
Abstract
Background:
We aimed to compare the efficacy and safety of irinotecan/S-1 (IRIS) therapy with S-1 monotherapy in patients with gemcitabine-refractory pancreatic cancer.
Methods:
Patients were treated with oral S-1 (80–120 mg for 14 days every 4 weeks) plus intravenous irinotecan (100 mg m−2 on days 1 and 15 every 4 weeks; IRIS group) or oral S-1 group (80–120 mg daily for 28 days every 6 weeks). The primary endpoint was progression-free survival (PFS).
Results:
Of 137 patients enrolled, 127 were eligible for efficacy. The median PFS in the IRIS group and S-1 monotherapy group were 3.5 and 1.9 months, respectively (hazard ratio (HR)=0.77; 95% confidence interval (CI), 0.53–1.11; P=0.18), while the median overall survival (OS) were 6.8 and 5.8 months, respectively (HR=0.75; 95% CI, 0.51–1.09; P=0.13). Response rate was significantly higher in the IRIS group than in the S-1 monotherapy group (18.3% vs 6.0%, P=0.03). Grade 3 or higher neutropenia and anorexia occurred more frequently in the IRIS group.
Conclusions:
There was a trend for better PFS and OS in the IRIS group that could be a treatment arm in the clinical trials for gemcitabine-refractory pancreatic cancer.
Similar content being viewed by others


Main
Pancreatic cancer is a poor-prognosis disease and the eighth leading cause of cancer-related deaths worldwide, with 266 000 deaths reported in 2008 (Jemal et al, 2011). Gemcitabine (GEM) monotherapy, GEM-containing combination regimens, and a regimen consisting of folinic acid, fluorouracil, irinotecan, and oxaliplatin (FOLFIRINOX) have been used as first-line standard treatments (Burris et al, 1997; Moore et al, 2007; Conroy et al, 2011; Von Hoff et al, 2013).
For second-line pancreatic cancer, three phase III studies have been recently reported (Oettle et al, 2014; Gill et al, 2016; Wang-Gillam et al, 2016). Two out of them demonstrated positive results (Oettle et al, 2014; Wang-Gillam et al, 2016).
S-1 is an oral anticancer fluropyrimidine derivative that combines tegafur, a pro-drug of 5-fluorouracil (FU), with 2 modulators, that is, gimeracil and oteracil. S-1 was reported to have antitumour effect in chemotherapy-naive pancreatic cancer in Japan (Okusaka et al, 2008). S-1 is also recommended as a second-line treatment for GEM-refractory pancreatic cancer in Japan (Morizane et al, 2009; Japan Pancreas Society, 2013). A phase II study (n=40) yielded promising outcomes, a median progression-free survival (PFS) of 2.0 months and overall survival (OS) of 4.5 months, respectively, with acceptable tolerability (Morizane et al, 2009). An in vitro study using pancreatic cancer cell lines indicated that irinotecan hydrochloride (CPT-11)/5-FU had significantly higher growth inhibition activity than single drug treatment (Piacentini et al, 2006); CPT-11 is a semi-synthetic camptothecin derivative that inhibits topoisomerase I. These data suggest that CPT-11/5-FU may have promising efficacy against pancreatic cancer.
Shitara et al (2008) conducted a pilot study to evaluate the efficacy and safety of CPT-11/S-1 (IRIS) therapy in patients with pancreatic cancer (first-line treatment, n=10, GEM-refractory second-line treatment: n=6). This study indicated that IRIS therapy improved the response rate (RR; 43.7%), median time to progression (4.9 months), and median OS (11.3 months). In colorectal cancer, efficacy of IRIS therapy was also reported in phase II (Komatsu et al, 2011) and phase III (Muro et al, 2010) studies (doses of CPT-11 were 100 and 125 mg m−2, respectively; dose of S-1 was 80–120 mg per day in common), and it is recommended as one of the standard therapy in European Society of Medical Oncology guidelines and Japanese Society for Cancer of the Colon and Rectum (Schmoll et al, 2012; Japanese Society for Cancer of the Colon and Rectum, 2014). Based on these results, as well as the need for treatment options in pancreatic cancer, we conducted a randomised phase II study to evaluate the efficacy and safety of IRIS therapy compared with S-1 monotherapy as a second-line treatment in patients with GEM-refractory pancreatic cancer.
Materials and methods
Patients
The major eligibility criteria were as follows: histologically or cytologically proven adenocarcinoma or adenosquamous carcinoma, previous GEM-refractory pancreatic cancer, disease progression confirmed by computed tomography (CT), an age of 20 years or more, at least one measurable lesion identified under the Response Evaluation Criteria in Solid Tumors (RECIST), Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0–1, and adequate organ function. GEM-refractory was defined as follows: (i) patients who experienced progression confirmed by the image during the first-line therapy including GEM or (ii) patients who relapsed during the GEM-adjuvant treatment or within 24 weeks after the last GEM administration. The major exclusion criteria were as follows: a treatment history of CPT-11 or fluorinated pyrimidines, a history of radiation therapy except for intraoperative radiation, moderate or severe pleural and peritoneal effusions, synchronous or metachronous cancer except for radically treated early cancer, clinically significant cardiac disease, and active infections.
All patients provided written informed consent before enrolment. This study was conducted in accordance with the Declaration of Helsinki (2008) and the Good Clinical Practice (GCP) guideline. Each hospital’s institutional review board approved this study protocol.
Study design
This study was a multicentre, randomised, comparative phase II study conducted at 16 centres in Japan. Eligible patients were randomly assigned at a 1 : 1 ratio to IRIS group or S-1 monotherapy group, considering PS (0, 1) and GEM treatment period as adjustment factors using a minimisation method. The study treatments were started within 8 days from the randomisation. An independent data and safety monitoring committee regularly reviewed the evaluation of efficacy and safety. The primary endpoint was PFS. The secondary endpoints were OS, time to treatment failure (TTF), RR, and safety.
Treatment
Patients assigned to the IRIS group received CPT-11 (100 mg m−2) on day 1 (Komatsu et al, 2011) and 15. S-1 administration was based on the body surface area (<1.25 m2, 40 mg; ⩾1.25 to <1.5 m2, 50 mg; ⩾1.5 m2, 60 mg). It was administered orally, twice a day (morning and evening) for 14 days, repeating every 4 weeks. Those assigned to S-1 monotherapy group received S-1 orally at the same dose as the IRIS group for 28 days, repeating every 6 weeks. The treatments were continued until disease progression, unacceptable toxicity, or withdrawal of consent.
Administration criteria for each cycle were neutrophil count ⩾1500 mm−3, platelet count ⩾100 000 mm−3, aspartate aminotransferase/alanine aminotransferase ⩽150 IU l−1, serum creatinine ⩽1.2 mg dl−1, diarrhoea/stomatitis ⩽grade 1, and no fever ⩾38.0 °C with suspicion of infection. S-1 was interrupted when the following events occurred during administration: neutrophil count ⩽1000 mm−3; platelet count ⩽50 000 mm−3; serum creatinine ⩾1.5 mg dl−1; diarrhoea/stomatitis ⩾grade 2; and fever ⩾38.0 °C with suspicion of infection. Patients could re-start the S-1 treatment when recovery was observed in the same treatment cycle (within 15 days in the IRIS group, or within 8 days in the S-1 monotherapy group).
The doses of CPT-11 and S-1 could be reduced by 20 mg m−2 and 10–30 mg per day, respectively, up to two levels. The dose of S-1 was reduced one level in the subsequent cycle when the following events occurred: neutrophil count <500 mm−3, febrile neutropenia (neutrophil count <1000 mm−3 and fever ⩾38.0 °C), platelet count<25 000 mm−3, platelet count decreased at grade 3 (<50 000 mm−3, ⩾25 000 mm−3) requiring platelet transfusion, diarrhoea ⩾grade 2, and stomatitis ⩾grade 3. The dose of CPT-11 was reduced one level in the subsequent cycle when diarrhoea ⩾grade 2 or diarrhoea including watery stools occurred. When febrile neutropenia occurred, the dose of both CPT-11 and S-1 was reduced.
Assessments
Haematological tests, biochemical tests, renal function tests, and assessments of symptoms and signs were performed every week. CT scans and test for tumour markers, including carbohydrate antigen 19–9 (CA19-9) and carcinoembryonic antigen (CEA), were performed every 4 weeks. Tumour responses were assessed at the baseline and every 4 weeks from the randomisation to the treatment discontinuation. All images for PFS and tumour reponses were reviewed by the independent extramural committee, in accordance with RECIST ver.1.0 (Therasse et al, 2000). RR was defined as the percentage of patients exhibiting a complete response (CR) and partial response (PR) within the full analysis set (FAS). The disease control rate (DCR) was defined as the percentage of CR, PR, and stable disease in the FAS. Toxicities were monitored in each cycle and assessed using the Common Terminology Criteria for Adverse Events (CTCAE v3.0.).
Classification of UGT1A1 polymorphisms
Among uridine diphosphate glucuronosyltransferase (UGT) isoforms, several polymorphisms of UGT1A1 have been well characterised. Studies have reported a correlation between UGT1A1*28 and UGT1A1*6 and CPT-11 toxicity (Han et al, 2006; Yamamoto et al, 2009; Satoh et al, 2011). In this study, we investigated the relationship between UGT1A1 polymorphisms and IRIS therapy-induced toxicities. Patients were divided into three groups according to their genotypes of two polymorphisms, UGT1A1*6 and UGT1A1*28: the homozygous group (UGT1A1*6/*6, UGT1A1*28/*28, or UGT1A1*6/*28); the heterozygous group (UGT1A1*6/*1, UGT1A1*28/*1); and the wild-type group (UGT1A1*1/*1).
Statistical considerations
In this study, the primary endpoint was PFS. PFS was defined as the time from randomisation to the first date of documented disease progression or death for any reason. The median PFS in S-1 monotherapy was assumed to be 2.0 months, based on the previous phase II study (Morizane et al, 2009). We assumed that the median PFS would be 3.5 months in the IRIS group (hazard ratio (HR) 0.571), 1.5 months longer than that of S-1 monotherapy group. To detect the difference using the log-rank test with a significance level of two-sided 5% and a power of 80%, 100 events were required for this study. Assuming the patient enrolment (2 years) and follow-up periods (1 year after the last patient randomisation), the required number of patients was estimated at 102 (51 patients for each group). Considering that some patients would be ineligible, we set the target number of enrolled patients at 120 in total (60 patients for each group). The primary endpoint was analysed in the FAS. The secondary endpoints were OS, TTF, RR, and safety. OS was defined as the time from randomisation to death for any reason. TTF was defined as the time from randomisation to the first date of treatment discontinuation, disease progression, or death for any reason. PFS, OS, and TTF were regarded as censored at the last assessment for patients without event.
To compare the difference in PFS between the groups, the stratified log-rank test was performed using the PS (0, 1) and the period of the first-line treatment (<90 days (including the postoperative adjuvant chemotherapy period) or ⩾90 days) as the stratified factors. The median PFS (95% confidence intervals (CIs)) was estimated using the Kaplan–Meier method. The HRs (95% CIs) were estimated using the stratified Cox proportional hazards model. The OS and TTF were compared between groups by a stratified analysis, and the HRs (95% CIs) and median survival times (95% CIs) were estimated. When an imbalance in baseline characteristics was found between groups, the PFS was analysed using a Cox proportional hazards model to investigate the influence of the imbalance, if applicable. In exploratory analyses, the subgroup analysis was performed on demographic factors in the stratified Cox proportional hazards model. The 95% CIs of RR and DCR were calculated, and a chi-square test was used to compare the differences in RR and DCR groups. Data analysis was performed using SAS version 9.1.3 (SAS Institute, Cary, NC, USA).
Results
Patient characteristics
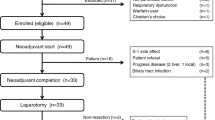
The trial profile is shown in Figure 1. From November 2008 through March 2011, a total of 137 patients were enrolled at 16 centres (IRIS group, n=67; S-1 monotherapy group, n=70). Four patients who did not receive the study treatment were excluded, 133 were included in the safety analysis population (IRIS group, n=64; S-1 monotherapy group, n=69). Six patients were later found ineligible and were excluded from FAS. Thus, 127 patients were included in the FAS (IRIS group, n=60; S-1 monotherapy group, n=67). There were imbalanced distributions in the groups regarding age and total bilirubin (Table 1).

Trial profile. *Represents that patients can be included in more than one category.
Treatment delivery
The median of treatment cycles in the IRIS group and the S-1 monotherapy group were 3.0 (range, 1–27) and 2.0 (range, 1–12), respectively. The relative dose intensity (RDI) was 79.5% (range, 46.3–100) for CPT-11 and 88.1% (range, 7.1–100) for S-1 in the IRIS group, and 78.6% (range, 17.9–100) in the S-1 monotherapy group. The RDI for S-1 was higher in the IRIS group than with the S-1 monotherapy group.
Disease progression was the leading cause of treatment discontinuation in both groups (65.6% in the IRIS group, 71.0% in the S-1 monotherapy group). Post-study therapy was given to 63.3% in the IRIS group and 62.7% in the S-1 monotherapy group (Table 2).
Efficacy
Data cut-off was on 29 February 2012 when 100 PFS events were confirmed. The median follow-up period was 13.1 months (range, 0–13.8) in the IRIS group and 11.4 months (range, 6.0–16.5) in the S-1 monotherapy group. The median PFS was 3.5 months (95% CI 2.1–4.6; 57 events) and 1.9 months (95% CI 1.8–2.1; 63 events) in the IRIS group and the S-1 monotherapy group, respectively, with an HR of 0.77 (95% CI 0.53–1.11; P=0.18; Figure 2A). Treatment with the IRIS group resulted in a median OS of 6.8 months (95% CI 5.8–9.3; 55 events), whereas the S-1 monotherapy group resulted in a median OS of 5.8 months (95% CI 5.1–8.0; 61 events), with an HR of 0.75 (95% CI 0.51–1.09; P=0.13; Figure 2B). The median TTF was 2.1 months (95% CI 1.6–4.1; 58 events) in the IRIS group and 1.8 months (95% CI 1.8–2.0; 65 events) in the S-1 monotherapy group, with an HR of 0.809 (95% CI 0.56–1.17; P=0.28). Importantly, there was a significant difference in RRs, with the IRIS group having an RR of 18.3% (95% CI 9.5–30.4) and the S-1 monotherapy group having an RR of 6.0% (95% CI 1.7–14.6; P=0.03). DCRs were 53.3% (95% CI 40.0–66.3) in the IRIS group and 35.8% (95% CI 24.5–48.5) in the S-1 monotherapy group (P=0.05). With respect to tumour markers, serum CA19-9 levels decreased in 53.3% of patients in the IRIS group and in 40.3% of patients in the S-1 monotherapy group. Serum CEA levels decreased in 40% of patients in the IRIS group and in 25.4% of patients in the S-1 monotherapy group.

Kaplan–Meier curves for progression-free survival assessed by RECIST (A) and overall survival (B). CI, confidence interval; HR, hazard ratio; IRIS, irinotecan plus S-1; OS=overall survival; PFS=progression-free survival.
The results of the pre-specified PFS and OS subgroups analyses were shown in Figure 3.

Subgroup analyses of progression-free survival assessed by RECIST (A) and overall survival (B). CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; IRIS, irinotecan plus S-1.
Safety
Table 3 shows the toxicities observed in the safety analysis population. Several grade 3 or higher haematological toxicities (IRIS group vs S-1 monotherapy group) were observed, including neutropenia (15.6% vs 4.3%), anaemia (15.6% vs 10.1%), and leucopenia (14.1% vs 2.9%). Most patients recovered from the toxicities without therapeutic treatment in both groups.
Common grade 3 or higher non-haematological toxicities (IRIS group vs S-1 monotherapy group) were anorexia (23.4% vs 17.4%), hyponatraemia (3.1% vs 10.1%), and nausea (6.3% vs 2.9%). The incidence of other events was <5%. Treatment-related death (TRD) due to infection associated with grade 3–4 neutropenia occurred in one patient in the IRIS group.
Relationships between UGT1A1 polymorphisms and toxicity of irinotecan
In the IRIS group of the safety analysis population, one patient did not provide genetic test data; hence 63 patients were included in the analysis of UGT1A1 polymorphisms (wild-type group (n=28), heterozygous group (n=29), and homozygous group (n=6)). Grade 3 or higher neutropenia occurred in 5 of 28 patients (17.9%) in the wild-type group, 4 of 29 patients (13.8%) in the heterozygous group, and 1 of 6 patients (16.7%) in the homozygous group. There was no relationship between UGT1A1 polymorphisms and grade 3 or higher neutropenia.
Discussion
This study was conducted to evaluate the efficacy and safety of IRIS therapy compared with S-1 monotherapy as a second-line treatment for patients with GEM-refractory pancreatic cancer.
In this study, longer median PFS in the IRIS group than in the S-1 monotherapy group was obtained closely to the assumed value in the sample size calculation. However, the HR for PFS resulted in 0.77 indicating smaller treatment effect than the assumed value, 0.571, as seen in the Kaplan–Meier plot in which consistent improvement between groups was not demonstrated over time. It may also attribute to the better prognosis by S-1 monotherapy than expected (Supplementary Figure 1).
Although the population of patients included in this study was all from a single country, Japan, the results of the IRIS group in our study were comparable to those observed in Western studies (Supplementary Table 1). The median PFS in the IRIS group (3.5 months) was longer than any other second-line treatment regimens for GEM pre-treated pancreatic cancer patients, as seen in the CONKO-003 study (oxaliplatin, folinic acid, and FU, 2.9 months; folinic acid and FU, 2.0 months; Oettle et al, 2014), the PANCREOX study (mFOLFOX, 3.1 months; FU/leucovorin (LV), 2.9 months; Gill et al, 2016), and the NAPOLI-1 study (5-FU/LV/nanoliposomal irinotecan, 3.1 months; FU/LV, 1.5 months; Wang-Gillam et al, 2016). The median OS in the IRIS group (6.8 months, 95% CI 5.8–9.3) were also comparable to those in 5-FU/LV/nanoliposomal irinotecan (6.1 months, 95% CI 4.8–8.9; Wang-Gillam et al, 2016) and in mFOLFOX6 (6.1 months, 95% CI 3.1–7.9 months; Gill et al, 2016). In addition, the patient background of our study was slightly worse than that in the PANCREOX and the NAPOLI-1 study. The study population in our study consisted of patients who had experienced progression during first-line GEM-based therapy. Patients in the PANCREOX and the NAPOLI-1 study had progressed after previous GEM-based therapy. It can be said that IRIS therapy deserves a standard treatment comparator in a randomised study for a second-line treatment of GEM-refractory pancreatic cancer.
The most frequently observed grade 3 or higher toxicity was anorexia in both groups. Anorexia occurred in 15 of 64 patients (23.4%) in the IRIS group and 12 of 69 patients (17.4%) in the S-1 monotherapy group. Although anorexia was considered to be related to the study treatments in most patients, most events were resolved within the same treatment cycle. The incidence of grade 3 and higher haematological toxicities was <20%. Patients recovered from these toxicities with no therapeutic treatment. Two drugs used in this study are known to suppress the bone marrow function (myelosuppression). However, the incidence of haematological toxicities of the IRIS group in this study, such as neutropenia, was lower than those in the previous studies (Shitara et al, 2008; Komatsu et al, 2011), suggesting that IRIS therapy has an acceptable safety profile. The incidence of grade 3 or higher nausea and vomiting was <10%, indicating that aggravation of these events was effectively controlled. The toxicities observed in this study were comparable to those previously reported, and none of them were clinically significant, except for one TRD. The tolerability of IRIS therapy was also comparable to that observed in the NAPOLI-1 study (Wang-Gillam et al, 2016). The incidence of grade 3 or higher neutropenia and thrombocytopenia was 20% and 2%, respectively, in 5-FU/LV/MM-398, which was higher than our study. The IRIS therapy is therefore considered to be manageable and tolerable.
No relationship was observed between UGT1A1 polymorphisms and toxicity in our study. Previous studies reported that the incidence of neutropenia was low in patients with homozygous UGT1A1 polymorphisms with low doses of CPT-11 (Hoskins et al, 2008; Yamamoto et al, 2009; Satoh et al, 2011). In this study, the dose of CPT-11 in IRIS therapy was as low as 100 mg m−2. Depending on the dose of CPT-11, the incidence of toxicities may not be affected by UGT1A1 mutation.
IRIS therapy was well tolerated and is expected to have survival benefits with the HR of 0.75. In conclusion, IRIS therapy could be a treatment arm in the clinical trials for the second-line treatment of GEM-refractory pancreatic cancer. Further clinical studies would be warranted.
Change history
14 February 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Burris HA, Moore MI, Anderson J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvement in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreatic cancer: a randomized trial. J Clin Oncol 15: 2403–2413.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup (2011) FOLFIRINOX versus gemcitabine for metastatic pancreas cancer. N Engl J Med 364: 1817–1825.
Gill S, Ko YJ, Cripps M, Beaudoin A, Dhesy-Thind SK, Zulfiqar M, Zalewski P, Do T, Cano PO, Lam W, Dowden SD, Grassin H, Stewart J, Moore MJ (2016) PANCREOX: a randomized Phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 34: 3914–3920.
Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Jang IJ, Lee DH, Lee JS (2006) Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non–small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol 24: 2237–2244.
Hoskins JM, Marcuello E, Altes A, Marsh S, Maxwell T, Van Booven DJ, Paré L, Culverhouse R, McLeod HL, Baiget M (2008) Irinotecan pharmacogenetics: influence of pharmacodynamic genes. Clin Cancer Res 14: 1788–1796.
Japan Pancreas Society (2013) Pancreatic Cancer Medical Guideline.
Japanese Society for Cancer of the Colon and Rectum (2014) JSCCR Guidelines for the treatment of colorectal cancer.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global Cancer Statistics. CA Cancer J Clin 61: 69–90.
Komatsu Y, Yuki S, Sogabe S, Fukushima H, Iwanaga I, Kudo M, Tateyama M, Meguro T, Uebayashi M, Saga A, Sakata Y, Asaka M (2011) Phase II study of combined treatment with irinotecan and S-1 (IRIS) in patients with inoperable or recurrent advanced colorectal cancer (HGCSG0302). Oncology 80: 70–75.
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W National Cancer Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966.
Morizane C, Okusaka T, Furuse J, Ishii H, Ueno H, Ikeda M, Nakachi K, Najima M, Ogura T, Suzuki E (2009) A phase II study of S-1 in gemcitabine-refractory metastatic pancreatic cancer. Cancer Chemother Pharmacol 63: 313–319.
Muro K, Boku N, Shimada Y, Tsuji A, Sameshima S, Baba H, Satoh T, Denda T, Ina K, Nishina T, Yamaguchi K, Takiuchi H, Esaki T, Tokunaga S, Kuwano H, Komatsu Y, Watanabe M, Hyodo I, Morita S, Sugihara K (2010) Irinotecan plus S-1 (IRIS) versus fluorouracil and folinic acid plus irinotecan (FOLFIRI) as second-line chemotherapy for metastatic colorectal cancer: a randomised phase 2/3 non-inferiority study (FIRIS study). Lancet Oncol 11: 853–860.
Oettle H, Riess H, Steiler JM, Heil G, Schwaner I, Seraphin J, Görner M, Mölle M, Greten TF, Lakner V, Bischoff S, Sinn M, Dörken B, Pelzer U (2014) Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from CONKO-003 trial. J Clin Oncol 32: 2423–2429.
Okusaka T, Funakoshi A, Furuse J, Boku N, Yamao K, Ohkawa S, Saito H (2008) A late phase II study of S-1 for metastatic pancreatic cancer. Cancer Chemother Pharmacol 61: 615–621.
Piacentini P, Donadelli M, Costanzo C, Moore PS, Palmieri M, Scarpa A (2006) Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch 448: 797–804.
Satoh T, Ura T, Yamada Y, Yamazaki K, Tsujinaka T, Munakata M, Nishina T, Okamura S, Esaki T, Sasaki Y, Koizumi W, Kakeji Y, Ishizuka N, Hyodo I, Sakata Y (2011) Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci 102: 1868–1873.
Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, Arnold D, Ciardiello F, Hoff P, Kerr D, Köhne CH, Labianca R, Price T, Scheithauer W, Sobrero A, Tabernero J, Aderka D, Barroso S, Bodoky G, Douillard JY, El Ghazaly H, Gallardo J, Garin A, Glynne-Jones R, Jordan K, Meshcheryakov A, Papamichail D, Pfeiffer P, Souglakos I, Turhal S, Cervantes A (2012) ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol 23: 2479–2516.
Shitara K, Komatsu Y, Yuki S, Munakata M, Muto O, Shimaya S, Sakata Y (2008) Pilot study of combination chemotherapy using irinotecan plus S-1 for metastatic pancreatic cancer. Oncology 75: 67–70.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92: 205–216.
Von Hoff DD, Ervin T, Arena PF, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691–1703.
Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, Macarulla T, Lee KH, Cunningham D, Blanc JF, Hubner RA, Chiu CF, Schwartsmann G, Siveke JT, Braiteh F, Moyo V, Belanger B, Dhindsa N, Bayever E, Von Hoff DD, Chen LT NAPOLI-1 Study Group (2016) Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 387: 545–557.
Yamamoto N, Takahashi T, Kunikane H, Masuda N, Eguchi K, Shibuya M, Takeda Y, Isobe H, Ogura T, Yokoyama A, Watanabe K (2009) Phase I/II pharmacokinetic and pharmacogenomic study of UGT1A1 polymorphism in elderly patients with advanced non-small cell lung cancer treated with irinotecan. Clin Pharmacol Ther 5: 149–154.
Acknowledgements
This study was supported by Yakult Honsha. We thank all the patients, clinicians, and support staff who participated in this study. We also thank Atsushi Sato, Kunihisa Miyakawa, and Hiroki Yoshikawa for performing extramural reviews to assess objective responses and PFS, as well as, Takushi Okusaka, Fumitaka Nagamura, Satoshi Morita, and Hironobu Minami for their helpful advice. We also thank Yukitoshi Yasuzawa for his helpful advice. We also thank Donald Chow for his medical writing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Tatsuya Ioka has received research fund and honoraria including Speakers from Bureau from Taiho Pharmaceutical and Yakult Honsha. Tatsuya Ioka held an advisory role for Taiho Pharmace utical. Yoshito Komatsu has received research fund and honoraria from Yakult Honsha and Taiho Pharmaceutical. Nobumasa Mizuno, Haruo Iguchi, and Junji Furuse have received honoraria from Taiho Pharmaceutical and Yakult Honsha. Nobumasa Mizuno and Atsushi Ishiguro have received research fund from Taiho Pharmaceutical. Akihito Tsuji and Junji Furuse have received research fund from Taiho Pharmaceutical and Yakult Honsha. Taroh Satoh has received research fund from Yakult Honsha. Junji Furuse and Yuh Sakata held an advisory role for Taiho Pharmaceutical and Yakult Honsha. Chikuma Hamada held an advisory role for Yakult Honsha. All the remaining authors have declared no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ioka, T., Komatsu, Y., Mizuno, N. et al. Randomised phase II trial of irinotecan plus S-1 in patients with gemcitabine-refractory pancreatic cancer. Br J Cancer 116, 464–471 (2017). https://doi.org/10.1038/bjc.2016.436
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.436
Keywords
This article is cited by
-
A retrospective comparative study of S-IROX and modified FOLFIRINOX for patients with advanced pancreatic cancer refractory to gemcitabine plus nab-paclitaxel
Investigational New Drugs (2021)
-
Post-progression survival following second-line chemotherapy in patients with advanced pancreatic cancer previously treated with gemcitabine: a meta-analysis
Investigational New Drugs (2018)

{kind=link}