391e1, –2 .
GTEx Consortium ( 2015 ) Human genomics. the genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans . Science 348 (6235) : 648 – 460 .
Hjelmborg JB, Scheike T, Holst K, Skytthe A, Penney KL, Graff RE, Pukkala E, Christensen K, Adami HO, Holm NV, Nuttall E, Hansen S, Hartman M, Czene K, Harris JR, Kaprio J, Mucci LA ( 2014 ) The heritability of prostate cancer in the Nordic Twin Study of Cancer . Cancer Epidemiol Biomarkers Prev 23 (11) : 2303 – 2310 .
Hoffmann TJ, Witte JS ( 2015 ) Strategies for imputing and analyzing rare variants in association studies . Trends Genet 31 (10) : 556 – 563 .
Howie BN, Donnelly P, Marchini J ( 2009 ) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies . PLoS Genet 5 (6) : e1000529 .
Ilic D, Neuberger MM, Djulbegovic M, Dahm P ( 2013 ) Screening for prostate cancer . Cochrane Database Syst Rev 1 : CD004720 .
Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X ( 2013 ) Sequence kernel association tests for the combined effect of rare and common variants . Am J Hum Genet 92 (6) : 841 – 853 .
Ji G, Long Y, Zhou Y, Huang C, Gu A, Wang X ( 2012 ) Common variants in mismatch repair genes associated with increased risk of sperm DNA damage and male infertility . BMC Med 10 : 49 .
Kang J, D'Andrea AD, Kozono D ( 2012 ) A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy . J Natl Cancer Inst 104 (9) : 670 – 681 .
Kazma R, Babron MC, Gaborieau V, Genin E, Brennan P, Hung RJ, McLaughlin JR, Krokan HE, Elvestad MB, Skorpen F, Anderssen E, Vooder T, Valk K, Metspalu A, Field JK, Lathrop M, Sarasin A, Benhamou S consortium I ( 2012 ) Lung cancer and DNA repair genes: multilevel association analysis from the International Lung Cancer Consortium . Carcinogenesis 33 (5) : 1059 – 1064 .
Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, Tymrakiewitcz M, O'Brien L, Hall A, Wilkinson R, Al Olama AA, Morrison J, Muir K, Neal D, Donovan J, Hamdy F, Easton DF, Eeles R Collaborators U, British Association of Urological Surgeons' Section of O ( 2009 ) A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer . Br J Cancer 100 (2) : 426 – 430 .
Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O'Brien L, Sawyer E, Hall A, Wilkinson R, Dadaev T, Goh C, Easton D Collaborators U, Goldgar D, Eeles R ( 2011 ) BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients . Br J Cancer 105 (8) : 1230 – 1234 .
Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA Team NGESP-ELP Christiani DC, Wurfel MM, Lin X ( 2012 ) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies . Am J Hum Genet 91 (2) : 224 – 237 .
Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M, O'Brien L, Sawyer E, Hall A, Wilkinson R, Easton D Collaborators U Goldgar D, Eeles R, Kote-Jarai Z ( 2012 ) Germline BRCA1 mutations increase prostate cancer risk . Br J Cancer 106 (10) : 1697 – 1701 .
Leongamornlert D, Saunders E, Dadaev T, Tymrakiewicz M, Goh C, Jugurnauth-Little S, Kozarewa I, Fenwick K, Assiotis I, Barrowdale D, Govindasami K, Guy M, Sawyer E, Wilkinson R Collaborators U Antoniou AC, Eeles R, Kote-Jarai Z ( 2014 ) Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease . Br J Cancer 110 (6) : 1663 – 1672 .
Luedeke M, Linnert CM, Hofer MD, Surowy HM, Rinckleb AE, Hoegel J, Kuefer R, Rubin MA, Vogel W, Maier C ( 2009 ) Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes . Cancer Epidemiol Biomarkers Prev 18 (11) : 3030 – 3035 .
Makridakis NM, Reichardt JK ( 2012 ) Translesion DNA polymerases and cancer . Front Genet 3 : 174 .
Quaresma M, Coleman MP, Rachet B ( 2015 ) 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971-2011: a population-based study . Lancet 385 (9974) : 1206 – 12018 .
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M ( 2015 ) Integrative analysis of 111 reference human epigenomes . Nature 518 (7539) : 317 – 330 .
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM ( 2015 ) Integrative clinical genomics of advanced prostate cancer . Cell 161 (5) : 1215 – 1228 .
Scarbrough PM, Weber RP, Iversen ES, Brhane Y, Amos CI, Kraft P, Hung RJ, Sellers TA, Witte JS, Pharoah P, Henderson BE, Gruber SB, Hunter DJ, Garber JE, Joshi AD, McDonnell K, Easton DF, Eeles R, Kote-Jarai Z, Muir K, Doherty JA, Schildkraut JM ( 2016 ) A cross-cancer genetic association analysis of the dna repair and dna damage signaling pathways for lung, ovary, prostate, breast, and colorectal cancer . Cancer Epidemiol Biomarkers Prev 25 (1) : 193 – 200 .
Wang K, Li M, Hakonarson H ( 2010 ) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data . Nucleic Acids Res 38 (16) : e164 .
Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, Qureshi M, Dong Q, Gu X, Chen WV, Spitz MR, Eisen T, Amos CI, Houlston RS ( 2008 ) Common 5p15.33 and 6p21.33 variants influence lung cancer risk . Nat Genet 40 (12) : 1407 – 1409 .
Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, Zong X, Laplana M, Wei Y, Han Y, Lloyd A, Delahaye-Sourdeix M, Chubb D, Gaborieau V, Wheeler W, Chatterjee N, Thorleifsson G, Sulem P, Liu G, Kaaks R, Henrion M, Kinnersley B, Vallee M, LeCalvez-Kelm F, Stevens VL, Gapstur SM, Chen WV, Zaridze D, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Krokan HE, Gabrielsen ME, Skorpen F, Vatten L, Njolstad I, Chen C, Goodman G, Benhamou S, Vooder T, Valk K, Nelis M, Metspalu A, Lener M, Lubinski J, Johansson M, Vineis P, Agudo A, Clavel-Chapelon F, Bueno-de-Mesquita HB, Trichopoulos D, Khaw KT, Johansson M, Weiderpass E, Tjonneland A, Riboli E, Lathrop M, Scelo G, Albanes D, Caporaso NE, Ye Y, Gu J, Wu X, Spitz MR, Dienemann H, Rosenberger A, Su L, Matakidou A, Eisen T, Stefansson K, Risch A, Chanock SJ, Christiani DC, Hung RJ, Brennan P, Landi MT, Houlston RS, Amos CI ( 2014 ) Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer . Nat Genet 46 (7) : 736 – 741 .
Ward LD, Kellis M ( 2016 ) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease . Nucleic Acids Res 44 (D1) : D877 – D881 .
Wood RD, Mitchell M, Lindahl T ( 2005 ) Human DNA repair genes, 2005 . Mutat Res 577 (1-2) : 275 – 283 .
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X ( 2011 ) Rare-variant association testing for sequencing data with the sequence kernel association test . Am J Hum Genet 89 (1) : 82 – 93 .
Wu X, Xu Y, Feng K, Tompkins JD, Her C ( 2013 ) MutS homologue hMSH5: recombinational DSB repair and non-synonymous polymorphic variants . PLoS One 8 (9) : e73284 .
Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G, Xu C, Wang X, Shao Q, Chen Z, Tao Z, Qi J, Zhou F, Wang Z, Fu Y, He D, Wei Q, Guo J, Wu D, Gao X, Yuan J, Wang G, Xu Y, Wang G, Yao H, Dong P, Jiao Y, Shen M, Yang J, Ou-Yang J, Jiang H, Zhu Y, Ren S, Zhang Z, Yin C, Gao X, Dai B, Hu Z, Yang Y, Wu Q, Chen H, Peng P, Zheng Y, Zheng X, Xiang Y, Long J, Gong J, Na R, Lin X, Yu H, Wang Z, Tao S, Feng J, Sun J, Liu W, Hsing A, Rao J, Ding Q, Wiklund F, Gronberg H, Shu XO, Zheng W, Shen H, Jin L, Shi R, Lu D, Zhang X, Sun J, Zheng SL, Sun Y ( 2012 ) Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4 . Nat Genet 44 (11) : 1231 – 1235 .
Yadav S, Mukhopadhyay S, Anbalagan M, Makridakis N ( 2015 ) Somatic mutations in catalytic core of POLK reported in prostate cancer alter translesion DNA synthesis . Hum Mutat 36 (9) : 873 – 880 .
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Mr Z Abbasi, Mr M Akhlil Abdul-Hamid, Mr Paul D Abel, Professor Paul H Abrams, Dr Fawzi A Adab, Mr Andrew Adamson, Mr A Adeyoju, Mr Naveed Afzal, Mr Ernest K N Ahiaku, Mr Munir Ahmed, Mr Mohammed L Al Sudani, Dr Christopher Alcock, Dr Zulfiqar Ali, Mr David J Almond, Dr Roberto Alonzi, Dr Amir S M Al-Samarraie, Dr Al-Samerraie, Mr Waleed Al-Singary, Mr Al-Sudani, Mr John Anderson, Mr Steven Andrews, Mr Henry Andrews, Mr Iqbal Anjum, Mr Ken Anson, Dr Nicola A Anyamene, Mr Ike Apakama, Dr F Aparcia, Mr J A A Archbold, Dr D Ash, Dr Richard F U Ashford, Dr A Azzabi, Mr David Badenoch, Dr Amit Bahl, Mr M J Bailey, Mrs Karen Bailey, Mr Andrew J Ball, Mr G Banerjee, Dr N Barber, Dr Jim Barber, Dr Baria, Mr Douglas G Barnes, Mr J Bashir, Mr Pradip Basu, Mr Christopher A Bates, Dr N A Bax, Mr D Baxter-Smith, Mr Amar Bdesha, Mr Christopher J M Beacock, Professor Ronald P Beaney, Mr Ralph Beard, Mr John D Beatty, Mr Rupert Beck, Ms Gail Beese, Dr Sharon Beesley, Mr C Richard W Bell, Mr James Bellringer, Dr Richard Benson, Dr Beresford, Mr Christopher R A Bevis, Dr Rajanee Bhana, Mr S Bhanot, Dr A Bhatnagar, Mr R I Bhatt, Mr Brian Birch, Dr Alison Birtle, Mr M Bishop, Mr C Shekhar Biyani, Mr A R E Blacklock, Miss Rosemary Blades, Dr Peter Bliss, Dr David J Bloomfield, Miss S Boddy, Professor C M Booth, Mr Pradeep Bose, Dr Michael C Bott, Dr David Bottomley, Mr Nigel R Boucher, Dr J Bowen, Dr Mark Bower, Mr W G Bowsher, Mr P J R Boyd, Mr F James Bramble, Mr Simon F Brewster, Mr Tim Briggs, Dr Cathryn Brock, Dr Sue Brock , Mr Stephen Bromage, Mr Richard Brough, Dr Richard Brown, Mr Stephen Brown, Mr Richard Brown, Mr Tony J Browning, Mr N Bryan, Mr Neil A Burgess, Mr Nicholas Burns-Cox, Mr Paul C Butterworth, Mr D Cahill, Mr P S Callaghan, Mr John Calleary, Dr M Calleja, Dr Frances Calman, Dr Philip Camilleri, Mr Alister Campbell, Miss Andrea Cannon, Dr Dawn M Carnell, Mr T W Carr, Mr Simon Carter, Mr Charles J M Carter, Dr Adam C Carter, Dr Bruce M Castle, Mr David Chadwick, Mr Rohit Chahal, Dr P Chakraborti, Mr Chappell, Mr C Charig, Dr Anula D Chetiyawardana, Mr Christopher Chilton, Mr F I Chinegwundoh, Dr Irene Chong, Dr Ananya Choudhury, Mr Wai-Man Chow, Mr Timothy J Christmas, Dr Mark J Churn, Mr Noel W Clarke, Mr Jorge Clavijo-Eisele, Dr M Coe, Mr N P Cohen, Mr C Coker, Dr Trevor Cole, Dr David J Cole, Mr O Cole, Mr Gerald Collins, Dr Matthew Collinson, Mr I Conn, Dr C Connell, Dr Audrey Cook, Mr Peter Cooke, Mr Graeme Cooksey, Mr L Coombs, Mr Robert F Copland, Mr Andrew J Cornaby, Mr P A Cornford, Mr Corolis, Mr John Corr, Mr C B Costello, Mrs N Coull, Dr Richard Cowan, Mr Robert Cox, Dr C Coyle, Mr Jeremy Crew, Mr John C Crisp, Dr W Cross, Mr W Cross, Dr Dorthe Cruger, Mr Malcolm Crundwell, Mr Cummings, Mr Nazeer Dahar, Dr Francis N Daniel, Mr J Darrad, Mr Pallon Daruwala, Mr Gautam Das, Mr Shibendra Datta, Dr S Davidson, Dr Joseph Davies, Mr Owen W Davison, Mr Guy Dawkins, Mr Chris Dawson, Mr Alan R De Bolla, Professor David Dearnaley, Mr Ken M Desai, Dr George P Deutsch, Mr John Dick, Mr Andrew J Dickinson, Dr Jeanette Dickson, Mr Michael Dinneen, Dr Sanjay Dixit, Dr H Jane Dobbs, Mr A Doble, Dr David Dodds, Mr Alan Doherty, Mr P Donaldson, Dr M Dooldeniya, Dr S Fiona Douglas, Mr Drake, Dr Gill M Duchesne, Mr Peter Duffy, Mr Michael Dunn, Mr W D Dunsmuir, Dr Sajid K Durrani, Mr Alan C Eaton, Professor Diane Eccles, Mr B Eddy, Mr C D Eden, Mr J Edwards, Mr Jeremy Elkabir, Dr P Tony Elliott, Mr B W Ellis, Dr R Ellis, Dr A El-Modir, Mr Andrew W S Elves, Dr Christine Elwell, Mr Mark Emberton, Dr Louise Emmerson, Mr Roland C D England, Mr R D Errington, Professor D Gareth Evans, Dr Alison Falconer, Mr Derek Fawcett, Dr C Featherston, Dr Carolyn J Featherstone, Mr Jeremy Feggetter, Dr C Ferguson, Dr D Fermont, Mr Michael Ferro, Mr Matthew Fletcher, Dr A Folkes, Mr Trevor F Ford, Mr Paul W Foster, Dr Kevin N Franks, Dr Olivera Frim, Dr Joanna Gale, Mr Christopher Gallegos, Mr James S Gelister, Dr Ghana, Dr Stephanie Gibbs, Mr Hugh Gilbert, Mr David Gillatt, Dr John Glaholm, Mr Jonathan M Glass, Mr James Glenister, Dr Thomas D Goode, Ms E M Gordon, Mr Richard L Gower, Dr John Graham, Mr Damian Green, Mr Jonathan Greenland, Dr Robert Grieve, Mr Thomas R L Griffiths, Mr Sandy Gujral, Dr Nishi Gupta, Mr Riza Murat Gurun, Mr Peter J Guy, Mr Neil Haldar, Mr N Halder, Professor F C Hamdy, Dr C Hamilton, Mr John Hammonds, Mr S J Hampson, Mr Damien C Hanbury, Dr P D John Hardman, Dr Stephen J Harland, Mr John M Harney, Dr Peter Harper, Dr Sarah Harris, Mr D Harris, Mr G S M Harrison, Mr D R Harriss, Mr N Harvey-Hills, Mr Simon Hawkyard, Dr Catherine M Heath, Mr Michael Hehir, Mr Giles O Hellawell, Mr David Hendry, Mr Mike Henley, Dr Ann Henry, Dr John Hetherington, Dr Tamas Hickish, Mr James A Hicks, Dr Serena Hilman, Mr Richard Hindley, Mr John R Hindmarsh, Mr John Hines, Dr M Hingorani, Mr Edwin T S Ho, Professor Shirley Hodgson, Dr U Hoffman, Mr David Holden, Dr A Hollingdale, Mr Graham W Hollins, Mr Simon A V Holmes, Dr Gail Horan, Professor Alan Horwich, Professor Peter Hoskin, Mr Graham P Howell, Mr D Hrouda, Dr Robert Huddart, Ms Liz Hudson, Dr Rob Hughes, Mr Michael Hughes, Mr Owen Hughes, Dr Caroline Humber, Mr John W Iacovou, Dr A Ibrahim, Mr John A Inglis, Mr Stuart Irving, Mr C Irwin, Dr Louise Izatt, Mr Victor Izegbu, Mr Basharat Jameel, Mr Michael J James, Professor N James, Mr R Lester James, Mr Pradip Javle, Dr P Jenkins, Dr Sameer Jhavar, Dr Gareth Jones, Mr Chris R Jones, Dr David A Jones, Mr J Joseph, Dr Shelagh Joss, Mr Amir Kaisary, Dr Alexandre L Kaliski, Dr G Kapur, Mr O Karim, Dr Stephen J Karp, Mr F X Keeley, Mr Anand R Kelkar, Mr J P Kelleher, Mr John Kelly, Dr Sue Kenwrick, Mr F Khan, Dr Vincent Khoo, Ms Rachel M Kimber, Mr R Kinder, Professor Roger S Kirby, Professor David Kirk, Dr Peter Kirkbride, Mr Magdi M Kirollos, Mr Roger Kockelbergh, Mr Philip C W C Koenig, Mr Gordon G Kooiman, Dr O Koreich, Mr Anthony Koupparis, Mr Mohamed Kourah, Dr Sigurd Kraus, Ms Magda L Kujawa, Mr Ravi Kulkarni, Mr M Kumar, Dr Ian H Kunkler, Professor H Kynaston, Dr Katherine L Lachlan, Dr Robert Laing, Dr Fiona Lalloo, Mr M Lancashire, Mr Stephen E M Langley, Mr Marc Laniado, Mr T R Larner, Mr Maurice W Lau, Mr W T Lawrence, Miss Anne Lawson, Mr Pieter J Le Roux, Professor Mary Leader, Mr J O Lee, Ms L Lee, Ms A Lee, Dr R John Lemburger, Dr Priscilla Leone, Dr Jason Lester, Mr Hing Leung, Mr J Lewis, Mr D Christopher Lewis, Mr Thomas Liston, Dr Jacqueline Livsey, Mr S Lloyd, Dr Imogen Locke, Mr Richard Lodge, Dr John Logue, Mr Mark Longmuir, Mr Malcolm G Lucas, Mr C J Luscombe, Dr Anna Lydon, Mr Michael Lynch, Mr Naing N K Lynn, Mr James P A MacDermott, Mr Ruaraidh P Macdonagh, Mr Macdonald, Mr Sanjeev Madaan, Dr Kudingila R Madhava, Dr Joseph Maguire, Professor E R Maher, Dr Rana Mahmood, Dr Graeme H M Mair, Mr Peter R Malone, Dr Stephen A Mangar, Mr Mark Mantle, Mr I Mark, Mr Robert Mason, Professor M D Mason, Mr Matanhelia, Mr Shyam Matenhelia, Mr Philip N Matthews, Dr J McAleese, Ms Donna McBride, Mr Jonathan McFarlane, Mr McGrath, Mr Craig McIlhenny, Mr Paul McInerney, Mr Gregor McIntosh, Dr F McKinna, Dr Duncan McLaren, Miss Esther McLarty, Dr Rhona McMenemin, Mr Alan McNeill, Mr T A McNicholas, Mr Robert N Meddings, Mr A David Mee, Dr Lucinda Melcher, Mr Memon, Mr Pravin Menzes, Mr Marek Miller, Mr Robert Mills, Mr S Mitchell, Dr Natasha Mithal, Dr Anita Mitra, Ms Gillian E Mobb, Mr Leslie E F Moffat, Mr Mokete, Dr Julian Money-Kyrle, Mr Bruce Montgomery, Mr Martin P Moody, Mr Roland Morley, Mr Sean B Morris, Professor Patrick Morrison, Dr Diana Mort, Mr Amir H Mostafid, Mr Hanif Motiwala, Mr Gulzar Mufti, Mr Gordon Muir, Mr Faiz Mumtaz, Mr Michael Murphy, Mr Keith W Murray, Dr Alexandra Murray, Dr Shirley Murrell, Dr D Muthukumar, Mr Harry Naerger, Mr Siva Namasivayam, Mr Vinod Nargund, Mr Nawrocki, Mr Donald Neilson, Dr A Nethersell, Mr Julian Barwell, Dr Jacqueline C Newby, Dr Hugh Newman, Dr R Newton, Mr Neil Oakley, Mr P J O'Boyle, Mr J O'Brien, Mr Tim S O'Brien, Dr H O'Donnell, Mr Neil O'Donoghue, Mr E O'Donoghue, Mr Chris Ogden, Mr Hemant Ohja, Professor Tim Oliver, Mr Eng K Ong, Mr P O'Reilly, Dr J S O'Rourke, Mr David Osborn, Dr Peter Ostler, Professor Joe O'Sullivan, Dr J Owen, Mr Edward Palfrey, Dr Miguel Panades, Dr Niki Panakis, Mr M Pancharatnam, Mr Michalakis L Pantelides, Dr U Panwar, Dr Omi Parikh, Dr Chris Parker, Mr Christopher H Parker, Mr Bohdan T Parys, Dr Sarah Pascoe, Mr Anup Patel, Dr Joan Paterson, Mr S Pathack, Ms Jhumur Pati, Dr Helen Patterson, Dr Pattu, Mr A Paul, Dr Heather Payne, Dr David Peake, Dr I Pedley, Mr A Pengelly, Mr Amjad M Peracha, Dr Matthew Perry, Mr Raj Persad, Mr John Peters, Mr N H Philp, Mr T Philp, Dr Lisa M Pickering, Dr Katharine Pigott, Mr R Plail, Dr P Nicholas Plowman, Mr Richard D Pocock, Mr A J Pope, Mr Rick Popert, Mr Tim Porter, Mr John M Potter, Mr Christopher Powell, Dr Thomas B Powles, Mr Krishna Prasad, Mr Seshadri Sri Prasad, Mr J W Prejbisz, Mr Stephen Prescott, Dr Andrew Protheroe, Mr Khaver N Qureshi, Dr Nigel Raby, Dr Narasimhan Ragavan, Mr Palaniappa G S Raju, Dr Prakash B Ramachandra, Dr R Raman, Mr Abhay Rane, Dr Julia Rankin, Mr Y Rao, Mr Hari L Ratan, Mr Ramachandran Ravi, Dr K Ravishankar, Dr Read, Mr Paul J Reddy, Mr Peter R Rimington, Dr Peter A Ritchie, Dr J Trevor Roberts, Mr Andrew Robertson, Dr Angus Robinson, Dr Anne C Robinson, Mr Lee Q Robinson, Mr Mark A Rochester, Mr P B Rogers, Mr Tomas P Rosenbaum, Mr Neil Rothwell, Mr Carl Rowbotham, Mr Rowe, Dr Kathryn Rowley, Dr Deborah Ruddy, Mr John Rundle, Dr John M Russell, Mr P G Ryan, Dr A Sabharwal, Dr Anand K Saggar, Dr Ali Samanci, Mr Vijay K Sangar, Mr M F Saxby, Mr Hartwig Schwaibold, Dr John E Scoble, Dr Christopher Scrase, Mr Selim, Mr Henry Sells, Mr Krishna K Sethia, Mr David C Shackley, Dr Shaffer, Dr Nihil Shah, Dr D Shakespeare, Dr Sue Shanley, Mr Neerah K Sharma, Dr Denise J Sheehan, Dr Elizabeth Sherwin, Dr Poh Lin Shum, Dr LucySide, Dr Norma Sidek, Professor Karol Sikora, Dr R Simcock, Mr Andrew M Sinclair, Mr Pravin Singh, Dr M Siva, Mr Michael F Smith, Mr James Smith, Dr Michael Sokal, Mr Graham M Sole, Mr Mark J Speakman, Dr Alexander Spiers, Dr Thiagarajan Sreenivasan, Dr Narayanan N Srihari, Mr Srinivasan, Mr Rajagopalan Sriram, Dr John N Staffurth, Dr D Stewart, Dr Andrew Stockdale, Mr Mark A Stott, Mr M J Stower, Mr John R Strachan, Professor Nicholas S A Stuart, Dr Elaine Sugden, Mr Duncan Summerton, Dr Santhanam Sundar, Mr S K Sundaram, Mr Gokarakonda Suresh, Mr Shabbir Susnerwala, Mr Kuchibhotla S Swami, Miss Stephanie J Symons, Dr Isabel Syndikus, Dr Saad Tahir, Dr J Tanquay, Dr John W Taylor, Dr J W Taylor, Mr T Terry, Dr Robert J Thomas, Mr Stephen A Thomas, Mr Alan Thompson, Dr Alastair H Thomson, Dr A Thurston, Dr Owen Tilsley, Mr Stuart F Tindall, Dr K Tipples, Dr Tong, Mr Hamid Toussi, Dr Elizabeth W Toy, Professor Richard C Trembath, Mr David N Tulloch, Mr Kevin J Turner, Mr James Tweedle, Dr C J Tyrell, Mr N Umez-Eronini, Mr Graeme H Urwin, Mr Justin A Vale, Dr Van As, Dr Nicholas Van As, Dr Subramaniam Vasanthan, Mr Sean Vesey, Dr Maria Vilarino-Varela, Dr John Violet, Mr Jaspal Virdi, Dr Robert Wade, Dr Katherine Waite, Mr E M Walker, Mr Roger Walker, Mr David M A Wallace, Mr Nicholas A Watkin, Mr M E Watson, Professor J H Waxman, Mr Brian Waymont, Dr Andrew Weaver, Mr Ralph J Webb, Mr Andrew Wedderburn, Dr Paula Wells, Mr G D Wemyss-Holden, Mr P M T Weston, Dr Duncan Wheatley, Mr P Whelan, Dr D Whillis, Mr Adam D Wilde, Dr Vicki Wiles, Dr Marie Wilkins, Mr John H Williams, Mr Simon Williams, Mr Michael Willis, Mr Michael I Wills, Mr Richard Wilson, Mr J R Wilson, Mr Mathias H Winkler, Dr Marcus Wise, Mr Simon Woodhams, Professor C Woodhouse, Dr Cathryn Woodward, Dr Woolf, Mr K A Woolfenden, Dr Jane Worlding, Mr Mark Wright, Dr WYLIE, Dr James P Wylie, Dr Chris Wynne, Ms Angelika Zang, Dr A Zarkar
Angela Cox, Paul M. Brown, Anne George, Gemma Marsden, Athene Lane, Michael DavisPrasad Bollina, Sue Bonnington, Lynne Bradshaw, James Catto, Debbie Cooper, Liz Down, Andrew Doble, Alan Doherty, Garrett Durkan, Emma Elliott, David Gillatt, Pippa Herbert, Peter Holding, Joanne Howson, Mandy Jones, Roger Kockelbergh, Rajeev Kumar, Howard Kynaston, Athene Lane, Teresa Lennon, Norma Lyons, Hing Leung, Malcolm Mason, Hilary Moody, Philip Powell, Alan Paul, Stephen Prescott, Derek Rosario, Patricia O'Sullivan, Pauline Thompson, Sarah Tidball
Margaret Cook 1, Angela Morgan 2, Artitaya Lophatananon 3, 4, Cyril Fisher 2, Malgorzata Tymrakiewicz 2, Michelle Guy 2, Rosemary Wilkinson 2, Sara Jugurnauth-Little 2, Steve Hazel 2, Melissa C. Southey 5, Liesel M. Fitzgerald 6, John Pedersen 7, John Hopper 8, Ami Karlsson 9, Carin Cavalli-Bjoerkman 9, Jan-Erik Johansson 9, Jan Adolfson 9, Markus Aly 9, 10, Michael Broms 9, Paer Stattin 9, Brian E. Henderson 11, Fredrick Schumacher 11, Anssi Auvinen 12, Kimmo Taari 13, Liisa Maeaettaenen 14, Paula Kujala 15, Teemu Murtola 16, 17, Teuvo LJ Tammela 17, Tiina Wahlfors 18, Andreas Roder 19, Peter Iversen 19, Peter Klarskov 20, Sune F. Nielsen 21, 22, Tim J. Key 23, Hans Wallinder 24, Sven Gustafsson 24, Jenny L. Donovan 25, Freddie Hamdy 26, Angela Cox 27, Anne George 28, Athene Lane 28, Gemma Marsden 26, Michael Davis 25, Paul Brown 25, Paul Pharoah 29, Lisa B. Signorello 31, 30, Wei Zheng 32, Shannon K. McDonnell 33, Daniel J. Schaid 33, Liang Wang 33, Lori Tillmans 33, Shaun Riska 33, Thomas Schnoeller 34, Kathleen Herkommer 35, Manuel Luedeke 34, Walther Vogel 36, Dominika Wokolorczyk 37, Jan Lubiski 37, Wojciech Kluzniak 37, Katja Butterbach 38, Christa Stegmaier 39, Bernd Holleczek 39, Hui-Yi Lin 40, Hyun Park 41, Julio Pow-Sang 41, Thomas Sellers 41, Chavdar Slavov 42, Aleksandrina Vlahova 43, Atanaska Mitkova 44, Darina Kachakova 44, Elenko Popov 42, Svetlana Christova 43, Tihomir Dikov 43, Vanio Mitev 44, Allison Eckert 45, Amanda Spurdle 46, Angus Collins 45, Glenn Wood 45, Greg Malone 45, Judith A. Clements 45, Kris Kerr 45, Megan Turner 45, Pamela Saunders 45, Peter Heathcote 45, Srilakshmi Srinivasan 45, Leire Moya 45, Trina Yeadon 45, Australian Prostate Cancer BioResource 45, Joana Santos 47, Carmen Jerónimo 47, Paula Paulo 47, Pedro Pinto 47, Rui Henrique 47, Sofia Maia 47, Agnieszka Michael 48, Andrzej Kierzek 48, Huihai Wu 48 1Centre for Cancer Genetic Epidemiology, Department of Public Health and Primary Care, University of Cambridge, Strangeways Laboratory, Worts Causeway, Cambridge CB1 8RN, UK, 2The Institute of Cancer Research, Sutton, UK, 3Institute of Population Health, University of Manchester, Manchester, UK, 4Warwick Medical School, University of Warwick, Coventry, UK, 5Genetic Epidemiology Laboratory, Department of Pathology, The University of Melbourne, Grattan Street, Parkville, Victoria 3010, Australia, 6Cancer Epidemiology Centre, The Cancer Council Victoria, 615 St Kilda Road, Melbourne, Victoria, Australia, 7Tissupath Pty Ltd., Melbourne, Victoria 3122, Australia, 8Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, The University of Melbourne, Victoria, Australia, 9Department of Medical Epidemiology and Biostatistics, Karolinska Institute, Stockholm, Sweden, 10Department of Clinical Sciences at Danderyds Hospital, Stockholm, Sweden, 11Department of Preventive Medicine, Keck School of Medicine, University of Southern California/Norris Comprehensive Cancer Center, Los Angeles, California, USA, 12Department of Epidemiology, School of Health Sciences, University of Tampere, Tampere, Finland, 13Department of Urology, Helsinki University Central Hospital and University of Helsinki, Helsinki, Finland, 14Finnish Cancer Registry, Helsinki, Finland, 15Fimlab Laboratories, Tampere University Hospital, Tampere, Finland, 16School of Medicine, University of Tampere, Tampere, Finland, 17Department of Urology, Tampere University Hospital and Medical School, University of Tampere, Finland, 18BioMediTech, University of Tampere and FimLab Laboratories, Tampere, Finland, 19Copenhagen Prostate Cancer Center, Department of Urology, Rigshospitalet, Copenhagen University Hospital, Tagensvej 20, 7521, DK-2200 Copenhagen, Denmark, 20Department of Urology, Herlev Hospital, Copenhagen University Hospital, Herlev Ringvej 75, DK-230 Herlev, Denmark, 21Department of Clinical Biochemistry, Herlev Hospital, Copenhagen University Hospital, Herlev Ringvej 75, DK-230 Herlev, Denmark, 22Faculty of Health and Medical Sciences, University of Copenhagen, 23Cancer Epidemiology Unit, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, UK, 24Department of Epidemiology and Biostatistics, School of Public Health, Imperial College, London, UK, 25School of Social and Community Medicine, University of Bristol, Canynge Hall, 39Whatley Road, Bristol, BS8 2PS, UK, 26Nuffield Department of Surgical Sciences, University of Oxford, Oxford, UK, Faculty of Medical Science, University of Oxford, John Radcliffe Hospital, Oxford, UK, 27CR-UK/YCR Sheffield Cancer Research Centre, University of Sheffield, Sheffield, UK, 28University of Cambridge, Department of Oncology, Box 279, Addenbrooke's Hospital, Hills Road Cambridge CB2 0QQ, UK, 29Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Strangeways Laboratory, Worts Causeway, Cambridge, UK, 30International Epidemiology Institute, 1555 Research Blvd., Suite 550, Rockville, MD 20850, USA, 31Department of Epidemiology, Harvard School of Public Health, 677 Huntington Avenue, Boston, MA 02115, USA, 32Division of Epidemiology, Department of Medicine, Vanderbilt University Medical Center, 2525 West End Avenue, Suite 800, Nashville, TN 37232 USA, 33Mayo Clinic, Rochester, Minnesota, USA, 34 Department of Urology, University Hospital Ulm, Germany, 35Department of Urology, Klinikum rechts der Isar der Technischen Universitaet Muenchen, Munich, Germany, 36Institute of Human Genetics, University Hospital Ulm, Germany, 37International Hereditary Cancer Center, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland, 38Division of Clinical Epidemiology and Aging Research, German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany, 39Saarland Cancer Registry, 66119 Saarbruecken, Germany, 40Biostatistics Program, Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL 33612, USA, 41Department of Cancer Epidemiology, Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL 33612, USA, 42Department of Urology and Alexandrovska University Hospital, Medical University, Sofia, Bulgaria, 43Department of General and Clinical Pathology, Medical University, Sofia, Bulgaria, 44Department of Medical Chemistry and Biochemistry, Molecular Medicine Center, Medical University, Sofia, 2 Zdrave Str., 1431 Sofia, Bulgaria, 45Australian Prostate Cancer Research Centre-Qld, Institute of Health and Biomedical Innovation and School of Biomedical Science, Queensland University of Technology, Brisbane, Australia, 46Molecular Cancer Epidemiology Laboratory, Queensland Institute of Medical Research, Brisbane, Australia, 47Department of Genetics, Portuguese Oncology Institute, Porto, Portugal, 48The University of Surrey, Guildford, Surrey, GU2 7XH, UK
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Saunders, E., Dadaev, T., Leongamornlert, D. et al. Gene and pathway level analyses of germline DNA-repair gene variants and prostate cancer susceptibility using the iCOGS-genotyping array. Br J Cancer 114, 945–952 (2016). https://doi.org/10.1038/bjc.2016.50
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.50

{kind=link}
{kind=link}
{kind=link}
{kind=link}