
Abstract
Background:
Current treatment strategies for head and neck cancer are associated with significant morbidity and up to 50% of patients relapse, highlighting the need for more specific and effective therapeutics. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and Smac mimetics (SMs) are promising anticancer agents, but their effect on head and neck squamous cell carcinoma (HNSCC) remains unknown.
Methods:
We examined the response of a panel of nine HNSCC cell lines to TRAIL and SMs and investigated the mechanism of cell type-specific response by functional analysis.
Results:
Head and neck cancer cell lines revealed a converse response pattern with three cell lines being highly sensitive to Smac-164 (SM) but resistant to TRAIL, whereas the other six were sensitive to TRAIL but resistant to SM. Distinct protein expression and activation patterns were found to be associated with susceptibility of HNSCC cell lines to TRAIL and SM. Tumour necrosis factor-related apoptosis-inducing ligand sensitivity was associated with high caspase-8 and Bid protein levels, and TRAIL-sensitive cell lines were killed via the type II extrinsic apoptotic pathway. Smac mimetic-sensitive cells expressed low levels of caspase-8 and Bid but had high TNF-α expression. Smac mimetic-induced cell death was associated with caspase-10 activation, suggesting that in the absence of caspase-8, caspase-10 mediates response to SM. Cotreatment with TNF-α sensitised the resistant cells to SM, demonstrating a decisive role for TNF-α-driven feedback loop in SM sensitivity.
Conclusions:
Tumour necrosis factor-related apoptosis-inducing ligand and SMs effectively kill HNSCC cell lines and therefore represent potential targeted therapeutics for head and neck cancer. Distinct molecular mechanisms determine the sensitivity to each agent, with levels of TNF-α, caspase-8, Bid and caspase-10 providing important predictive biomarkers of response to these agents.
Similar content being viewed by others


Main
Surgery and radiotherapy alone or in combination with chemotherapy are the common treatment modalities for head and neck cancer. However, they have severe side effects and more than 50% of patients relapse (Suh et al, 2014). This highlights the need for more effective, targeted therapies and for the development of strategies to select patients who could benefit from specific treatment regimens.
In recent years, novel therapeutic agents have emerged that target specific molecules involved in apoptotic pathways. The tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) shows selective cytotoxicity in several types of malignant cell lines (Falschlehner et al, 2009) and recombinant TRAIL is currently used in phase I/II clinical trials for the treatment of patients with advanced tumours (Herbst et al, 2010). Tumour necrosis factor-related apoptosis-inducing ligand induces cell death by binding to its corresponding cell surface receptors TRAIL-R1 or -R2. Upon clustering, the activated receptors recruit several cytosolic proteins to form the death-inducing signalling complex (DISC) (Wang and El-Deiry, 2003). This leads to the activation of the initiator caspase-8 and/or -10 (Juo et al, 1999), which can in turn trigger two different extrinsic apoptotic pathways. In the type I pathway, caspase-8 directly activates the effector caspases (caspase-3, -6 and -7) resulting in the execution of apoptotic cell death, whereas the type II pathway involves caspase-8-dependent cleavage of Bid. Cleaved Bid initiates the permeabilisation of the outer mitochondrial membrane, leading to cytochrome c release, activation of the initiator caspase-9 and the caspase cascade involving caspase-3 (Kruyt, 2008; Kantari and Walczak, 2011).
Smac mimetics (SMs) are a class of targeted anticancer drugs that have been developed to mimic functionally the endogenous proapoptotic protein Smac/Diablo (Chen and Huerta, 2009). Smac/Diablo is a mitochondrial protein that is released into the cytoplasm following permeabilisation of the outer mitochondrial membrane in response to an intrinsic death stimulus (Du et al, 2000; Yang and Yu, 2003). This protein is a natural inhibitor of a family of inhibitor of apoptosis proteins (IAPs) (Du et al, 2000), including cIAP-1, cIAP-2 and XIAP. So far, XIAP has been the only IAP family member for which direct binding to and inhibition of caspases-3, -7 and -9 by proteasomal degradation has been described (Deveraux et al, 1997; Petersen et al, 2007; Varfolomeev et al, 2007). In contrast to this, the precise roles of cIAP-1 and -2 are currently not known. Accumulating evidence suggests that cIAP-1 and -2 are involved in various signal-transduction pathways, including NF-κB activation and TNF-α secretion (Mahoney et al, 2008), leading to the induction of apoptotic (Tenev et al, 2011) or necroptotic cell death (McComb et al, 2012). Different types of SMs have been developed as potential anticancer agents with varying modes of action, for example, induction of TNF-α-dependent apoptosis (Petersen et al, 2007) and blocking of IAP activity (Creagh et al, 2004; Bockbrader et al, 2005).
Both TRAIL and SMs have been tested in several cancer models, but very little data are available on their therapeutic potential in HNSCC (Kruyt, 2008; Chen and Huerta, 2009; Gasparini et al, 2013). In this study, we have tested the sensitivity of a panel of HNSCC cell lines to TRAIL and Smac-164 (SM) and identified a cell line-specific response. The sensitivity of a cell line to TRAIL or SM was found to be determined by the expression pattern of components of apoptotic signalling pathways and TNF-α levels. Importantly, in HNSCC cell lines with low caspase-8 levels, SM treatment induced caspase-10 activation. These findings identify cell type-specific mechanisms of TRAIL and SM action and provide potential biomarkers for selecting tumours that are likely to benefit from such treatments.
Materials and methods
Cell lines
The cell lines HSC3 and HSC3M3 were a gift from Dr Kazuya Tominaga, Department of Oral Pathology, Osaka Dental University (Hirakata, Osaka, Japan). The HN5 cell line was provided by Dr Barry Gusterson, Department of Pathology, University of Glasgow (Glasgow, UK). The HN30 cell line was a gift from Dr Andrew Yeudall, Philips Institute of Oral and Craniofacial Molecular Biology (Richmond, VA, USA). The H357 cell line was a gift from Dr Stephen Prime, Department of Oral and Dental Science, University of Bristol (Bristol, UK). UMSCC74A, UMSCC74B, UMSCC11B and UMSCC22B were provided by Dr Thomas E Carey, University of Michigan (Ann Arbor, MI, USA). All cell lines except H357 were cultured in DMEM supplemented with 10% FCS, 50 μg ml−1 streptomycin, 100 μg ml−1 penicillin and 1 mM sodium pyruvate and incubated at 37 °C and 5% CO2. H357 cells were cultured in DMEM-F12 supplemented with 5% FCS, 4 mM L-glutamine, 69 nM hydrocortisone, 5 μg ml−1 streptomycin, 5 μg ml−1 penicillin and 1 mM sodium pyruvate. For selection of Bcl-2-overexpressing cells, G418 was added to a final concentration of 400 μg ml−1. Selection of sh-Bid and sh-scrambled control, as well as sh-caspase-8 and sh-scrambled control cells was performed using 2 μg ml−1 puromycin.
Reagents and antibodies
The Smac-164 (SM) was a gift from Prof Pierfausto Seneci (University of Milan, Milan, Italy). Recombinant human isoleucine zipper trimerised TRAIL (izTRAIL), necrostatin-1, HS101 and HS201 mouse monoclonal anti-TRAIL receptor 1 and 2 antibodies were provided by Prof Henning Walczak (University College London, London, UK). The caspase inhibitor z-VAD-fmk (z-VAD) was purchased from Promega (Madison, WI, USA), the human TNF-α ELISA Kit from Life Technologies (Paisley, UK), XIAP siRNA oligonucleotide (5′-AUCCAUCCAUGGCAGAUUA-3′) from MWG Biotechnology (Ebersberg, Germany), the neutralising IgA monoclonal antibody to human TNF-α from InvivoGen (San Diego, CA, USA) and mouse monoclonal anti-human CD120a (TNF-R1), clone H398 from ABD Serotec (Puchheim, Germany). Antibodies used for immunoblotting were: β-actin (1 : 10 000), tubulin (1 : 10 000) (Sigma-Aldrich, St Louis, MO, USA), Bcl-2 (1 : 1000; Santa Cruz Biotechnology, Dallas, TX, USA), caspase-9 (1 : 1000), caspase-3 (1 : 1000), Mcl-1 (1 : 1000), PARP (1 : 1000), Bid (1 : 1000), CoxIV (1 : 1000) (Cell Signaling, Danvers, MA, USA), caspase-8 (1 : 500), c-FLIP (1 : 500) (Enzo Life Sciences, Exeter, UK), cIAP-1 (0.5 μg ml−1; R&D System, Minneapolis, MN, USA), cIAP-2 (4 μg ml−1; BD Pharmingen, Oxford, UK), Smac/Diablo (1 : 1000; Epitomics, Burlingame, CA, USA), XIAP (1 : 4000; BD Transduction Laboratories, Oxford, UK), cytochrome c (1 : 1000; Abcam, Cambridge, UK) and caspase-10 (1 : 1000; MBL International, Woburn, MA, USA). Secondary HRP-coupled anti-rabbit (1 : 2000) and anti-mouse antibodies (1 : 1000) were obtained from Fisher Scientific (Loughborough, UK) and Sigma-Aldrich, respectively. The p50 and p52 antibodies (1 : 1000) were provided by Dr Dagmar Kulms, Centre for Regenerative Therapies (Dresden, Germany).
MTT cell viability assay
Cells were seeded in 96-well plates at a density of 2–4 × 103 cells one day before SM or TRAIL treatment. In case of the inhibitor studies, 30 μ M necrostatin-1 (nec-1) was included in the treatment, whereas z-VAD-fmk (20 μ M) was added 1 h in advance. The cells were incubated for 24–72 h (endpoint measurement) or 2–8 h (time kinetic). Twenty microlitres of MTT reagent (5 mg ml−1) and, after 3 h, 150 μl of solubilisation solution (50% dimethylformamide, 0.2% glacial acetic acid, 20 mM HCl, 10% SDS) was added to each well. After 16–24 h, OD595 was measured using the LT-400 microplate reader (Labtech, Uckfield, East Sussex, UK). The statistical analysis of the results was performed using Student’s unpaired two-tailed t-test (GraphPad Prism 5 software, La Jolla, CA, USA).
Immunoblot analysis
Cells were trypsinised, washed in 1 × PBS and then resuspended in lysis buffer (1 mM MgCl2, 12.5 mM HEPES/KOH, pH 7.4, 1 mM EGTA, 0.1% Triton X-100) including protease inhibitors. After 30 min incubation on ice, the protein lysates were obtained by centrifugation (13 200 r.p.m. for 15 min at 4 °C) and the protein concentration was determined by the Bradford assay (Bradford, 1976). Thirty micrograms of protein was separated on 10–15% 1.5-mm-thick SDS gels, then transferred to nitrocellulose membranes (400 mA for 90 min) and subsequently probed with the respective antibodies. The Mini-PROTEAN electrophoresis system combined with the Mini-Trans Blot module (Bio-Rad, Hercules, CA, USA) was used for protein electrophoresis and tank transfer.
Lentiviral knockdown
Lentiviral constructs were produced in HEK 293T cells. HEK 293T cells were transfected with the construct of interest as well as plasmids coding for gag/pol and env using calcium phosphate as described previously (Jordan and Wurm, 2004). At 24, 36 and 48 h after transfection, viral supernatants were harvested and filtered through a 0.45 μm filter. For the storage of the virus at -80 °C, 5 μg ml−1 polybrene was added. Infection of target cells with virus was performed overnight. Cells were selected depending on their newly acquired antibiotic resistance. A control of nontransduced cells was treated in parallel to ensure toxicity of the antibiotic. The pGIPZ-sh-Bid constructs including the sh-scrambled control were kindly provided by Prof Simone Fulda (Institute for Experimental Cancer Research in Pediatrics, JW Goethe University Hospital Frankfurt, Frankfurt, Germany). A mixture of five Bid sh-RNAs was used for transduction. The lentiviral, doxycycline-inducible TRIPZ caspase-8 and scrambled control sh-RNAs were a gift from Prof Pascal Meier (the Breakthrough Toby Robins Breast Cancer Research Centre, Institute of Cancer Research, London, UK). Expression of the sh-RNAs was induced by adding 1 μg ml−1 doxycycline for 36 h.
ELISA
Equal numbers of cells were seeded in 12-well plates in 1 ml medium and incubated for 24 h. The next day, fresh medium including 50 ng ml−1 TRAIL or 50 nM SM was applied. After 24 h, the medium was collected and stored at −80 °C until further analysis. The level of secreted TNF-α was measured by ELISA using a 96-well plate. The capture/coating antibody (anti-human TNF-α) was used at a final concentration of 2 μg ml−1. After an overnight incubation followed by several washing steps, unspecific binding sites were blocked for 1–2 h at room temperature using assay buffer (0.5% BSA : PBS : 0.1% Tween-20). One hundred microlitres of the samples (medium of cells) or of a standard dilution series (recombinant human TNF-α) was added to designated wells, followed by 100 μl of detection antibody (1 : 1250 in assay buffer) and 100 μl of strept-HRP solution (diluted 1 : 1000 in assay buffer). Wells were washed three times, 50 μl TMB substrate and 20 min later 50 μl of stop solution (2 N H2SO4) was added. The colour intensity was measured at 450 nm using an LT-4000 microplate reader.
Transient XIAP knockdown using siRNA
HSC3M3 and UMSCC11B cells were transfected with XIAP siRNA oligonucleotide (5′-AUCCAUCCAUGGCAGAUUA-3′) or a scrambled control using X-tremeGENE siRNA transfection reagent (Roche Applied Science, West Sussex, UK) according to the manufacturer’s manual. The cells were incubated for 24–48 h before treatment with either TRAIL (50 ng ml−1) or SM (50 nM).
Generation of stable cell lines
Bcl-2 stably overexpressing HSC3M3 and H357 cell lines were established using pcDNA3-Bcl-2. pcDNA3.1 was used as a negative control. Cells were transfected with the construct of interest using X-tremeGENE HP DNA transfection reagent (Roche Applied Science) as recommended by the manufacturer. The following day, the medium was replaced with medium containing the respective selection antibiotic (G418). The cells were incubated for 1–2 weeks and the medium was changed every 2 days. Stable colonies were collected by trypsinisation and transferred to T25 flasks for expansion. Cells were tested for selection efficiency by immunoblotting.
FACS analysis of TRAIL receptors
In 96-well plates, cells were seeded at a density of 4 × 105 cells per well. After centrifugation, cells were resuspended in the respective primary antibody (HS101 anti-TR1 as well as HS201 anti-TR2). The antibodies were used at a concentration of 5 μg ml−1 diluted in FACS buffer. Cells were incubated on ice for 30 min, followed by centrifugation and three washing steps with ice-cold FACS buffer. The secondary antibody (1 : 200) was added to the cells and incubated for 20 min on ice. Cells were centrifuged and washed three times with ice-cold FACS buffer. Afterwards, they were incubated for 20 min on ice in the presence of Strept-PE-coupled antibody (protected from light), centrifuged and washed three times with ice-cold FACS buffer. The cells were resuspended in 100 μl of PI solution (1 μg ml−1) and analysed by flow cytometry with a FACS Calibur (BD Biosciences, San Jose, CA, USA).
Preparation of subcellular fractions
Mitochondrial and cytosolic/ER fractions were isolated as described previously (Frezza et al, 2007). Briefly, cells of four confluent 15 cm plates were harvested for each condition, cells were washed two times with 1 × PBS and resuspended 1 : 1 in ice-cold IBc buffer (10 mM Tris, pH 7.5, 1 mM EGTA, 200 mM sucrose, plus protease inhibitor cocktail). After 30 min, incubation cells were lysed using a 26 G syringe until ∼60% of the cells were Trypan blue positive. Several centrifugation steps were performed at low speed to remove unlysed, intact cells (700–1200 g for 5 min at 4 °C). To obtain a mitochondrial and cytosolic fraction, the supernatant was centrifuged at 7200 g for 20 min (4 °C). The supernatant represented the cytosolic/ER fraction. The pellet (mitochondria) was washed two times and then lysed in lysis buffer. Thirty micrograms mitochondrial protein was used for immunoblot analysis. Corresponding volumes of the cytosolic fraction were loaded. Purity of each fraction was analysed using specific marker proteins.
Results
Head and neck cancer cell lines show distinct responses to TRAIL and SMs
To assess the therapeutic potential of TRAIL and SMs in head and neck cancer, a panel of nine HPV-negative HNSCC cell lines (HSC3, HSC3M3, HN5, HN30, UMSCC74A, UMSCC74B, H357, UMSCC11B and UMSCC22B) was tested for the cytotoxic effect of izTRAIL and Smac-164 (SM). The molecular characteristics of these cell lines are shown in the Supplementary Table S1. Treatment with lethal concentrations of both drugs revealed a distinct pattern of sensitivity of the cell lines to either TRAIL or SM (Figure 1A). While HSC3, HSC3M3 and HN5 cells were very sensitive to SM, they displayed complete resistance to TRAIL. On the other hand, HN30, UMSCC74A, UMSCC74B, H357, UMSCC11B and UMSCC22B were highly sensitive to TRAIL, but fully resistant to SM (Figure 1A). The majority of responsive cells were highly sensitive to even low concentrations of the respective drug and responded in a dose-dependent manner (IC50 values; Supplementary Table S2).

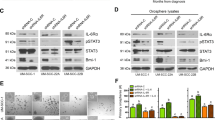
Head and neck cancer cell lines show distinct sensitivity/resistance to TRAIL and Smac-164 (SM). (A) Nine HNSCC cell lines were treated with TRAIL (50 ng ml−1) or SM (28.33 nM) and cell viability was measured 72 h later by MTT assay. Values of treated cells were normalised with respect to the untreated controls. Smac mimetic-sensitive (TRAIL-resistant) HSC3 and TRAIL-sensitive (SM-resistant) H357 cells were treated with 50 ng ml−1 TRAIL (B) or 200 nM SM (C) alone or combined with the caspase inhibitor z-VAD-fmk (20 μM) and/or the RIP1 kinase inhibitor necrostatin-1 (30 μM) and cell survival was analysed 24 h later by MTT assay. All values indicate means±s.d. of three independent experiments (n=3). ***P<0.0001. (D) The expression level of caspase-8, c-FLIP, Bcl-2, cIAP-2, XIAP, cIAP-1, Mcl-1, Smac/DIABLO and caspase-3 protein was analysed in nine HNSCC cell lines.
Smac mimetics induce both apoptotic and necroptotic cell death in the responsive HNSCC cell lines
To determine the mode of cell death, representative cell lines were treated with TRAIL or SM in combination with either the caspase inhibitor z-VAD and/or the RIP1 kinase inhibitor necrostatin-1 (nec-1) (Figures 1B and C). Addition of z-VAD substantially increased cell viability, ranging from 40 to 90%, in the TRAIL-responsive H357 cells (Figure 1B). Treatment with nec-1 had no additional effect on TRAIL-induced cell death when used either alone or in combination with z-VAD, suggesting a solely apoptotic response of sensitive HNSCC cells to TRAIL. Adding z-VAD increased cell survival in response to SM in the sensitive cells by ∼60%. Also, nec-1 treatment alone or in combination with z-VAD further increased cell survival by a small but significant amount (Figure 1C). These data suggest that SM mainly induces caspase-dependent cell death in the responsive HNSCC cell lines with evidence of partial RIPK1-dependent necroptotic cell death in response to SM.
Distinct protein signature predicts TRAIL and SM sensitivity
The differential responses to TRAIL or SM among the HNSCC cell lines provided an ideal model to identify a predictive gene expression signature for tumour cell susceptibility to these drugs. Evaluation of several protein markers involved in cell death pathways indicated clear differences between several proteins: caspase-8 level was significantly higher in TRAIL-sensitive cell lines, and also increased expression levels of c-FLIP, cIAP-2 and Bcl-2 proteins were observed in TRAIL-sensitive (SM-resistant) cell lines in contrast to SM-sensitive (TRAIL-resistant) cell lines. Expression levels of XIAP, cIAP-1, Mcl-1, Smac/DIABLO and caspase-3 did not show any clear differences between the various sensitive and resistant cells (Figure 1D). Similarly, the cell surface expression levels of TRAIL receptors DR4 and DR5 did not correlate with TRAIL sensitivity in HNSCC cell lines (Supplementary Figure S3).
TRAIL induces type II extrinsic apoptosis in responsive HNSCC cells
To characterise the apoptotic pathway induced by TRAIL in sensitive HNSCC cell lines, the kinetics of caspase activation were analysed in representative TRAIL-responsive (H357) and resistant (HSC3) cell lines. Cleavage of caspase-8 into the active p18 fragment and caspase-3 into the active p17 fragment were observed only in TRAIL-sensitive H357 cells. Cleavage occurred as early as 2 h after TRAIL treatment. The presence of cleaved PARP protein, indicative of caspase-3 activity, was detected in H357 cells only. Additionally, cleavage of the full-length Bid protein to generate tBid was observed in conjunction with caspase-8 activation at an early stage of apoptosis in H357 cells, indicating a role for Bid in mediating TRAIL-induced cell death (Figure 2A).

TRAIL induces the type II extrinsic apoptotic pathway in sensitive cells. (A) TRAIL-resistant HSC3 and TRAIL-sensitive H357 cells were incubated in the presence of 50 ng ml−1 TRAIL for 2, 4 and 6 h. Cleavage of caspase-8, caspase-3, Bid and PARP was analysed. (B) sh-scrambled (scr) and sh-Bid HSC3 and H357 cells were generated. The efficiency of the knockdown was assessed via immunoblotting. Sh-scr-infected cells served as control. (C) Cell viability of the knockdown and control cells in the presence of increasing concentrations of TRAIL was determined by MTT assay after 16 h. Values are means±s.d. of three independent experiments (n=3). (D) Cleavage of caspase-9 was analysed in response to 3 h of 100 ng ml−1 TRAIL treatment in representative sensitive (H357) and resistant (HSC3) cell lines. (E) H357, H357-GFP and H357-Bcl-2 cells were treated with TRAIL (50 ng ml−1) for 24 h and cell viability was determined by MTT assay. All values are means±s.d. of three independent experiments (n=3). (F) Immunoblotting showing Bcl-2 expression in the H357, H357-GFP and H357-Bcl-2 cells. (G) Caspase-3 and PARP cleavage as well as cIAP-1 and XIAP protein levels were analysed by immunoblotting in UMSCC11B cells treated between 2 and 12 h with 50 ng ml−1 TRAIL or 50 nM SM. (H) The effect of the pancaspase inhibitor z-VAD-fmk (20 μM) on XIAP cleavage in UMSCC11B cells incubated with 50 ng ml−1 TRAIL or 50 nM SM was analysed by immunoblotting. (I) XIAP was downregulated by siRNA in UMSCC11B cells. Cell viability was tested by MTT assay after treatment with 50 ng ml−1 TRAIL or 50 nM SM for 24 h. The results are shown as means±s.d. of three independent experiments (n=3). Reduction in XIAP protein level after 24 and 48 h was confirmed by immunoblotting.
To determine the importance of Bid in TRAIL-induced death, H357 and HSC3 Bid knockdown and control cells were generated (Figure 2B) and analysed for response to TRAIL. Over 30% increased cell survival was observed in Bid-knockdown H357 cells as compared with sh-scrambled control after treatment with 100 ng ml−1 TRAIL (Figure 2C). In addition, cleavage and activation of caspase-9 correlated with cell type-specific sensitivity (Figure 2D) and further supported a mitochondrial involvement during TRAIL-induced apoptosis in HNSCC cell lines. Additionally, Bcl-2 overexpression resulted in increased resistance to TRAIL in sensitive H357 cells (Figures 2E and F), suggesting a type II extrinsic apoptosis mechanism involving Bid, Bcl-2 and mitochondria.
IAP family members such as XIAP have been implicated in cell death in response to TRAIL treatment (Lu et al, 2008; Ndozangue-Touriguine et al, 2008; Allensworth et al, 2012). We investigated cIAP-1 and XIAP protein levels in TRAIL-sensitive UMSCC11B cells following 2 to 12 h of TRAIL treatment. Both proteins showed reduced levels with increasing duration of TRAIL treatment (Figure 2G). However, XIAP downregulation was caspase-mediated as it was blocked by z-VAD (Figure 2H). Additionally, downregulation of XIAP by siRNA neither had an additional sensitisation effect of TRAIL-responsive cells to TRAIL (Figure 2I) nor sensitised resistant HSC3M3 cells to TRAIL (Figure 2H). Therefore, XIAP is not critical for sensitivity/resistance of HNSCC cell lines to TRAIL.
Smac mimetics induce cell death in HNSCC cells through caspase-10 activation
To examine the importance of caspase-8 in response to SM, doxycycline-inducible caspase-8 knockdown HSC3 cells were generated and tested for sensitivity to SM. Depletion of caspase-8 only marginally protected HSC3 cells from SM-induced cell death (Figures 3A and B). We then investigated whether caspase-10 is involved in SM-induced cell death and showed cleavage and activation of caspase-10 in response to SM treatment (Figure 3C). Similar levels of caspase-10 cleavage and activation were observed in caspase-8 knockdown compared with sh-scrambled control HSC3 cells in response to SM treatment (Figure 3A and Supplementary Figure S4A), indicating a caspase-8-independent mechanism of caspase-10 activation. These results suggest that caspase-10 may substitute for the lack of caspase-8 or cooperate with low caspase-8 activity to induce cell death in response to SM. Importantly, there was no cleavage of caspase-10 in response to SM or TRAIL treatment in TRAIL-sensitive (SM-resistant) H357 cell line (Figure 3C and Supplementary Figure S4B).

Smac mimetic-induced cell death is mainly caspase-8 independent and associated with cytochrome c release. (A) HSC3 cells were either infected with an inducible lentiviral sh-caspase-8 or a scrambled (scr) sh-RNA control. Expression of the sh-RNA was induced by addition of 1 μg ml−1 doxycycline for 36 h. The efficiency of the caspase-8 knockdown in HSC3 cells was determined by immunoblotting. Non-induced cells served as control. (B) Induced HSC3 caspase-8 knockdown or sh-scr control cells were treated with increasing concentrations of SM. Cell viability was analysed 24 h later by MTT assay. Values indicate means±s.d. of three independent experiments (n=3). (C) Immunoblotting for full-length and cleaved caspase-10 and -3 in response to treatment with 200 nM SM for up to 6 h. Caspase-10 fragments that were specifically detected in SM-sensitive HSC3 cells are marked. (D) HSC3 and H357 cells were incubated with 200 nM SM for 3 h. Cytochrome c release was analysed by subcellular fractionation. Purity of each fraction was assessed using characteristic marker proteins. Thirty micrograms of mitochondrial fractions were loaded. (E) Cleavage of caspase-9 was analysed in response to 3 h of 200 nM SM treatment in representative sensitive (HSC3) and resistant (H357) cell lines. (F) HSC3M3 cells were treated with 50 nM SM. After 2, 4, 8 and 12 h capase-3 activation, PARP cleavage, cIAP-1 and XIAP protein levels were analysed. (G) Treatment of HSC3M3 with 50 ng ml−1 TRAIL or 50 nM SM±the pancaspase inhibitor z-VAD-fmk (20 μM). After 8 h of treatment, cIAP-1 and XIAP protein levels were analysed by immunoblotting. (H) Transient knockdown of XIAP in HSC3M3 cells. Cell viability in response to 50 ng ml−1 TRAIL or 50 nM SM was analysed after 24 h by MTT assay. Values are shown as means±s.d. of three independent experiments (n=3). Efficient knockdown was confirmed by immunoblotting.
We then examined the mechanism of cell death downstream of initiator caspases and observed cytochrome c release, as well as cleavage and activation of caspase-9 at 3 h after SM treatment. This result suggests a role for the intrinsic mitochondrial apoptosis pathway in sensitivity of cells to SM (Figures 3D and E). As a role for caspase-10 in Bid cleavage has been previously reported (Fischer et al, 2006), we investigated whether caspase-10 activation led to mitochondrial dysfunction via proapoptotic Bid protein. SM treatment reduced full-length Bid protein in sensitive HSC3 cells (Supplementary Figure S5A). Furthermore, Bid knockdown (Figure 2B) diminished the level of cytochrome c release in response to SM treatment (Supplementary Figure S5B). However, Bid knockdown did not inhibit the effect of SM in the sensitive HSC3 cells (Supplementary Figure S5C), suggesting a Bid-independent caspase-10-mediated cell death by SM.
Further we investigated the role of IAPs in SM sensitivity of HNSCC cells (Figure 3F). Smac mimetic induced cell death as evident by caspase-3 activation in the responsive cells as early as 4 h after treatment and also resulted in early cIAP-1 depletion and downregulation of XIAP. Downregulation of XIAP but not cIAP-1 was caspase-mediated as the effect was blocked by the addition of z-VAD (Figure 3G). These data suggest that XIAP does not have a decisive role in SM-induced apoptosis in HNSCC. In agreement with these data, XIAP knockdown did not significantly increase sensitivity of responsive cells to SM (Figure 3H). Importantly, XIAP knockdown also did not sensitise SM-resistant cells to SM, indicating different processes other than XIAP being responsible for SM resistance (Figure 2I). By contrast, depletion of cIAP-1 occurred in both sensitive and resistance cell lines after SM treatment, implying that the observed distinct SM response in HNSCC cell lines was not solely mediated by cIAP-1.
TNF-α levels determine SM sensitivity
IAP antagonists are known to induce NF-κB-mediated production of TNF-α resulting in killing of the cells through an autocrine mechanism (Vince et al, 2007). We therefore investigated the expression of endogenous TNF-α and its induction in response to SM and TRAIL in HNSCC cell lines. A direct correlation between TNF-α secretion levels and SM sensitivity in HNSCC cell lines was detected. Non-stimulated SM-sensitive HSC3 and HSC3M3 cells showed significantly higher levels of secreted TNF-α than resistant HN30, UMSCC11B and H357 cells (Figure 4A). Smac mimetics further induced autocrine TNF-α secretion by up to six-fold over endogenous levels in SM-sensitive but not -resistant cell lines. TRAIL treatment had no significant effect on TNF-α secretion in all cell lines tested. Importantly, addition of TNF-α-blocking antibody rescued SM-sensitive HSC3 cells from SM-induced cell death (Figure 4B) and combining SM with rhTNF-α sensitised resistant UMSCC11B cells to SM inducing 60% cell death (Figures 4C and D). These data show a decisive role for autocrine TNF-α secretion in SM-mediated cell death in HNSCC cell lines.

TNF- α levels correlate with SM sensitivity and partially sensitise resistant cells to SM. (A) Three SM-sensitive cell lines (HSC3, HSC3M3 and HN5) and three SM-resistant cell lines (HN30, UMSCC11B and H357) were treated with 50 ng ml−1 TRAIL or 50 nM SM for 24 h. The amount of secreted TNF-α was determined using ELISA. The experiment was performed in duplicate; the results depicted are the average of the TNF-α concentration in the tested samples. One of two independent experiments is shown. (B) HSC3 cells were treated with 50 nM SM alone or in combination with 200 μg ml−1 TNF-α blocking antibody (ab) for 4 h. Cell viability was measured by MTT assay. Values are means±s.d. of three independent experiments (n=3). (C) Cell viability of HSC3M3 and UMSCC11B cells was determined by MTT assay after 24 h treatment with 10 ng ml−1 rhTNF-α, 50 nM SM or 50 ng ml−1 TRAIL. Values indicate means±s.d. of three independent experiments (n=3). (D) UMSCC11B cells were incubated in the presence of increasing concentrations of rhTNF-α alone or combined with 50 ng ml−1 TRAIL or 50 nM SM. Cell viability was measured 24 h later by MTT assay. Values are means±s.d. of three independent experiments (n=3). (E) The effect of 200 nM SM on the noncanonical NF-κB signalling pathway was analysed in SM-sensitive HSC3 and -resistant H357 cells. Processing of p100 to p52 was assessed by immunoblotting.
The difference in TNF-α secretion levels between SM-sensitive and -resistant cell lines was concomitant with the activity of the noncanonical NF-κB signalling pathway (Figure 4E). The SM-sensitive HSC3 cells demonstrated baseline level of p100 processing and p52 generation before treatment, which was further increased in response to SM. This effect was observed at early time points, suggesting a role for p100 processing in the initiation stages of cell death and coincided with a clear caspase-3 cleavage and activation (Supplementary Figure S5A). By contrast, SM-resistant H357 cells lacked baseline p100 processing and did not generate p52 in response to SM treatment.
Collectively, our data indicate a cell line-specific response to the cytotoxic effect of TRAIL and SM in HNSCC. The pathways used, such as type I or II extrinsic apoptosis and/or necroptosis, are determined by the cellular levels of the proapoptotic proteins such as caspase-8, Bid and TNF-α. Our results also identify caspase-10 as an important initiator caspase in response to SM in cell lines with low endogenous caspase-8 activity.
Discussion
Treatment modalities for head and neck cancers remain to be surgery and radiotherapy alone or in combination with chemotherapy. These treatment modalities are ineffective in most patients and are associated with severe side effects. Currently, no effective targeted drugs are available for this type of cancer. Targeted agents, however, such as TRAIL and SMs have been reported to kill tumour cells while sparing non-immortalised cells (Walczak et al, 1999; Muller-Sienerth et al, 2011). Additionally, TRAIL and SMs have shown promising anticancer activity in phase I and phase II clinical trials for certain types of advanced cancers (Lu et al, 2008; Cai et al, 2011; Mahalingam et al, 2011; Gasparini et al, 2013). We therefore investigated their potential as anticancer drugs for head and neck cancers by testing their effect in a panel of HNSCC cell lines. We then sought to identify biomarkers that could predict the response to TRAIL and SM treatment.
Smac mimetics have been reported to sensitise HNSCC cells to radiotherapy, chemotherapy or TRAIL treatment (Ren et al, 2007; Sun et al, 2011; Xu et al, 2011; Yang et al, 2011), but could not induce cell death as a single agent in HNSCC cell lines such as 1483 and JHU-012 (Sun et al, 2011). Here we show that izTRAIL and Smac-164 (SM) effectively kill specific HNSCC cell lines as single agents when used at very low concentrations. Uniquely, the HNSCC cell lines in the panel were either sensitive to TRAIL or SM with no cell line being clearly sensitive to both agents. This selective response led to the investigation of the molecular pathways involved in response to each drug as such knowledge could help in the future stratification of patients for suitable treatment option.
Protein expression analysis identified caspase-8 as an important biomarker for TRAIL sensitivity as well as an indicator of the type of apoptotic pathway involved. TRAIL-resistant HNSCC cells are characterised by almost undetectable levels of caspase-8, whereas TRAIL-sensitive cells express either high or moderate levels and use the type I and type II extrinsic apoptotic pathway, respectively. Caspase-8 protein levels have been associated with survival and prognosis of different solid and haematological malignancies (Pingoud-Meier et al, 2003). Upregulation of caspase-8 has been shown to sensitise acute lymphoblastic leukaemia cells to chemotherapeutic drugs, whereas low caspase-8 levels were associated with poor prognosis.
Recently, procaspase-8 mutations have been identified in HNSCC primary tumour samples as well as in HNSCC cell lines (Agrawal et al, 2011; Stransky et al, 2011; Pickering et al, 2013). Importantly, heterozygous procaspase-8 mutations have been described for HN30, HN5 (Pickering et al, 2013) and HSC3 cells, which have been used in this study (Catalogue of Somatic Mutations in Cancer, COSMIC, Cell Lines Project). As these cell lines differ in their sensitivity to TRAIL treatment, procaspase-8 mutagenesis analyses do not seem to be a useful tool to predict TRAIL sensitivity. Instead, measuring caspase-8 protein levels might be a more useful biomarker of predicting treatment response. However, in head and neck cancers further investigations are needed to determine the differences in caspase-8 protein levels and its association with clinical outcome.
Analysis of the mechanism of SM-induced cell death in HNSCC cells demonstrated that RIPK1-dependent necroptosis may have a role in SM sensitivity in certain HNSCC cell lines as treatment with nec-1 increased cell survival by a small but significant amount (Figure 1C). Cell death induced by SMs has been shown to be dependent on caspase-8 but not caspase-9 activity (Petersen et al, 2007). In this HNSCC cell line panel, SM sensitivity was demonstrated to be caspase-8 independent as its knockdown had minor effect on response to SMs. Interestingly, caspase-10 was shown to become activated as detected by its cleavage in response to SM, suggesting that in the absence or low caspase-8 levels, caspase-10 may substitute and initiate the apoptotic signalling by SM. Caspase-10 was originally discovered as a protein recruited to both the CD95 and p55 tumour necrosis factor receptor signalling complexes, positively regulating CD95 and p55 signal transduction (Vincenz and Dixit, 1997). However, in recent years the functional role of caspase-10, especially in TRAIL signalling, has been contested. Sprick et al (2002) reported the recruitment of caspase-10 to the native TRAIL and CD95 DISC alongside its cleavage and activation; however, they found that caspase-10 was unable to functionally substitute caspase-8. Furthermore, caspase-8-deficient Jurkat cells that express wild-type caspase-10 have shown to be resistant to TRAIL (Seol et al, 2001). In contrast to this, caspase-10 is mutated in hereditary autoimmune diseases such as autoimmune lymphoproliferative syndrome type II with pleiotropic apoptosis defects (Wang et al, 1999). Caspase-8 and -10 share the mutual substrate Bid (Milhas et al, 2005; Fischer et al, 2006; Wachmann et al, 2010) and death receptor recruitment of caspase-10, as well as apoptosis induction in the absence of caspase-8 has been shown (Kischkel et al, 2001). Therefore, numerous evidence supports a role for caspase-10 in acting as an initiator caspase independent of caspase-8 activity; however, this may depend on the nature of cell stress and cell type.
Here we show that SM treatment resulted in cytochrome c release as well as cleavage and activation of caspase-9, which suggests a role of the intrinsic mitochondrial pathway in SM-induced cell death. However, Bid knockdown and Bcl-2 overexpression (data not shown) did not substantively block cell death induced by SM. As described above, capase-9 activity has shown not to be required for SM-induced cell death, thus the importance of the intrinsic pathway in SM-induced cell death in HNSCC remains to be elucidated.
Overexpression of XIAP has been associated with TRAIL resistance in different tumour types (Gillissen et al, 2013). Additionally, combined XIAP and cIAP-1 knockdown was shown to increase the sensitivity of prostate cancer cells to TRAIL treatment (Dai et al, 2009). In TRAIL-resistant HNSCC cells depletion of XIAP did not affect the response to TRAIL, suggesting that XIAP is not important in conferring resistance to TRAIL. As TRAIL treatment induced both cIAP-1 and XIAP downregulation in TRAIL-sensitive HNSCC cells, it is conceivable that inhibition of both cIAP-1 and XIAP is required to sensitise resistant HNSCC cells to TRAIL, and this needs further investigation.
TNF-α has been proposed as the main culprit in cancer cell apoptosis induced by SMs (Petersen et al, 2007; Varfolomeev et al, 2007; Vince et al, 2007). Notably, the basal levels of TNF-α secretion were much higher in SM-sensitive HNSCC cells than in -resistant cell lines. Smac mimetics treatment further increased the levels of secreted TNF-α in responsive HNSCC cell lines but not in the resistant ones. TNF-α serum levels might thus be a predictive marker for SM sensitivity in head and neck cancer. TNF-α serum levels are widely used indicators for the efficiency of different treatment strategies and outcome of disease in diverse cancer types (Kurokawa et al, 1998; D'Silva and Ward, 2007; Su et al, 2011; Wang et al, 2013; Tripsianis et al, 2014), their analysis is therefore already established and could provide a noninvasive future strategy to predict SM or TRAIL treatment response in head and neck cancer.
Upregulation of TNF-α in response to SM treatment has previously been reported to induce TNF-α-dependent cell death in responsive cell lines. However, TNF-α-independent cell death is also possible in this setting (Allensworth et al, 2013). In this study, the addition of TNF-α alone to SM-sensitive HNSCC cell lines resulted in apoptosis but at a significantly lower level as compared with SM alone treatment, indicating that TNF-α upregulation by itself is insufficient to initiate SM-dependent response in HNSCC cells; cIAP protein downregulation (e.g. by SM) is additionally required supporting the notion of a ‘dual-hit’ mechanism. Importantly, SM-resistant cells already show a decrease in cIAP-1 protein expression in the presence of SM (Figure 2G) and can then be sensitised to SM-induced cell death by the addition of TNF-α.
Smac mimetics target cIAPs in particular cIAP-1 to stimulate their autoubiquitination and degradation, which results in the activation of NF-κB and TNF-α secretion and consequently activation of caspase-8 and cell death. Here we demonstrated that SM-sensitive cell lines have an active noncanonical NF-κB signalling pathway in the absence of cellular stress, this was further induced by treatment with SM corresponding to increased TNF-α secretion.
In conclusion, this study shows that TRAIL and Smac-164 are highly effective in killing all HNSCC cell lines tested and have important potential as future targeted treatments of head and neck cancers. Caspase-8 expression was identified as a biomarker for predicting TRAIL sensitivity, whereas high TNF-α level corresponded with SM sensitivity with TNF-α being able to sensitise SM-resistant cells to SMs. Interestingly, caspase-10 was found to contribute to SM-induced cell death in the absence of caspase-8. Further investigations including other HNSCC cell lines as well as in vivo studies will determine the future application of TRAIL and SMs as novel therapeutics for HNSCC patients.
Change history
11 November 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, Muzny DM, Wu Y, Wood LD, Hruban RH, Westra WH, Koch WM, Califano JA, Gibbs RA, Sidransky D, Vogelstein B, Velculescu VE, Papadopoulos N, Wheeler DA, Kinzler KW, Myers JN (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333 (6046): 1154–1157.
Allensworth JL, Aird KM, Aldrich AJ, Batinic-Haberle I, Devi GR (2012) XIAP inhibition and generation of reactive oxygen species enhances TRAIL sensitivity in inflammatory breast cancer cells. Mol Cancer Therap 11 (7): 1518–1527.
Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR (2013) Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat 137 (2): 359–371.
Bockbrader KM, Tan M, Sun Y (2005) A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene 24 (49): 7381–7388.
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254.
Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, Yi H, Zhang T, Sun D, Kang S, Guo M, Leopold L, Yang D, Wang S (2011) A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem 54 (8): 2714–2726.
Chen DJ, Huerta S (2009) Smac mimetics as new cancer therapeutics. Anti-Cancer Drugs 20 (8): 646–658.
Creagh EM, Murphy BM, Duriez PJ, Duckett CS, Martin SJ (2004) Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J Biol Chem 279 (26): 26906–26914.
D'Silva NJ, Ward BB (2007) Tissue biomarkers for diagnosis & management of oral squamous cell carcinoma. AlphaOmegan 100 (4): 182–189.
Dai Y, Liu M, Tang W, Li Y, Lian J, Lawrence TS, Xu L (2009) A Smac-mimetic sensitizes prostate cancer cells to TRAIL-induced apoptosis via modulating both IAPs and NF-kappaB. BMC Cancer 9: 392.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388 (6639): 300–304.
Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102 (1): 33–42.
Falschlehner C, Ganten TM, Koschny R, Schaefer U, Walczak H (2009) TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv Exp Med Biol 647: 195–206.
Fischer U, Stroh C, Schulze-Osthoff K (2006) Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 25 (1): 152–159.
Frezza C, Cipolat S, Scorrano L (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protocols 2 (2): 287–295.
Gasparini C, Vecchi Brumatti L, Monasta L, Zauli G (2013) TRAIL-based therapeutic approaches for the treatment of pediatric malignancies. Curr Med Chem 20 (17): 2254–2271.
Gillissen B, Richter A, Richter A, Overkamp T, Essmann F, Hemmati PG, Preissner R, Belka C, Daniel PT (2013) Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent 'type I' mode. Cell Death Dis 4: e643.
Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, Ashkenazi A, Jubb AM, Mendelson DS (2010) Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol 28 (17): 2839–2846.
Jordan M, Wurm F (2004) Transfection of adherent and suspended cells by calcium phosphate. Methods 33 (2): 136–143.
Juo P, Woo MS, Kuo CJ, Signorelli P, Biemann HP, Hannun YA, Blenis J (1999) FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ 10 (12): 797–804.
Kantari C, Walczak H (2011) Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta 1813 (4): 558–563.
Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, Gazdar A, Blenis J, Arnott D, Ashkenazi A (2001) Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem 276 (49): 46639–46646.
Kruyt FA (2008) TRAIL and cancer therapy. Cancer Lett 263 (1): 14–25.
Kurokawa H, Yamashita M, Yamashita Y, Murata T, Miura K, Kajiyama M (1998) Estimation of tumor necrosis factor-alpha in the diagnosis, the prognosis and the treatment follow-up of oral squamous cell carcinoma. Fukuoka Igaku Zasshi 89 (11): 312–320.
Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, Meagher JL, Stuckey JA, Wang S (2008) SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res 68 (22): 9384–9393.
Mahalingam D, Oldenhuis CN, Szegezdi E, Giles FJ, de Vries EG, de Jong S, Nawrocki ST (2011) Targeting TRAIL towards the clinic. Curr Drug Targets 12 (14): 2079–2090.
Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG (2008) Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 105 (33): 11778–11783.
McComb S, Cheung HH, Korneluk RG, Wang S, Krishnan L, Sad S (2012) cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ 19 (11): 1791–1801.
Milhas D, Cuvillier O, Therville N, Clave P, Thomsen M, Levade T, Benoist H, Segui B (2005) Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem 280 (20): 19836–19842.
Muller-Sienerth N, Dietz L, Holtz P, Kapp M, Grigoleit GU, Schmuck C, Wajant H, Siegmund D (2011) SMAC mimetic BV6 induces cell death in monocytes and maturation of monocyte-derived dendritic cells. PLoS One 6 (6): e21556.
Ndozangue-Touriguine O, Sebbagh M, Merino D, Micheau O, Bertoglio J, Breard J (2008) A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 27 (46): 6012–6022.
Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X (2007) Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12 (5): 445–456.
Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, Chung W, Issa JP, Zweidler-McKay PA, Wu X, El-Naggar AK, Weinstein JN, Wang J, Muzny DM, Gibbs RA, Wheeler DA, Myers JN, Frederick MJ (2013) Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov 3 (7): 770–781.
Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, Shalaby T, Grotzer MA (2003) Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin Cancer Res 9 (17): 6401–6409.
Ren X, Xu Z, Myers JN, Wu X (2007) Bypass NFkappaB-mediated survival pathways by TRAIL and Smac. Cancer Biol Ther 6 (7): 1031–1035.
Seol DW, Li J, Seol MH, Park SY, Talanian RV, Billiar TR (2001) Signaling events triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL): caspase-8 is required for TRAIL-induced apoptosis. Cancer Res 61 (3): 1138–1143.
Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H (2002) Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J 21 (17): 4520–4530.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio RC, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, Grandis JR (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333 (6046): 1157–1160.
Su C, Zhou C, Zhou S, Xu J (2011) Serum cytokine levels in patients with advanced non-small cell lung cancer: correlation with treatment response and survival. Med Oncol 28 (4): 1453–1457.
Suh Y, Amelio I, Guerrero Urbano T, Tavassoli M (2014) Clinical update on cancer: molecular oncology of head and neck cancer. Cell Death Dis 5: e1018.
Sun Q, Zheng X, Zhang L, Yu J (2011) Smac modulates chemosensitivity in head and neck cancer cells through the mitochondrial apoptotic pathway. Clin Cancer Res 17 (8): 2361–2372.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43 (3): 432–448.
Tripsianis G, Papadopoulou E, Anagnostopoulos K, Botaitis S, Katotomichelakis M, Romanidis K, Kontomanolis E, Tentes I, Kortsaris A (2014) Coexpression of IL-6 and TNF-alpha: prognostic significance on breast cancer outcome. Neoplasma 61 (2): 205–212.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131 (4): 669–681.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J (2007) IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131 (4): 682–693.
Vincenz C, Dixit VM (1997) Fas-associated death domain protein interleukin-1beta-converting enzyme 2 (FLICE2), an ICE/Ced-3 homologue, is proximally involved in CD95- and p55-mediated death signaling. J Biol Chem 272 (10): 6578–6583.
Wachmann K, Pop C, van Raam BJ, Drag M, Mace PD, Snipas SJ, Zmasek C, Schwarzenbacher R, Salvesen GS, Riedl SJ (2010) Activation and specificity of human caspase-10. Biochemistry 49 (38): 8307–8315.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5 (2): 157–163.
Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ (1999) Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98 (1): 47–58.
Wang S, El-Deiry WS (2003) TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 22 (53): 8628–8633.
Wang YS, Miao LY, Liu L, Cai HR, Ding JJ, Ren SX, Zhou CC, Schmid-Bindert G (2013) Serum cytokine levels in patients with advanced non-small cell lung cancer: correlation with clinical outcome of erlotinib treatment. Chin Med J 126 (20): 3931–3935.
Xu Y, Zhou L, Huang J, Liu F, Yu J, Zhan Q, Zhang L, Zhao X (2011) Role of Smac in determining the chemotherapeutic response of esophageal squamous cell carcinoma. Clin Cancer Res 17 (16): 5412–5422.
Yang J, McEachern D, Li W, Davis MA, Li H, Morgan MA, Bai L, Sebolt JT, Sun H, Lawrence TS, Wang S, Sun Y (2011) Radiosensitization of head and neck squamous cell carcinoma by a SMAC-mimetic compound, SM-164, requires activation of caspases. Mol Cancer Ther 10 (4): 658–669.
Yang Y, Yu X (2003) Regulation of apoptosis: the ubiquitous way. FASEB J 17 (8): 790–799.
Acknowledgements
We thank Professors Pascal Meier, the Breakthrough Toby Robins Breast Cancer Research Centre, Institute of Cancer Research, London, UK, for TRIPZ-inducible lentiviral sh-caspase-8 and Simone Fulda, JW Goethe University Hospital Frankfurt, Germany for pGIPZ-shBid. We also thank Sebastian Kupka, UCL Cancer Institute, London, UK for helpful discussions. Nina Raulf was supported by a research grant from the Rosetrees Trust and Radien El-Attar was supported by a scholarship from the Egyptian Ministry of Education.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Raulf, N., El-Attar, R., Kulms, D. et al. Differential response of head and neck cancer cell lines to TRAIL or Smac mimetics is associated with the cellular levels and activity of caspase-8 and caspase-10. Br J Cancer 111, 1955–1964 (2014). https://doi.org/10.1038/bjc.2014.521
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.521
Keywords
This article is cited by
-
Systemic network analysis identifies XIAP and IκBα as potential drug targets in TRAIL resistant BRAF mutated melanoma
npj Systems Biology and Applications (2018)
-
Cytotoxic effects of SMAC-mimetic compound LCL161 in head and neck cancer cell lines
Clinical Oral Investigations (2016)
-
Caspase-8 activation by TRAIL monotherapy predicts responses to IAPi and TRAIL combination treatment in breast cancer cell lines
Cell Death & Disease (2015)
