
Abstract
Background:
DT01 is a DNA-repair inhibitor preventing recruitment of DNA-repair enzymes at damage sites. Safety, pharmacokinetics and preliminary efficacy through intratumoural and peritumoural injections of DT01 were evaluated in combination with radiotherapy in a first-in-human phase I trial in patients with unresectable skin metastases from melanoma.
Methods:
Twenty-three patients were included and received radiotherapy (30 Gy in 10 sessions) on all selected tumour lesions, comprising of two lesions injected with DT01 three times a week during the 2 weeks of radiotherapy. DT01 dose levels of 16, 32, 48, 64 and 96 mg were used, in a 3+3 dose escalation design, with an expansion cohort at 96 mg.
Results:
The median follow-up was 180 days. All patients were evaluable for safety and pharmacokinetics. No dose-limiting toxicity was observed and the maximum-tolerated dose was not reached. Most frequent adverse events were reversible grades 1 and 2 injection site reactions. Pharmacokinetic analyses demonstrated a systemic passage of DT01. Twenty-one patients were evaluable for efficacy on 76 lesions. Objective response was observed in 45 lesions (59%), including 23 complete responses (30%).
Conclusions:
Intratumoural and peritumoural DT01 in combination with radiotherapy is safe and pharmacokinetic analyses suggest a systemic passage of DT01.
Similar content being viewed by others

Main
Malignant melanoma has a rising incidence and a mortality that remains extremely high for stages III and IV, accounting for 75% of skin cancer deaths (Lo and Fisher, 2014). Approximately 20% of melanoma patients develop skin metastasis (Savoia et al, 2009). Recent advances have led to the development and approval of promising immunotherapies for the treatment of melanoma such as ipilimumab, which targets CTLA4 (Fellner, 2012) monoclonal antibodies blocking the programmed death-1 (PD-1) receptor or its ligand (PD-L1) (Hamid et al, 2013) as well as molecular-targeted therapies affecting the mitogen-activated protein/extracellular signal-regulated kinase (MEK) (Kirkwood et al, 2012) and BRAF (Robert et al, 2015). These new therapies demonstrate high response rates and durable responses. However some tumours are still resistant to these treatments, therefore new approaches must be developed to overcome these limitations. Although conventional therapies such as chemotherapy and radiotherapy (RT) display poor antitumour activities (Kim et al, 2010; Kirkwood et al, 2012), RT still remains a treatment option for patients with skin metastases of melanoma. Up to 25% of complete response (CR) rates were reported with high-dose RT (Sause et al, 1991), and <10% was observed with low-dose RT (⩽5 Gy per fraction) (Konefal et al, 1987; Olivier et al, 2007).
RT cytotoxicity is mainly due to DNA damage. DNA breaks are the most severe RT-induced DNA damage and are lethal to the cell if not repaired (Radford, 1985). The ability of cancer cells to recognise DNA damage and initiate repair is an important mechanism of radioresistance (Bradbury and Middleton, 2004). Inhibition of DNA repair makes cancer cells more vulnerable to DNA-damaging therapies such as RT in preclinical models and could provide a means to improve treatment efficacy (Helleday et al, 2008).
DT01 is a signal interfering DNA designed to counteract DNA repair. It consists of 32 base-pair deoxyribonucleotides forming an intramolecular DNA double helix that mimics DNA lesions. It acts as a bait for DNA-damage signalling enzymes, such as the poly(ADP-ribose) polymerase (PARP) and DNA-dependent protein kinase (DNA-PK), and induces a ‘false’ DNA-damage signal, ultimately inhibiting recruitment of many proteins involved in the DNA-break repair pathways, and consequently disabling DNA-repair activity at damage sites (Quanz et al, 2009a, 2009b). As a result, DT01 synergistically increased the efficacy of different DNA-damaging therapies in several preclinical models of cancer (Quanz et al, 2009a; Devun et al, 2012, 2014; Herath et al, 2016). The combination of intratumoural (IT) and peritumoural (PT) subcutaneous injection of DT01 with RT showed supra-additive efficacy in human melanoma xenografted in mice (Biau et al, 2014). As concomitant chloroquine appeared to increase the cellular uptake of DT01 (Berthault et al, 2011), a daily dose of chloroquine was administered to these patients.
The tissue inhibitor of metalloproteinases 3 (TIMP3) expression, inversely correlated with disease progression and the angiogenic capacity of melanoma (Das et al, 2014), was reported to be deregulated in high-stage melanoma (Liu et al, 2008). The extracellular concentration of the TIMP3 protein was identified as a potential biomarker of DT01 activity in melanoma cells, in vitro (unpublished data).
Since DT01 sensitises RT and is injected locally, we decided to perform a first-in-human phase I trial of DT01 in patients with skin metastasis of melanoma in combination with RT.
Patients and methods
All patients provided written informed consent. The study was approved by an independent ethics committee (Comité de Protection des Personnes Ile de France III) and was conducted in accordance with the Declaration of Helsinki, the ICH Harmonised Tripartite Guideline for Good Clinical Practice.
Patient selection
Adult patients with histologically confirmed skin metastases from cutaneous melanoma (with at least two measurable tumour lesions of ⩽4 cm in largest diameter), including melanoma-in-transit, who were not eligible for immediate surgery and refractory to conventional treatment were eligible for the study. The lesions had to be in a non-previously irradiated field. Patients had to have adequate hematopoietic, renal and liver functions and an Eastern Cooperative Oncology Group performance status of 0 or 1.
Exclusion criteria included: any serious concomitant systemic disorders incompatible with the study (e.g., active infection), known central nervous system metastases, history of epilepsy, history of porphyria, active psoriasis, clinically significant hepatic disease or renal disease, severe gastrointestinal, neurological and blood disorders. Patients receiving anti-vitamin K or cyclosporin therapy within 10 days before the first dose of study treatment, anticancer therapy within 4 weeks before the first dose of study treatment (3 months for ipilimumab) were also excluded, as were patients with significant coagulation abnormalities, hypersensitivity to 4-aminoquinoline compounds (chloroquine) or to any of its derivatives, retinal or visual field changes attributable to previous chloroquine administration or any other aetiology, positive for HIV and Hepatitis B or C.
Study design and treatment
This first-in-human phase I trial (NCT01469455) was an open label, non-randomised, multi-centre study. Patients were sequentially assigned to escalating daily total doses of DT01 (16, 32, 48, 64 and 96 mg) using a traditional 3+3 design. An expansion cohort was planned at the recommended phase II dose. The primary objective was to evaluate the safety and tolerability profiles of DT01 in combination with RT and concomitant dosing of chloroquine. The secondary objectives were to determine the dose-limiting toxicities (DLTs), the recommended phase II dose, the pharmacokinetic (PK) parameters of DT01, pharmacodynamic biomarkers and to identify preliminary signs of efficacy.
DT01 was administered three times a week (every other day) over 2 weeks (six DT01 administrations in total) with two tumours per patient, each treated by IT and PT subcutaneous injections between 3 and 5 h before the RT sessions. In the irradiated field, the two injected tumours were chosen by the investigators in an unblinded fashion (Supplementary Figure 1). The total daily doses administered per patient were 16, 32, 48, 64 and 96 mg. Each of the two lesions received half the total dose of DT01. At doses of 16, 32, 48 and 64 mg, DT01 was administered per lesion by two injections (1 × IT and 1 × PT), whereas at the highest dose of 96 mg, three injections were administered per lesion (1 × IT and two or three PT) with constant volumes of 0.4 ml per injection. At the 96 mg dose level, nine additional patients were included. Three of these were treated with IT and PT injections of DT01 as in the escalating dose study and six of these with only PT injections after protocol amendment to compare the efficacy of different routes at the same dose (i.e., 96 mg), with the same number of patients (i.e., six per route).
RT was administered on all the DT01-injected tumours and on all other nodules in the involved field. RT was delivered by orthovoltage irradiation, or using photons, electrons or combined electron and photon irradiation to a total dose of 30 Gy in 10 fractions in 2 weeks. Details of the RT technique used for each patient can be found in Supplementary Table 1.
As concomitant chloroquine increased cellular uptake of DT01 in vitro (Berthault et al, 2011), although not confirmed in animal models, patients were administered a daily oral dose of chloroquine (100 mg) from day −7 pre-dose to day 12 (last day of RT).
Assessments
Adverse events were graded using NCI CTCAE version 4.03. DLTs were defined as any grades 3 or 4 adverse events at least possibly related to DT01, chloroquine or RT occurring between day −7 and day 26.
Blood samples for PK analysis were obtained on PK day 1 at time points 0 (pre-dose), 1, 2.5, 4.5, 6.5, 9 and 24 h, after a single dose of DT01. Plasma DT01 quantification was performed by Bertin Pharma (Orléans, France) using a validated ELISA sandwich method. PK analysis by trapezoidal method with a non-compartmental model was performed using Phoenix WinNonlin (version 6.3; Pharsight Corporation, St Louis, MO, USA).
As siRNA, the closest pharmaceutical class, are known to be activators of the mammalian innate immune system (Robbins et al, 2009), inflammatory cytokines and interferon were quantified. Blood samples were collected for inflammatory proteins (IL-6, IL-12, IP10, MCP1, INFa2, CRP and LDH) on day 1 (pre-dose and 4.5 h post dosing), day 2 (24 h day 1 post dosing) and day 12 (4.5 h post dosing), before the session of RT. Cytokine quantification was performed by Bertin Pharma.
Plasma samples for biomarkers analysis were obtained on PK time points. Plasma TIMP3 concentration was determined with the TIMP3 (MIG-5) human ELISA kit (Abcam, Cambridge, MA, USA) on non-diluted plasma samples.
The tumour response was clinically evaluated by investigators by measuring the longest diameter of the target lesions over time. A partial response (PR) was defined as a decrease of >30%, while progressive disease was defined as an increase of >20% compared with the baseline. Complete response corresponded to the disappearance of all target lesions. Each target lesion was measured by clinical examination at baseline and days 26, 40, 54, 90, 180, 270 and 360, or at patient last visit. Baseline tumour burden was assessed during pre-visits occurring between 1 and 3 weeks before day 1. Patient response was estimated by the variation of the sum of the target lesion diameter.
Statistical methods
Descriptive analyses were performed for results of this phase I trial. However, a posteriori explorative tests were performed for some analyses. Statistical comparison between sub-groups was assessed using a two-sided Fisher’s exact test to determine the effect of administration route on response rate. The correlations between patient responses and AUC or basal TIMP3 level and patient response were analysed by Pearson’s r and Spearman rank coefficient. Comparison of responses between patients<3.6 μg.h ml−1 and patients>3.6 μg.h ml−1 was performed using Student t-test. The dose-proportionality analysis was considered with AUC0–24h and Cmax and was analysed by using a regression model.
Results
Patient characteristics
Twenty-three patients were included in the current trial (Table 1) between October 2011 and June 2014. The median age was 72 years old, ranging from 40 to 85 years. All patients had undergone prior surgery, while a few had received immunotherapy (22%), chemotherapy (13%) or RT (9%). Seven patients (30%) had distant skin metastases. Majority of the treated lesions (74%) were located on the leg. Other lesions were located on the head (17%), the arm (4%), or the chest (4%). The median size of tumour lesions injected with DT01 was 9 mm (range: 3–36), compared with 6 mm (range: 2–20) for non-injected lesions. The median number of irradiated lesions per patient was 4 (range: 1–7). Of these, two tumour lesions were injected with DT01, with the exception of one patient in the 32 mg cohort where only half the dose was received as only one lesion was injected. This patient was still included in the 32 mg cohort as the DT01 dose administered per lesion was the same as the other lesions in this cohort.
Safety
All patients received the full course of treatment and were evaluable for safety. None of the patients displayed DLT in this trial. Therefore, the maximum-tolerated dose (MTD) was not reached. The most common related adverse events were grades 1 and 2 local site injection reactions with pain at the site of injection and erythema occurring in 12 (52%) and 7 (30%) patients, respectively. The most common related general adverse events were pain in extremities and paraesthesia, both reported in two patients. Grades 1- and 2-related adverse events were not dose-dependent and occurred in 17 (74%) and 7 (30%) patients, respectively (Supplementary Table 2). Fourteen serious adverse events occurred in seven patients (Supplementary Table 3); however, none of these were considered related to DT01.
Pharmacokinetics
Pharmacokinetic analyses demonstrated a systemic passage of DT01 (Table 2). The half-life ranged from 2.4 to 4.9 h. The DT01 concentration over time increased in a nonlinear manner (Figure 1A). In the 96 mg cohort, the systemic exposure was higher in patients who were treated with a combination of IT and PT injection of DT01, compared with patients receiving PT injections alone (Figure 1B).

Average DT01 concentration in plasma over time after a single administration.(A) Patients treated with escalating doses of DT01 through PT and IT injection. (B) All patients treated with the highest dose of DT01 (96 mg), including the five patients treated with PT and intratumour DT01 (IT+PT) and the six patients treated with only PT DT01 (PT).
Efficacy
Two out of 23 patients were not evaluable for efficacy as one patient did not receive the treatment as per protocol with a 2-week interruption of RT and no valid tumour assessment was performed in the second patient.
Patient response was evaluated by considering all target lesions from each patient. Of the 21 evaluable patients, one patient (5%) experienced a CR to treatment, and 13 patients (62%) had a PR, resulting in an overall response rate (ORR) of 67% (Figure 2A). Ten out of 14 (71%) responding patients did not show local relapse (with a median follow-up of 6 months after the recorded response) (Figure 2B). Overall, 18 patients (86%) had no local disease progression at exit.

Waterfall plots of best overall response according to modified RECIST criteria in the 21 evaluable patients for efficacy.(A) Overall response per patient. (B) Patient responses over time. The dots represent patient exit from the study. (C) Best overall response per treated tumour lesion in the 76 lesions treated with RT with or without DT01 injection. *DT01-injected lesions (both IT+PT and PT-treated lesions). A full colour version of this figure is available at the British Journal of Cancer journal online.
A total of 76 tumour lesions were treated in the 21 evaluable patients (Supplementary Table 4). All target lesions were irradiated, including 41 lesions injected with DT01. Ten out of the 41 injected lesions were only treated with PT injections of DT01 in the 96 mg expansion cohort. Among the 76 evaluable lesions, a CR was observed in 23 lesions (30%), PR in 22 lesions (29%), and SD in 17 lesions (22%) resulting in an ORR of 59% (Figure 2C). The dose level and the route of administration (direct injection or not) did not significantly correlate with efficacy. Overall response rate in the 41 lesions injected with DT01 was 68% whereas in the 35 non-injected lesions it was 49% (P=0.103).
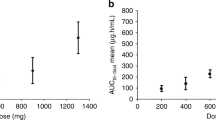
A statistically significant relationship between DT01 blood exposure and patient best response was identified (Figure 3). The average best response in patients where the AUC exceeded 3.6 h.μg ml−1 (the median AUC) was −62.3% compared with −25.6% in patients with a lower AUC (P=0.049).

Dot plot representing the percentage change of lesion diameter.The correlation between the best overall responses of the 16 evaluable patients treated with IT and PT DT01 and their plasma concentrations of DT01. The patients treated with only PT DT01 were excluded from this analysis.
Pharmacodynamic biomarkers
No changes in inflammatory protein or cytokine levels (IL-6, IL-12, IP10, MCP1, INFa2, CRP and LDH) were observed over treatment time. No significant change in the TIMP3 level was seen during the 2 weeks of treatment. However, baseline plasma concentration of TIMP3 positively correlated with ORR (r=0.423) (Figure 4).

Dot plot demonstrating the correlation between the overall responses of the 21 evaluable patients and their pre-treatment plasma concentrations of TIMP3.
Discussion
DT01 is a first-in-class DNA-repair targeting agent. The results of this first-in-human trial demonstrate that DT01 can be safely administered in combination with RT in patients with skin metastases from melanoma. As no DLT was reported, no MTD was reached. These observations are consistent with the preclinical safety data reported in animal models (Schlegel et al, 2012; Biau et al, 2014). As nucleotides are known to induce an inflammatory response due to their CpG content (Robbins et al, 2009), such motifs were avoided in the DT01 sequence. The monitoring of the cytokine markers specific of this response (IL-6 and IL-12) (Quanz et al, 2009a) in treated patients confirmed the absence of a Toll-like response.
Pharmacokinetic analyses showed a systemic passage of DT01 in a nonlinear fashion. The lack of linearity in the dose/AUC relationship may be explained by: (i) the use of a flat dose not corrected for the patient weight; and (ii) the high variability of absorption because of the mode of IT administration in this trial. Blood exposure after PT administration was 40% lower, with less variation than after combination IT and PT administration at the same dose (96 mg) (Table 2). This suggests that the blood exposure in patients after DT01 IT administration may be highly dependent on their tumour vascularisation. Indeed, pharmacokinetic studies in animal models indicate that AUC is dose-dependent after subcutaneous or intravenous administration (Schlegel et al, 2012).
Complete responses were observed in 30% of treated lesions, which compares favourably to the 9% rate reported in the literature with similar RT schemes (Konefal et al, 1987; Olivier et al, 2007). Strikingly, no significant difference in the ORR was observed in DT01-injected and non-injected lesions. This may be partly explained by the systemic passage of DT01 and/or a potential abscopal effect through immunogenicity. The systemic passage is supported by the significant correlation between DT01 systemic exposure and efficacy. The RT-induced abscopal effect is infrequently reported in melanoma (Kingsley, 1975; Postow et al, 2012), suggesting that this effect is rare with RT alone. Durable stable disease (followed by a slow, steady decline in total tumour burden in some patients) and response after an increase in total tumour burden were observed in more than 40% of the patients. These types of response are rarely observed after RT (Wolchok et al, 2009).
Pre-treatment plasmatic concentrations of TIMP3 positively correlated with efficacy, making TIMP3 a possible predictive biomarker of efficacy that would need further investigation.
As the addition of chloroquine failed to demonstrate any benefit and its absence did not impact DT01 efficacy in the latest preclinical studies (Devun et al, 2014; Herath et al, 2016), this will not be used in subsequent clinical trials.
In conclusion, our study demonstrates that DT01 in combination with RT is safe in patients with skin metastases from melanoma. The antitumour activity observed in DT01-injected as well as in non-injected tumour lesions may be related to the systemic distribution of DT01 and/or stimulation of the immune response. Although antitumour activity needs to be interpreted with caution due to the small patient population, these data support further development of DT01.
Change history
24 May 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Berthault N, Maury B, Agrario C, Herbette A, Sun JS, Peyrieras N, Dutreix M (2011) Comparison of distribution and activity of nanoparticles with short interfering DNA (Dbait) in various living systems. Cancer Gene Ther 18 (10): 695–706.
Biau J, Devun F, Jdey W, Kotula E, Quanz M, Chautard E, Sayarath M, Sun JS, Verrelle P, Dutreix M (2014) A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia 16 (10): 835–844.
Bradbury PA, Middleton MR (2004) DNA repair pathways in drug resistance in melanoma. Anticancer Drugs 15 (5): 421–426.
Das AM, Seynhaeve AL, Rens JA, Vermeulen CE, Koning GA, Eggermont AM, Ten Hagen TL (2014) Differential TIMP3 expression affects tumor progression and angiogenesis in melanomas through regulation of directionally persistent endothelial cell migration. Angiogenesis 17 (1): 163–177.
Devun F, Biau J, Huerre M, Croset A, Sun JS, Denys A, Dutreix M (2014) Colorectal cancer metastasis: the DNA repair inhibitor Dbait increases sensitivity to hyperthermia and improves efficacy of radiofrequency ablation. Radiology 270 (3): 736–746.
Devun F, Bousquet G, Biau J, Herbette A, Roulin C, Berger F, Sun JS, Robine S, Dutreix M (2012) Preclinical study of the DNA repair inhibitor Dbait in combination with chemotherapy in colorectal cancer. J Gastroenterol 47 (3): 266–275.
Fellner C (2012) Ipilimumab (yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P T 37 (9): 503–530.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369 (2): 134–144.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8 (3): 193–204.
Herath NI, Devun F, Lienafa MC, Herbette A, Denys A, Sun JS, Dutreix M (2016) The DNA repair inhibitor DT01 as a novel therapeutic strategy for chemosensitization of colorectal liver metastasis. Mol Cancer Ther 15 (1): 15–22.
Kim C, Lee CW, Kovacic L, Shah A, Klasa R, Savage KJ (2010) Long-term survival in patients with metastatic melanoma treated with DTIC or temozolomide. Oncologist 15 (7): 765–771.
Kingsley DP (1975) An interesting case of possible abscopal effect in malignant melanoma. Br J Radiol 48 (574): 863–866.
Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V, Kemsley K, Dummer R (2012) Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy vs temozolomide in patients with advanced melanoma. Clin Cancer Res 18 (2): 555–567.
Konefal JB, Emami B, Pilepich MV (1987) Malignant melanoma: analysis of dose fractionation in radiation therapy. Radiology 164 (3): 607–610.
Liu S, Ren S, Howell P, Fodstad O, Riker AI (2008) Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res 21 (5): 545–558.
Lo JA, Fisher DE (2014) The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 346 (6212): 945–949.
Olivier KR, Schild SE, Morris CG, Brown PD, Markovic SN (2007) A higher radiotherapy dose is associated with more durable palliation and longer survival in patients with metastatic melanoma. Cancer 110 (8): 1791–1795.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, Sedrak C, Jungbluth AA, Chua R, Yang AS, Roman RA, Rosner S, Benson B, Allison JP, Lesokhin AM, Gnjatic S, Wolchok JD (2012) Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 366 (10): 925–931.
Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C (2009a) Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res 15: 1308–1316.
Quanz M, Chassoux D, Berthault N, Agrario C, Sun JS, Dutreix M (2009b) Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One 4 (7): e6298.
Radford IR (1985) The level of induced DNA double-strand breakage correlates with cell killing after X-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med 48 (1): 45–54.
Robbins M, Judge A, MacLachlan I (2009) siRNA and innate immunity. Oligonucleotides 19 (2): 89–102.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372 (1): 30–39.
Sause WT, Cooper JS, Rush S, Ago CT, Cosmatos D, Coughlin CT, JanJan N, Lipsett J (1991) Fraction size in external beam radiation therapy in the treatment of melanoma. Int J Radiat Oncol Biol Phys 20 (3): 429–432.
Savoia P, Fava P, Nardo T, Osella-Abate S, Quaglino P, Bernengo MG (2009) Skin metastases of malignant melanoma: a clinical and prognostic survey. Melanoma Res 19 (5): 321–326.
Schlegel A, Buhler C, Devun F, Agrario C, Urien S, Lokiec F, Sun JS, Dutreix M (2012) Pharmacokinetics and toxicity in rats and monkeys of coDbait: a therapeutic double-stranded DNA oligonucleotide conjugated to cholesterol. Mol Ther Nucleic Acids 1: e33.
Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, Humphrey R, Hodi FS (2009) Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 15 (23): 7412–7420.
Acknowledgements
We thank the patients, their families, the study teams involved in the trial, as well as the researchers involved in the preclinical work, for making this trial possible. We thank Nirmitha Herath for manuscript correction. This study was designed and funded by the study sponsor (DNA Therapeutics SA) and monitored by a clinical research organisation (ORION Santé SARL). The clinical research organisation collected all data and the study sponsor was responsible for data analysis and interpretation, with input from the authors and investigators. The data set was available to all authors on request, and the corresponding author had final responsibility for submission for publication. L’Agence Nationale pour la Recherche partly funded this study and had no involvement in the study design, the collection, analysis and interpretation of data, or in the writing of the report.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
BD reports grants and personal fees from Roche, grants and personal fees from Bristol-Myers Squibb, grants and personal fees from GlaxoSmithKline, outside the submitted work. CL reports personal fees from Roche, GlaxoSmithKline, Merck Sharp and Dhome, Bristol-Myers Squibb outside the submitted work. FD is employee of DNA Therapeutics; JMC, JSS and MD are cofounders of DNA Therapeutics, the company which holds the patent for DT01. MEM served as a consultant for DNA Therapeutics, Oncoethics, Merck Sharp and Dhome, Pfizer and Roche laboratories. The other authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Le Tourneau, C., Dreno, B., Kirova, Y. et al. First-in-human phase I study of the DNA-repair inhibitor DT01 in combination with radiotherapy in patients with skin metastases from melanoma. Br J Cancer 114, 1199–1205 (2016). https://doi.org/10.1038/bjc.2016.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.120
Keywords
This article is cited by
-
A Phase 1 dose-escalation study to evaluate safety, pharmacokinetics and pharmacodynamics of AsiDNA, a first-in-class DNA repair inhibitor, administered intravenously in patients with advanced solid tumours
British Journal of Cancer (2020)
-
DT01 DNA-repair inhibitor shows promise
Nature Reviews Clinical Oncology (2016)
