
Abstract
The past few years have witnessed major advances in the understanding of the molecular landscape of uveal melanoma (UM). The discovery of a mutational background that is completely different from the one of skin melanoma has granted to UM a stand-alone status. The absence of effective therapy for metastatic disease offers now a chessboard for targeted therapy but at the same time urges preclinical science to develop accordingly, to guide the use of economical resources to the best profit of patients. This review describes the current knowledge on the biology of this disease and discusses the challenges that must be undertaken to translate this knowledge into real benefit for patients.
Similar content being viewed by others

Introduction: a melanoma of the eye
Uveal melanoma (UM) is a rare malignant tumour accounting for two to eight new cases per million per year in western countries. The tumour arises from resident melanocytes of the uvea, a pigmented vascular layer located in the eye between the sclera and the retina (Singh et al, 2011).
Metastatic spread occurs in about half of patients. Owing to the absence in the eye of lymphatic drainage, blood vessels are the main route for dissemination. The liver is the most frequent site of metastasis (>80%), followed by the lungs, bone and skin.
Up to 50% of patients develop metastases within a median time of 2.4 years and the median survival with metastasis ranges from 3 to 12 months because of the lack of effective treatment options (Mariani et al, 2012).
Molecular features
Uveal melanoma is characterised by a relative genetic stability, with a low rate of structural variations at the chromosomal and sub-chromosomal level compared with other tumours or other types of melanoma.
Few recurrent mutations have been identified in primary tumours. GNAQ and GNA11 genes have been found mutated in the vast majority of tumours (83–96%), in a mutually exclusive way, and with identical biological effects (Van Raamsdonk et al, 2010). These mutations are thought to represent early drivers in tumorigenesis. A second step in tumoural progression involves BAP1, SF3B1 or EF1AX genes, which also are mutated in an almost mutually exclusive way (Harbour et al, 2010; Furney et al, 2013; Martin et al, 2013). BAP1 mutations are inactivating events and were shown to be coupled with the loss of the remaining allele on chromosome 3, more often through the loss of the entire chromosome (loss of heterozygosity or LOH). Loss of chromosome 3 had been also correlated with worse prognosis (Prescher et al, 1990) and was found to be enriched in metastatic samples. BAP1 mutations have been indeed shown to be enriched in tumours, which eventually gave rise to metastasis (Harbour et al, 2010). Therefore, BAP1 mutations and LOH of chromosome 3 define a class of primary tumours with a more aggressive biological behaviour. On the other hand, tumours with heterozygosity of chromosome 3 have better prognosis and are often mutated in EIF1AX (15%) or SF3B1 (14–29%) (Furney et al, 2013; Martin et al, 2013).
Other chromosomal aberrations, mainly gain of 8q and gain of 6p, have been independently correlated with bad prognosis (Aalto et al, 2001) and are currently used in clinical evaluation, together with chromosome 3 status, as markers of high-risk for metastasis. This suggests that the overexpression of genes located in 8q, such as MYC, NBS1, DDEF1 and LZTS1, and 6p might have a role in tumoural evolution (Ehlers et al, 2005; Ehlers and Harbour, 2005; Onken et al, 2008).
The definition of two subgroups of tumours with different propensity for metastatic recurrence was also confirmed by RNA analysis. Unsupervised hierarchical cluster analysis of primary UMs identifies two transcriptional subgroups characterised respectively by a high (class I) or low (class II) risk of metastasis. Based on these findings, RNA-based risk evaluation in patients with localised disease has been patented in the United States under the name of DecisionDx-UM (Harbour, 2014). Recently, a third prognostic class characterised by intermediate prognosis (class Ib) has been introduced in Decision-Dx UM, increasing the resolution of the test. However, the functional implications that lay behind the pure prognostic value of differential RNA expression signatures have not been fully evaluated.
The study of epigenetic deregulation through the alteration of DNA methylation patterns has been performed in a non-systematic way and in a very limited number of studies (Van der Velden and Maat, 2009). However, these data suggest that promoter hypermethylation may have a central role in UM pathogenesis. In the same way, studies underlining the role of particular miRNA in the pathogenesis of the disease are very limited, display low concordance and need cross-validation (Li et al, 2015).
Indeed, whereas GNAQ/11 mutant proteins are directly involved in the activation of signalling cascades, BAP1, SF3B1 and EF1AX have a role in the transcriptional and posttranscriptional regulation of the cell machinery. BAP1 is a nuclear deubiquitinase and was shown to interact with several nuclear transcription factors (Yu et al, 2010). Moreover, BAP1 is a component of the polycomb repressive deubiquitinase complex, a transcriptional modulator that cooperates with polycomb complexes to regulate the expression of a wide series of genes with roles in developmental processes and stem cell properties (Scheuermann et al, 2010). Finally, also a role of BAP1 in homologous recombination (HR) of DNA double-strand breaks has been proposed, suggesting a function for BAP1 in DNA repair and maintenance of genome integrity (Yu et al, 2014b). However, BAP1 loss in UM does not increase genetic instability and does not potentiate UM cell sensitivity to DNA-targeting agents as is the case in other types of cancers mutated in HR pathway components. In UM, BAP1 mutations and the consequent lack of BAP1 protein expression in the nucleus correlate with an increased risk of metastatic dissemination. However, no functional explanation of this correlation has so far been possible due in part to the reduced viability induced by artificial silencing of the protein and the lack of UM models harbouring BAP1 mutation or loss of the protein expression. The recent establishment of BAP1-mutated UM cell lines and patient-derived xenografts (PDXs) will probably foster the advancement in the understanding of the role of BAP1 mutations in UM (Némati et al, 2010; Amirouchene-Angelozzi et al, 2014). BAP1 is also mutated in other tumour types, such as clear cell renal carcinoma and mesothelioma, as well as in the germline of patients affected by familial cancer susceptibility (Carbone et al, 2013). Cross-comparison between models of these different tumours will also be needed to spread some light into the tumorigenic properties of BAP1 loss.
Transcriptional deregulation has been demonstrated in SF3B1-mutated UM. SF3B1 encodes for the subunit 1 of splicing factor 3B, a component of the spliceosome, which processes pre-mRNA into mature mRNA. SF3B1 mutations in UM are associated with alternative splicing patterns (Furney et al, 2013). Interestingly, investigation of splicing patterns in other SF3B1-mutated neoplasias (chronic lymphocytic leukemia and myelodysplastic syndrome) demonstrates that different hotspot mutations lead to the same splicing signature (Gentien et al, 2014). These data suggest the existence of common features in functional alterations caused by different SF3B1 mutations.
EIF1AX, whose product is involved in the initiation of protein translation, might also, when mutated, alter the balance of transcripts in neoplastic cells towards a pattern of gene expression favouring cell proliferation and survival. Such hypothesis remains still to be assessed in a pathologic situation such as UM. EF1AX was recently found mutated, although at low rates (⩽1.5%), also in papillary thyroid carcinoma, endometrial carcinoma and other cancers (The Catalogue Of Somatic Mutations In Cancer database, The Cancer Genome Atlas database).
Deregulated pathways
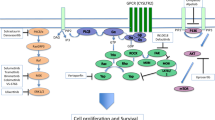
GNAQ and GNA11 encode members of the Gαq family of heterotrimeric G protein α, subunits of G proteins that mediate the cellular response to extracellular stimuli via interaction with membrane receptors (G protein-coupled receptors). Gαq proteins are known to activate via phospholipase C and protein kinase C (PKC) the mitogen-activated protein kinase (MAPK) pathway, which was shown in several reports to be constitutively upregulated in UM bearing GNAQ/11 mutations (Van Raamsdonk et al, 2010; Vaqué et al, 2013; Chen et al, 2014; Musi et al, 2014).
Mutant GNAQ/11 were also shown to activate the guanine nucleotide exchange factor Trio and to induce, via the small GTPases RhoA and Rac1, its release from the associated protein AMOT. Unbound, dephosphorylated YAP localises in the nucleus and is responsible for activation of different nuclear transcription factors including TEADs. Moreover, the activation of Trio by Gαq mutants is also suggested to have a role in the activation of the AP-1 transcription factor and possibly also of JNK and p38-dependent cascades (Vaqué et al, 2013; Feng et al, 2014; Yu et al, 2014a).
The importance of the MAPK, PKC and YAP pathways for UM survival and proliferation has been demonstrated by preclinical studies using selective PKC and MEK inhibitors, as well as the YAP inhibitor Verteporfin in vitro and in vivo (Chen et al, 2014; Yu et al, 2014a).
Although BAP1, SF3B1 and EIF1AX mutations have not been correlated with alterations in druggable signalling pathways, other intracellular signalling cascades have been implicated in survival and proliferation of UM.
Immunohistochemistry on tumour biopsies coupled with functional assessment of cellular sensitivity to specific inhibitors suggests a role of the PI3K/mTOR pathway in UM cells. Even if genetic alterations such as deletions in PTEN and mutations of PI3K appear to be rare events, the PI3K/mTOR pathway was suggested to be activated in UM by immunohistochemical detection of phosphorylated AKT in several tumour samples. This hypothesis was further confirmed by the strong inhibition of tumour growth in vitro and in vivo using PI3K and ATP-competitive and noncompetitive mTOR inhibitors (Khalili et al, 2012; Amirouchene-Angelozzi et al, 2014).
Immunohistochemical evaluation of IGF1 receptor (IGF1R) has shown increased expression of this receptor in metastatic compared with primary tumour samples. IGF1R is an upstream regulator of MAPK and PI3K/mTOR pathways. Its inhibition in UM cell lines results in decreased cell viability, confirming the role of these pathways in the UM tumour progression (All-Ericsson et al, 2004).
Similarly, c-MET, the membrane receptor of the hepatocyte growth factor, was found by immunohistochemistry as preferentially expressed in metastatic tissue and its inhibitor Crizotinib was demonstrated in vivo to delay the growth of metastasis (Surriga et al, 2013). However, the precise role of this signalling pathway in UM progression and metastasis development remains to be addressed.
Overexpression of the receptor c-KIT, shown by immunohistochemistry, has also been implicated in the survival of UM. c-KIT overexpression can activate the MAPK pathway in UM cell lines but clinical studies failed to demonstrate any efficacy of Imatinib in the absence of KIT mutations (Hofmann et al, 2009).
It is important to notice that the correlation between chromosome imbalance (mainly loss of chromosome 3 and gain of 8q) and poor prognosis suggests that potential therapeutic targets could be identified in these regions.
Therapy
Primary tumours are effectively treated with enucleation or conservative radiotherapy with the same overall survival rate (The Collaborative Ocular Melanoma Study).
On the contrary, options for metastatic patients are limited. Local treatment of oligometastatic liver disease by surgical complete resection is considered the best strategy in resectable patients, but based only on a limited number of non-randomised studies (Mariani et al, 2012). Other locoregional treatment possibilities for liver-limited disease such as chemo-embolisation, immuno- or radio-embolisation or isolated hepatic perfusion have shown only limited efficacy in non-randomised phase II studies and have not progressed into further clinical validation (Augsburger et al, 2009).
Systemic administration of classic chemotherapeutic agents has not shown to improve patients’ survival (Augsburger et al, 2009); in a randomised phase III trial, intra-arterial hepatic Fotemustine has demonstrated a slight increase in response rate and progression-free survival but no difference in survival in 171 patients (EORTC trial 18021).
The discovery of targetable pathways deregulated in UM, the increasing number of targeted molecules validated in preclinical development by the industry and the absence of a standard of care of the metastatic setting have fostered the clinical evaluation of kinase inhibitors and immunotherapy. To date, single-agent therapies (monotherapies) have not yet shown clear clinical benefit.
In the absence of compounds directly targeting GNAQ/11-mutated proteins, drugs acting on their downstream effectors remain among the most promising molecules. These compounds have been shown to be more selective for GNAQ/11-mutated models in preclinical studies; however, results obtained in clinical trials are quite limited to date. Treatment with the MEK inhibitor Selumitinib proved a significant advantage in progression-free survival but with only a non-significant trend towards improved overall survival (Carvajal et al, 2014). Similarly, the PKC inhibitor Sotrastaurin seems to show only a limited efficacy in the ongoing phase I (Piperno-Neumann et al, 2014).
Many other targeted agents have been tested in the clinics and others are still undergoing clinical evaluation (Table 1). B-Raf inhibitor Sorafenib, the anti-VEGF Bevacizumab and the VEGFR/PDGFR/c-KIT inhibitor Sunitinib showed limited responses when tested in non-randomised clinical trials. The immunomodulator agent anti-CTLA4 Ipilimumab demonstrated limited activity in retrospective and prospective studies, although some long-term disease stabilisations had been reported (Zimmer et al, 2015) and anti-PD1 antibodies are currently under clinical testing (NCT02359851). The use of HDAC inhibitors to induce differentiation and switch from aggressive ‘class II’ to milder ‘class I’ behaviour is being tested in the clinics but antitumoural activity in preclinical models seems quite limited (Landreville et al, 2012). In recent times, it has been reported that dendritic cell vaccination could potentially result of overall survival in patients with metastatic UM (Bol et al, 2014).
Adjuvant therapy also lacks a proof of efficacy in UM. A randomised trial with adjuvant Fotemustine is ongoing in clinical and/or genomic high-risk patients (EudraCT: 2008-005691-27). A trial with Crizotinib is also undergoing (NCT02223819). Given that (i) Crizotinib alone was shown in mouse models to be quite ineffective on already established tumours, and (ii) that after primary treatment (and probably at diagnosis) tumoural cells might have already spread to distant sites, an association of Crizotinib with a second molecule could be required to demonstrate a clinical benefit.
This limited activity obtained with single-agent targeted therapies underlines the necessity to refine the therapeutic strategy using drug combinations. The association of MEK and PKC inhibitors provided the first proof of tumour regression in preclinical UM models and suggests that drug combination might have a key role in improving treatment possibilities for this disease (Chen et al, 2014). Preclinical proof of concept of the superiority of drug combinations targeting simultaneously the PKC/MEK and PI3K axis has also been reported (Khalili et al, 2012; Musi et al, 2014). Indeed, trials testing the combination of MEK inhibitors with PKC (NCT01801358), PI3K (NCT02273219), AKT (NCT01979523) and chemotherapy such as Dacarbazine (NCT01974752) have been started. It is important to point out that toxicity and resistance will be two major challenges to address in the combinatorial approaches.
Translational challenges
New discoveries on the biology of UM need to be quickly translated into real improvement in the survival of patients diagnosed with metastatic lesions and of those with high risk for distant recurrences. We would like to discuss a few translational challenges concerning prognosis, follow-up and therapeutic approaches.
The growing interest in testing new therapies will call for a closer comparison of the available prognostic algorithms, which will be used to select patients who might benefit from the therapy. The definition of high-risk (class II) and low-risk (class I) patients based on gene expression profile appears to correlate strongly with copy number status of chromosome 3 and BAP1 loss of function, which is easily tested with immunohistochemical staining. However, the extent of this overlap has not yet been investigated. This leaves as an open question the superiority of RNA-based versus genetic approaches for prognostic evaluation of patients. The definition of the mutational landscape of UM now paves the way for the evaluation of algorithms integrating the evaluation of chromosomal aberrations, immunohistochemistry for BAP1 and mutational analysis of the other recurrently mutated genes. The combination of genetic and transcriptional analysis, which also integrates data from the heterogeneous environment of the cancer niche, might be an option worth to be tested.
The presence in almost all UMs of hotspot mutations in GNAQ/11 genes and the dependence on blood circulation for the spread of metastatic cells could make of liquid biopsies, and circulating tumour DNA in particular, a valuable tool for surveillance and treatment monitoring (Bidard et al, 2014). However, the detection of circulating markers still needs to be prospectively compared with standard imaging techniques. New technological advances need to be applied before the advantages of liquid biopsies on conventional diagnostic techniques could be evaluated.
The lack of efficacy displayed by single agents in clinical trials, often after successful functional validation in preclinical models, pinpoints the necessity to perform more robust preclinical validation in highly relevant models. It is also crucial to identify more efficient drug combination regimens. High-throughput screens and more in-depth preclinical studies on well-characterised cell lines and cohorts of PDXs will be required to test different combinations and adequate administration protocols. Importantly, such an approach will allow the identification of biomarkers of response already at the preclinical stage, which will be highly beneficial for the quick translation into clinical trials.
With the emergence of many new therapeutic agents and a plethora of potential drug combinations, drug sensitivity assessment in vitro is still a mandatory first step. It is very important to use in such a rare cancer a panel of representative UM cell lines, which harbour most of the genetic alterations and mutations observed in UM tumours. We have recently established a series of cell lines derived from both patients and PDXs of primary and metastatic UM. Importantly, some of these models harbour not only GNAQ/11 mutations but also BAP1 mutations. We have also identified cell lines with SF3B1 and EF1AX mutations among our panel and other cell lines established by other laboratories (Amirouchene-Angelozzi et al, 2014).
Patient-derived xenografts are a powerful tool to test drug efficacy before clinical assessment, as they maintain the characteristics of patients’ tumours (Laurent et al, 2013). Our experience in UM models and similar studies in other types of cancer suggest that tumour response to therapy in PDXs is often very variable as in patients. This observation implies the need not only to have representative models of the disease but also to test a large number of them in order to assess for a statistically significant effect as in clinical studies.
Immunomodulatory agents such as Ipilimumab have shown very limited results in UM patients compared with results obtained in skin melanoma (Zimmer et al, 2015). The very low mutational rate of UM and, consequently, the putatively low number of neoepitopes available for an immune response (Furney et al, 2013) might in part account for these results. The expression profile of immune checkpoint molecules in UM has not yet been addressed. However, the immune system is aware of UM, as immune cell infiltrates are found in a subgroup of primary tumours with bad prognosis (Bronkhorst and Jager, 2013), whereas expanded effector CD4+ and CD8+ T cells are found in the blood of metastatic patients (Péguillet et al, 2014). It would be interesting to evaluate the possibility to change this inefficient or even deleterious immune response to a productive one, for example, by the combination with cytotoxic (chemo and radio) therapies, which might increase the exposure to neoantigens, or by the association with vaccines. Several combinations and administration schedules might be possible in the field of immunotherapy, but relevant preclinical models are still urgently needed to test these novel approaches: a major limitation of PDXs is the absence of a functional immune system, which prevents their use to evaluate immunotherapeutic strategies. Today, no transgenic model matching UM disease progression is currently available but new technologies in genome editing might boost the development of such models. Moreover, establishing genetically modified mice displaying the genetic alterations of UM will also be an avenue for better understanding of the oncogenesis and progression, but for this purpose, along with mutated GNAQ/11 alleles, genome editing on BAP1, SF3B1 or EIF1AX will probably be necessary, as GNAQ/11 mutations are found in benign proliferations, such as blue nevi, and are therefore considered not sufficient for the progression into malignancy.
Finally, most of the studies on UM biology have been performed on primary tumours, because therapeutic enucleation provided the easiest and most common access to tumour samples. Our knowledge of the molecular features of the metastatic disease needs to be strongly improved. It would be also very important to establish UM animal models that develop liver metastases. In fact, metastases are not observed after subcutaneous injection of cell lines or tumour samples from primary or metastatic tumours. In recent times, UM cell lines have been shown to migrate to the liver after retro-orbital injection (Surriga et al, 2013), suggesting the feasibility to develop orthotopic models to evaluate the possibility of metastatic dissemination to the liver.
To conclude, we think that, in front of these challenges, collaborations between pharmaceutical companies and academic institutions will be mandatory in order to efficiently address our resources to the translation of knowledge into real benefit for UM patients.
References
Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S (2001) Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci 42: 313–317.
Amirouchene-Angelozzi N, Nemati F, Gentien D, Nicolas A, Dumont A, Carita G, Camonis J, Desjardins L, Cassoux N, Piperno-Neumann S, Mariani P, Sastre X, Decaudin D, Roman-Roman S (2014) Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol 8: 1508–1520.
Augsburger JJ, Corrêa ZM, Shaikh AH (2009) Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol 148: 119–127.
Bidard F-C, Madic J, Mariani P, Piperno-Neumann S, Rampanou A, Servois V, Cassoux N, Desjardins L, Milder M, Vaucher I, Pierga J-Y, Lebofsky R, Stern M-H, Lantz O (2014) Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int J Cancer 134: 1207–1213.
Bol KF, Mensink HW, Aarntzen EHJG, Schreibelt G, Keunen JEE, Coulie PG, de Klein A, Punt CJA, Paridaens D, Figdor CG, de Vries IJM (2014) Long overall survival after dendritic cell vaccination in metastatic uveal melanoma patients. Am J Ophthalmol 158: 939–947.
Bronkhorst IHG, Jager MJ (2013) Inflammation in uveal melanoma. Eye 27: 217–223.
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G (2013) BAP1 and cancer. Nat Rev Cancer 13: 153.
Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, Linette GP, Gajewski TF, Lutzky J, Lawson DH, Lao CD, Flynn PJ, Albertini MR, Sato T, Lewis K, Doyle A, Ancell K, Panageas KS, Bluth M, Hedvat C, Erinjeri J, Ambrosini G, Marr B, Abramson DH, Dickson MA, Wolchok JD, Chapman PB, Schwartz GK (2014) Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 311: 2397–2405.
Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, Bastian BC (2014) Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 33: 4724–4734.
Ehlers JP, Harbour JW (2005) NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res 11: 1849–1853.
Ehlers JP, Worley L, Onken MD, Harbour JW (2005) DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res 11: 3609–3613.
All-Ericsson C, Girnita L, Müller-Brunotte A, Brodin B, Seregard S, Ostman A, Larsson O (2004) c-Kit-dependent growth of uveal melanoma cells: a potential therapeutic target? Invest Ophthalmol Vis Sci 45: 2075–2082.
Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q, Gutkind JS (2014) Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a Trio-regulated Rho GTPase signaling circuitry. Cancer Cell 25: 831–845.
Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, Stern M-H, Marais R (2013) SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3: 1122–1129.
Gentien D, Kosmider O, Nguyen-Khac F, Albaud B, Rapinat A, Dumont AG, Damm F, Popova T, Marais R, Fontenay M, Roman-Roman S, Bernard OA, Stern M-H (2014) A common alternative splicing signature is associated with SF3B1 mutations in malignancies from different cell lineages. Leukemia 28: 1355–1357.
Harbour JW (2014) A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol Biol 1102: 427–440.
Harbour JW, Onken MD, Roberson EDO, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM (2010) Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330: 1410–1413.
Hofmann UB, Kauczok-Vetter CS, Houben R, Becker JC (2009) Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. Clin Cancer Res 15: 324–329.
Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE (2012) Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res 18: 4345–4355.
Landreville S, Agapova OA, Matatall KA, Kneass ZT, Onken MD, Lee RS, Bowcock AM, Harbour JW (2012) Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res 18: 408–416.
Laurent C, Gentien D, Piperno-Neumann S, Némati F, Nicolas A, Tesson B, Desjardins L, Mariani P, Rapinat A, Sastre-Garau X, Couturier J, Hupé P, de Koning L, Dubois T, Roman-Roman S, Stern M-H, Barillot E, Harbour JW, Saule S, Decaudin D (2013) Patient-derived xenografts recapitulate molecular features of human uveal melanomas. Mol Oncol 7: 625–636.
Li Z, Yu X, Shen J, Jiang Y (2015) MicroRNA dysregulation in uveal melanoma: a new player enters the game. Oncotarget 6: 4562–4568.
Mariani P, Servois V, Piperno-Neumann S (2012) Therapeutic options in metastatic uveal melanoma. Dev Ophthalmol 49: 166–181.
Martin M, Maßhöfer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, Lohmann DR, Zeschnigk M (2013) Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet 45: 933–936.
Musi E, Ambrosini G, de Stanchina E, Schwartz GK (2014) The phosphoinositide 3-kinase α selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol Cancer Ther 13: 1044–1053.
Némati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, Robert D, Péguillet I, Donnadieu M-H, Dahmani A, Bessard M-A, Gentien D, Reyes C, Saule S, Barillot E, Roman-Roman S, Decaudin D (2010) Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clin Cancer Res 16: 2352–2362.
Onken MD, Worley LA, Harbour JW (2008) A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res 14: 3737–3745.
Péguillet I, Milder M, Louis D, Vincent-Salomon A, Dorval T, Piperno-Neumann S, Scholl SM, Lantz O (2014) High numbers of differentiated effector CD4 T cells are found in patients with cancer and correlate with clinical response after neoadjuvant therapy of breast cancer. Cancer Res 74: 2204–2216.
Piperno-Neumann S, Kapiteijn E, Larkin JMG, Carvajal RD, Luke JJ, Seifert H, Roozen I, Zoubir M, Yang L, Choudhury S, Yerramilli-Rao P, Hodi FS, Schwartz GK (2014) Phase I dose-escalation study of the protein kinase C (PKC) inhibitor AEB071 in patients with metastatic uveal melanoma. J Clin Oncol 32: 5s.
Prescher G, Bornfeld N, Becher R (1990) Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst 82: 1765–1769.
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O’Brien J, Bastian BC (2010) Mutations in GNA11 in uveal melanoma. N Engl J Med 363: 2191–2199.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Müller J (2010) Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465: 243–247.
Singh AD, Turell ME, Topham AK (2011) Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology 118: 1881–1885.
Surriga O, Rajasekhar VK, Ambrosini G, Dogan Y, Huang R, Schwartz GK (2013) Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther 12: 2817–2826.
Vaqué JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, Debant A, Seeger MA, Ksander BR, Teramoto H, Gutkind JS (2013) A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol Cell 49: 94–108.
Van der Velden PA, Maat W (2009) Methylation in uveal melanoma. Br J Ophthalmol 93: 132.
Yu F-X, Luo J, Mo J-S, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J, Chen X, Zhang L, Wang C-Y, Bastian BC, Zhang K, Guan K-L (2014a) Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25: 822–830.
Yu H, Mashtalir N, Daou S, Hammond-Martel I, Ross J, Sui G, Hart GW, Rauscher FJ, Drobetsky E, Milot E, Shi Y, Affar EB (2010) The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol 30: 5071–5085.
Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, Barbour H, Corbeil L, Hébert J, Drobetsky E, Masson JY, Di Noia JM, Affar EB (2014b) Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA 111: 285–290.
Zimmer L, Vaubel J, Mohr P, Hauschild A, Utikal J, Simon J, Garbe C, Herbst R, Enk A, Kämpgen E, Livingstone E, Bluhm L, Rompel R, Griewank KG, Fluck M, Schilling B, Schadendorf D (2015) Phase II DeCOG-study of ipilimumab in pretreated and treatment-naïve patients with metastatic uveal melanoma. PLoS One 10: e0118564.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Nabil, AA., Marie, S., Marc-Henri, S. et al. Upcoming translational challenges for uveal melanoma. Br J Cancer 113, 1249–1253 (2015). https://doi.org/10.1038/bjc.2015.269
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.269
Keywords
This article is cited by
-
Single-cell RNA sequencing reveals intratumoral heterogeneity in primary uveal melanomas and identifies HES6 as a driver of the metastatic disease
Cell Death & Differentiation (2021)
-
Uveal melanoma: physiopathology and new in situ-specific therapies
Cancer Chemotherapy and Pharmacology (2019)
-
Identification of novel chemotherapeutic strategies for metastatic uveal melanoma
Scientific Reports (2017)
-
Update on Metastatic Uveal Melanoma: Progress and Challenges
BioDrugs (2016)
