
Abstract
Objective:
This study aims to investigate the using of bone marrow mesenchymal stem cells (BMSCs) genetically engineered to produce interferon-β (IFN-β) as a gene delivery system to treat hepatocellular carcinoma (HCC) in vitro and in vivo.
Methods:
To measure the effects on tumour cell growth in vitro, IFN-β-producing BMSCs (BMSC/IFN-β) were co-cultured with the HCC cell line HepG2 and Huh7. Enzyme-linked immunosorbent assay (ELISA) was used to detect the IFN-β secretion in the BMSC culture condition medium (CM). The effect of BMSC/IFN-β on HCC cells proliferation was examined both in vitro and in vivo by using MTT, colony formation assay, BrdU staining, cell cycle analysis, and xenografted NOD/SCID mouse tumour model. To examine the impact of BMSC/IFN-β on the AKT/FOXO3a signalling, RT–PCR and western blotting were performed.
Results:
The BMSC/IFN-β cells can stably secrete high levels of IFN-β. Both MTT and colony forming assay showed that HCC cells had a lower growth rate when cultured in BMSC/IFN-β-CM as compared with that in BMSC/vector-CM or DMEM culture group. Co-culture with BMSC/IFN-β-CM dramatically decreased the percentages of cells with incorporated BrdUrd. In BMSC/IFN-β-CM-treated HCC cells, the proportion of G1-phase cells increased but it decreased in the S phase of the cell. The BMSC/IFN-β inhibited HCC growth in NOD/SCID mice and proved the survival period of these mice. Compared with the control group, p21 and p27 expression of hepatoma cells increased, whereas cyclin D1 and phosphorylation of Rb expression decreased when co-cultured with BMSC/IFN-β-CM. It was associated with suppression of Akt activity and enhanced transcriptional activity of FOXO3a.
Conclusion:
The IFN-β gene-modified BMSCs can effectively inhibit the proliferation of HCC cells in vitro and in vivo through inhibiting AKT/FOXO3a pathway. These results indicate that BMSC/IFN-β are a powerful anticancer cytotherapeutic tool for HCC.
Similar content being viewed by others
Main
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third major cause of cancer-related death in the world (Parkin et al, 2005). Despite the improvement made in the treatment strategies for HCC during the past decades, the disease still has high mortality rate, mainly because of late diagnosis and lack of effective therapies for advanced HCC (Aravalli et al, 2008).
Interferon-β (IFN-β) is proved to have a significant growth inhibition effect on tumour cells in vitro (Wong et al, 1989). However, the short half-life and higher effective dose with multiple side effects of IFN-β hinder its use in in vivo studies. Previous studies showed that IFN-β gene therapy using adenoviral vectors is effective in several cancers including ovarian cancer (Xu et al, 1998), bladder cancer (Izawa et al, 2002), glioma (Natsume et al, 1999; Nakahara et al, 2003), and lung cancer (Wilderman et al, 2005). However, viral vector-based gene delivery administration has been limited by vector safety, organ toxicity, and difficult to selective delivery of the therapeutic gene to metastatic sites. To overcome this problem, human bone marrow-derived mesenchymal stem cells (BMSCs) have been utilised as biological vehicles for gene delivery.
The BMSCs have the advantage of being easily harvested and proliferated and they have lower immunogenicity. Our previously study has demonstrated that BMSCs have nontumourigenicity in cancer environment that proved that BMSC transplantation is safe for tumour therapy (Chan et al, 2010). Moreover, BMSC-based IFN-β therapy via systemic administration successfully migrated to tumour sites and attenuated growth of melanoma (Studeny et al, 2004) and glioma (Studeny et al, 2002; Nakamizo et al, 2005). These observations demonstrate that BMSCs have a high potential as biological vehicles for tumour tissue-targeted delivery of therapeutic agents or genes. However, this novel therapy has never been applied to HCC. Here we report that administered BMSC/IFN-β are capable of decreasing tumour formation of HCC cells through producing IFN-β and inhibiting cell cycle via AKT/FOXO3a pathways.
Materials and methods
Cell lines and bone marrow separation for primary human BMSC cultures
The HCC cell lines HepG2 and Huh7 were obtained from the Sun Yat-sen University Cancer Center (Guangzhou, China) and grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Invitrogen), 2 mM L-glutamine, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The BMSC samples were collected from six healthy bone marrow donors at the Department of Infectious Disease, the Third Affiliated Hospital of Sun Yat-sen University (Guangzhou, China) with prior written informed consents from the donors and approval from the Institutional Research Ethics Committees of Sun Yat-sen University and its Third Affiliated Hospital. Three primary BMSC cell lines were chosen randomly for the extensive experiments. Each experiment was performed in triplicate. The BMSC isolation and culture have been described in our previous study (Peng et al, 2011). Briefly, 100–120 ml of human bone marrow was obtained and anticoagulated with 1000 U ml−1 Liquaemin. Density gradient centrifugation was conducted in a laminar air-flow hood; bone marrow was diluted with normal saline and gently added to Percoll separating medium (Sigma-Aldrich, St Louis, MO, USA) of equal volume, followed by centrifugation at 2500 r.p.m. per min for 30 min. The interphase containing cells were obtained and washed three times with 10 ml normal saline. The cell suspension was collected and preserved in 10 ml normal saline, with 0.2 ml used to seed L-DMEM (Gibco BRL, Grand Island, NY, USA) culture medium supplemented with 10% FBS (Gibco BRL), incubated at 37 °C in a 25 cm2 culture flask. The culture medium was changed after 3 days and every 2 days thereafter. The BMSCs were digested with 0.25% Trypsin and 0.1% EDTA and passaged (1 : 2) when 70–80% cell fusion had occurred. Before experimental use, the third- to fifth-generation BMSCs were determined to be CD44 and CD90 positive and lack expression of CD45 and CD34 by flow cytometry analysis.
Plasmids and infection of BMSCs
The IFN-β expression construct was generated by subcloning PCR-amplified full-length human IFN-β cDNA into the pMSCV-retro-puro vector using forward primer 5′-GATCCGATTGAATTCATGACCAACAAG-3′ and reverse primer 5′-GCTAGGCTCGAGTCAGTTTCGGAGGTA-3′. Retroviral production and infection were performed as standard protocol. Briefly, the third-passage BMSCs were plated at a density of 1 × 105 cells per well in six-well culture plates. After 24 h, the retroviral pMSCV-IFN-β was added to infect the cells and incubated for 4 h. The cultures were then washed with 10 mM phosphate-buffered saline (PBS), pH 7.4, and the culture medium replaced. Parallel control cultures of BMSCs (pMSCV-BMSCs) were transfected with blank vector. Effective transfection was confirmed by IFN-β enzyme-linked immunosorbent assay (ELISA) of the culture supernatant (Invitrogen/PBL, Carlsbad, CA, USA) at 4, 24, 48, 72, and 96 h after transfection. The reporter plasmid for quantitatively detecting the transcriptional activity of FOXO was generated in the pGL3-Enhancer plasmid (Promega, Madison, WI, USA) as described previously (Tang et al, 1999).
Collection of BMSCs and BMSC/IFN-β conditioned media (CM)
The normal growth media were conditioned by culturing BMSC/IFN-β or BMSC/vector for 24 h. The CM were applied to the liver cancer cell lines that were seeded 24 h before.
The MTT (3-(4, 5-Dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide) assay
Cells were seeded in 96-well plates at a density of 1 × 103 cells per well. The CM were applied to the liver cancer cell lines that were seeded 24 h before. At each time point, cells were stained with 100 μl sterile MTT dye (0.5 mg ml−1, Sigma, St Louis, MO, USA) for 4 h at 37 °C, followed by removal of the culture medium and addition of 150 μl of dimethyl sulphoxide (Sigma). The absorbance was measured at 570 nm, with 655 nm as the reference wavelength. All experiments were performed in triplicate.
Colony formation assay
Cells were plated at density of 1 × 103 cells per dish and cultured for 7 days. The CM were applied to the liver cancer cell lines and changed every day. The colonies were stained with 1% crystal violet for 10 min after fixation with 10% formaldehyde for 5 min. Number of colonies was scored on an Artek Model 880 colony counter (Artek, Farmingdale, NY, USA).
Bromodeoxyuridine labelling and immunofluorescence
Cells were plated on coverslips (Fisher, Pittsburgh, PA, USA). The CM were applied to the HCC cell lines. After 24 h, cells were incubated with bromodeoxyuridine (BrdUrd) for 1 h and stained with anti-BrdUrd antibody (Upstate, Temecula, CA, USA) according to the manufacturer’s instruction. Grey-level images were acquired under a laser scanning microscope (Axioskop 2 plus, Carl Zeiss Co. Ltd, Jena, Germany).
Flow cytometry for cell cycle analysis
Cells were harvested, washed with cold PBS, and processed for cell cycle analysis using flow cytometry. Briefly, the cells were fixed in 75% ethanol and stored at −20 °C for later analysis. The fixed cells were centrifuged at 800 r.p.m. and washed with cold PBS twice. RNase A (20 μg ml−1 final concentration) and propidium iodide staining solution (50 μg ml−1 final concentration) were added to the cells and incubated for 30 min at 37 °C in the dark. One hundred thousand cells were analysed using a FACSCalibur instrument (BD Biosciences, San Jose, CA, USA) equipped with CellQuest 3.3 software. ModFit LT 3.1 trial cell cycle analysis software was used to determine the percentage of cells in the different phases of the cell cycle.
Xenografted tumour model
A total of 21 male NOD/SCID mice (4 weeks of age, 12–14 g) were purchased from the Institute of Materia Medica (Chinese Academy of Sciences, Beijing, China). All experimental procedures involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publications Nos. 80–23, revised 1996) and according to the institutional ethical guidelines for animal experiments. The protocol was approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University. The Huh7 cells were subcutaneously inoculated on the left flank of each mouse. Before inoculation, all cells were suspended in PBS at a concentration of 1 × 107/100 μl PBS. Then, the mice were divided into three groups. For the BMSC/IFN-β cell treatment group (n=7), BMSC/vector cell treatment group (n=7), and PBS treatment group (n=7), 3 × 105 BMSC cells or 100 μl PBS were injected intro tail vein in each group of mouse 3 days after HCC inoculation. After inoculation, tumour growth was examined every 3 days by measuring the length and width with a caliper, and tumour volumes were calculated as length × width2 × 0.52. On day 36, animals were killed, and tumours were excised and weighed.
RNA extraction, reverse transcription (RT), and real-time PCR
Total RNA from cultured cells was extracted using the Trizol reagent (Invitrogen) following the manufacturer’s instructions. The cDNAs were amplified and quantified in ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using dye SYBR Green I (Molecular Probes, Invitrogen). The primers designed using the Primer Express v 2.0 software (Applied Biosystems) and the primer sequences are provided as following: IFN-β forward, 5′-GAAGGAGGACGCCGCATTG-3′ and reverse, 5′-CTTGGAGGACCGATTACAGATAGTAGT-3′; p21Cip1 forward, 5′-CGATGCCAACCTCCTCAACGA-3′ and reverse, 5′-TCGCAGACCTCCAGCATCCA-3′; p27 Kip1 forward, 5′-TGCAACCGACGATTCTTCTACTCAA-3′ and reverse, 5′-CAAGCAGTGATGTATCTGATAAACAAGGA-3′; and cyclin D1 forward, 5′-AACTACCTGGACCGCTTCCT-3′ and reverse, 5′-CCACTTGAGCTTGTTCACCA-3′. Expression data were normalised to the geometric mean of housekeeping gene GAPDH (forward, 5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCACCA C CCTG TTGCTGTA-3′) to control the variability in expression levels and calculated as 2-((Ct of IFN-β or p21 or p27)—(Ct of GAPDH)), where Ct represents the threshold cycle for each transcript.
Western blotting
Western blotting was performed according to standard methods, using anti-Akt, anti-phosphorylated-Akt, anti-GSK-3β, anti-phosphorylated-GSK-3β, anti-phosphorylated-Rb, anti-Rb, anti-total FOXO3a, and anti-phosphorylated-FOXO3a (Ser253) antibodies (all from Cell Signaling, Danvers, MA, USA), and anti-p27 Kip1 and anti-p21Cip1 antibodies (BD, Franklin Lakes, NJ, USA). Blotted membranes were stripped and re-probed with an anti-GAPDH antibody (Sigma) as a loading control.
Luciferase assay
For the luciferase reporter assay, cells were co-transfected with FOXO luciferase reporter. Transfection efficiency was normalised by co-transfection with a pB-actin-RL reporter containing a Renilla luciferase gene under the control of a human β-actin promoter. The activities of firefly luciferase and Renilla luciferase were quantified using the dual-luciferase reporter assay system (Promega).
Immunohistochemistry (IHC)
The IHC procedure to detect Ki67 was performed and analysed with the AxioVision Rel.4.6 computerised image analysis system assisted with automatic measurement program (Carl Zeiss, Oberkochen, Germany). Briefly, the stained sections were evaluated at × 200 magnification, and 10 representative staining fields of each section were analysed to produce mean optical density value (MOD), which represents the strength of staining signals as measured per positive pixel. The mean absorbance data were statistically analysed using t-test to compare the average mean absorbance difference between different groups of tissues, and P<0.05 was considered significant.
Enzyme-linked immunosorbent assay
Culture supernatant was collected at different time points and the amount of IFN-β protein was quantitated using ELISA for IFN-β (Invitrogen/PBL) in accordance with the manufacturer’s protocol and analysed at an absorbance of 450 nm. The minimum sensitivity of detection was 15.6 pg ml−1.
Statistical analysis
All statistical analyses were carried out using the SPSS version 13.0 statistical software packages (Armonk, NY, USA). Comparisons between groups for statistical significance were performed with a two-tailed paired Student’s t-test. A P-value of <0.05 was considered statistically significant in all cases.
Results
High-level expression and bioactivity of IFN-β produced by BMSCs
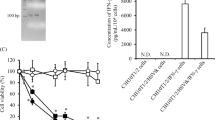
The BMSCs obtained by bone marrow aspiration and expanded in vitro appeared similar to fibroblasts, with a characteristic spindle-shaped fusiform morphology (Figure 1A). In cultured cells of the third generation analysed by flow cytometry, >97% of the cells expressed CD44 and CD90, a surface marker characteristic of BMSCs. The absence of contaminating haematopoietic cells in the BMSC population was verified by the lack of surface antigens CD34 or CD45 defining haematopoietic progenitor cells. Thus, the bone marrow cells after the third passage were of high purity, and expressed CD44+, CD90+, CD34−, and CD45−, which are markers for BMSCs. There was no change for IFN-β-transduced BMSCs from the cell shape.

Production of interferon-β (IFN-β) protein by human bone mesenchymal stem cells (BMSCs) after infection with adenoviral vectors. (A) The BMSCs were separated from bone marrow and purified by removing the nonadherent cells at least three times. The third-generation BMSCs were fusiform and fibroblast-like (left, × 100). The BMSCs infected with IFN-β (right, × 100). (B) Relative mRNA expression of IFN-β in BMSC/ IFN-β cell. All data are shown as mean±s.d. from three independent experiments. *P<0.05. (C) Relative protein expression of IFN-β in BMSC/ IFN-β cell culture medium. All data are shown as mean±s.d. from three independent experiments *P<0.05.
To determine the transduction efficiency of IFN-β in BMSCs, cells and condition medium were collected and detected by RT–PCR and ELISA for IFN-β at 1 week after infection. Reverse transcription–PCR showed that there was 2.35±2.3 times promotion of IFN-β expression in BMSC/IFN-β group as compared with BMSC/vector cells (t=−10.04, P=0.001, Figure 1B). Interferon-β concentration was 163±11.5 pg ml−1 for BMSC/IFN-β as compared with 96±8.9 pg ml−1 for BMSC/vector cells detected by ELISA (t=−7.97, P<0. 01, Figure 1C). The results indicated the stable expression of IFN-β in BMSCs.
BMSC/IFN-β significantly attenuated HCC cell growth and migration in vitro
To evaluate the bioactivity of BMSC/IFN-β on cancer cell proliferation, MTT assay was performed in HepG2 and Huh7 HCC cells. A drastic reduction of cell proliferation was observed in HepG2 and Huh7 cells in BMSC/IFN-β-CM, showing more than a two-fold decrease in cell number compared with the vector-control cells after a 5-day culture (Figure 2A). Such a proliferation inhibitory activity of BMSC/IFN-β was further demonstrated with the colony formation assay (Figure 2B). The number of colony formation detected in BMSC/IFN-β-CM-cultured cells was 0.18±0.05 for HepG2 cells (t=9.53P=0.001) and 0.20±0.05 for Huh7 cells (t=30.86 P<0.01) as compared with the DMEM-cultured groups. However, less colons were seen in the BMSC/vector control group (0.95±0.03 for HepG2 cells and 0.97±0.03 for Huh7 cells; Figure 2B).

Effect of BMSC/IFN-β on HCC proliferation. (A) Co-culture with BMSC/IFN-β significantly reduced HepG2 and Huh7 cell growth as determined by MTT assay (*P<0.05, compared with all other groups; analysis of variance and Student’s t-test; mean±s.d. of triplicate experiments). (B) Representative micrographs (left panel) and quantification (right panel) of crystal violet-stained cells. (C) Representative micrographs (left panel; magnification × 100) and quantification of BrdU incorporating cells after co-culture with BMSC/IFN-β or control vector. Each bar represents the mean of three independent experiments *P<0.05.
To investigate the mechanism that mediates the proliferation inhibition function of BMSC/IFN-β, a BrdUrd incorporation assay was performed. As shown in Figure 2C, HepG2 and Huh7cells cultured with BMSC/IFN-β-CM showed a lower BrdUrd incorporation (14.0±3.6% for HepG2 cells and 15.0±1.0% for Huh7 cells; Figure 2C) as compared with the BMSC/vector-CM-cultured cell group (49.3±1.5% for HepG2 cells and 54.3±2.5% for Huh7). There was no significant difference in BrdUrd incorporation rate between BMSC/vector-CM-cultured cells and DMEM-cultured cells.
Flow cytometry analysis showed that BMSC/IFN-β significantly increased the percentage of cells in the G0/G1 peak but decreased that in the S peak (Figure 3A). As shown in Figure 3A and B, BMSC/IFN-β-CM-treated cells displayed a significant increase in the percentages of cells in the G1 phase but decreased proportions in the S phase compared with BMSC/vector-CM- and DMEM-treated cells, indicating that BMSC/IFN-β might induce G1- to S-phase arrest of liver cancer cells.

The BMSC/IFN-β inhibits proliferation through decreasing the proportion of S-phase cells. Flow cytometric analysis of indicated HepG2 (A) and Huh7 (B) cell co-culture with BMSC/IFN-β or control vector, and quantification of G0/G1- and S-stage cells in HepG2 (right panel; A) and Huh7 cells (right panel; B). Each bar represents the mean of three independent experiments *P<0.05.
Systemic administration of BMSC/IFN-β significantly attenuated tumour formation in vivo
Having examined the effect of BMSC/IFN-β on HepG2 and Huh7 cell lines in vitro, the effect of systemic administration of BMSC/IFN-β on the growth of Huh7in HCC xenografts was evaluated using a NOD/SCID mouse model. The HE staining showed that the HCC model was established (Figure 4A). In this study, each experimental mouse bearing HCC treated with BMSC/vector, PBS, or BMSC/IFN-β, respectively, began to exhibit conspicuous difference in tumour growth within the first week, and the difference continued to expand through the experimental end point (Figure 4B). In parallel, this trend was also confirmed by the sizes and weights of dissected tumours (Figure 4C and D), strongly suggesting a markedly decelerated proliferation of the tumour cells by BMSC/IFN-β. In consistence, the proportions of proliferative Ki-67-positive cells in BMSC/IFN-β-treated tumours were substantially lower than those in control tumours (Figure 4E).

The BMSC/IFN-β inhibits HCC cell proliferation in vivo. (A) The HE staining for xenografted tumour nodules excised from experimental mice is shown in the picture. (B) Huh7 cells were injected into the groins of athymic mice. Tumour volumes were measured on the indicated days. Representative graph of tumour growth and mean tumour 36 days after inoculation are shown. All data are shown as mean±s.d. (C) At the experimental end point, mice were anaesthetised and killed, and tumours were dissected and photographed. (D) Each tumour formed by indicated cells was weighed. (E) Immunohistochemical (IHC) staining and quantification of the expression of Ki-67 in HCC lesion. All data are shown as mean±s.d. Each bar represents the mean of three independent experiments *P<0.05.
BMSC/IFN-β upregulated cell cycle inhibitors p21Cip1 and p27Kip1 and downregulated cell cycle regulator cyclin D1
Having determined that BMSC/IFN-β significantly attenuated HCC cell growth, we sought to determine the mechanism. For this purpose the levels of cell cycle regulators in BMSC/IFN-β-treated HCC cells were measured. It has been reported that activation of the Akt and the major effectors downstream of Akt, glycogen synthase kinase (GSK)-3β, is critical for cell proliferation. The effect of AKT is at least in part because of its role as a negative regulator of Transcriptional factor FOXO3a (Kops et al, 2002; Massague et al, 2005). The FOXO3a induces transcription of target genes involved in cell cycle arrest (p27Kip1, p21Cip1) by binding to promoters of these genes (You et al, 2006; Huang and Tindall, 2007; Ho et al, 2008; Chiacchiera et al, 2009; Matrone et al, 2010). In addition, the gene coding for cyclin D is reported to be negatively regulated by FOXOs in a direct manner (Schmidt et al, 2002; You et al, 2006; Huang and Tindall, 2007). To explore the role of FOXO3a in BMSC/IFN-β-treated cells, the transactivating activity of FOXO3a was examined in BMSC/IFN-β-treated cells and found to be drastically increased (Figure 5A). In parallel, there was an increased expression of p27 Kip1 and p21Cip1, accompanied by a decrease of cyclin D1 and decreased phosphorylation of FOXO3a and its upstream kinase Akt in BMSC/IFN-β-treated cells at both protein and mRNA levels (Figure 5B–E), suggesting that the Akt/FOXO3a axis is involved in the proproliferative effect of BMSC/IFN-β. Meanwhile, decreased phosphorylation of Rb, an important transcriptional repressor of G1/S progression, was found to be associated with the changes in the above-mentioned cell cycle regulators (Figure 5C). These findings suggest that the proliferative effect of BMSC/IFN-β on HCC may be through regulation of CDK and CDK regulators.

The BMSC/IFN-β inhibited proliferation through AKT/FOXO3a signal pathway. (A) Relative FOXO3a reporter activity in cells co-cultured with BMSC/IFN-β or control. (B) Real-time PCR analysis of expression of p21Cip1, p27Kip1, and cyclin D1 in indicated cells. Expression levels were normalised with GAPDH. (C) Western blotting analysis of expression of p21Cip1, p27Kip1, cyclin D1, phosphorylated Rb (p-Rb), and total Rb protein in indicated HCC cells. As a loading control, GAPDH was used. (D) Western blotting analysis of phosphorylated Akt (p-Akt), total Akt, phosphorylated GSK-3β (p-GSK-3β), total GSK-3β, phosphorylated FOXO3a (p-FOXO3a-Ser253), and total FOXO3a proteins in indicated HCC cells. As a loading control, GAPDH was used. (E) Western blotting analysis of phosphorylated FOXO3a (p-FOXO3a-Ser253) and total FOXO3a proteins in indicated HCC cells. As a loading control, GAPDH was used.
Discussion
Our data presented in this report suggest a pivotal role of BMSCs; that is, BMSCs engineered to express IFN-β produced sufficient amounts of IFN-β to induce proliferation inhibition of HCC cells in vitro and in vivo. Systemic transplantation of BMSC/IFN-β caused significant reduction of tumour burden in Huh7 HCC-bearing NOD/SCID mice. The effect of BMSC/IFN-β is associated with Akt-mediated phosphorylation of the transcription factor FOXO3a that in turn stimulates the cell cycle. Thus, the BMSC/IFN-β may represent a new therapeutic modality for the treatment of HCC and will have important implications for patients with HCC or other types of cancers.
Human IFN-β can be used to inhibit the proliferation of multiple tumour cells for the potent antiangiogenic and antimetastatic effect (Zhang et al, 2002; Angelucci et al, 2007; Ren et al, 2008). Liver cancer cells and several other types of tumour cells are sensitive to IFN-β (Sasaki et al, 2011). However, it is not effective to use IFN-β in treating cancer because of its short half-life and also because the maximally tolerated dose is not high enough to attain these effects (Salmon et al, 1996; Einhorn and Grander, 1996; Buchwalder et al, 2000). One study (Studeny et al, 2004) has shown that the local production of IFN-β in breast carcinoma tissues is the key for IFN-β-based cancer therapy. We wonder whether increasing local IFN-β concentration can also improve the treatment effect of HCC. We used BMSCs as a delivery system into the tumour microenvironment as BMSCs are capable of long-term transgene expression and have previously been shown to exert antitumour effects when transfected with adenoviruses expressing a variety of transgenes. Many studies have reported successful engraftment of ex vivo transduced BMSCs at the site of gliomas (Nakamura et al, 2004), melanoma (Elzaouk et al, 2006), and lung cancer in animal models (Mohr et al, 2008). Accordingly, the present study aim to test our hypothesis that IFN-β gene delivery by BMSCs should be effective in attenuating liver cancer.
In the first study, the effect of BMSC/IFN-β on the growth of two types of HCC cells was evaluated using a MTT and clone forming assay. The results clearly indicated that BMSC/IFN-β, but not BMSCs, significantly inhibit the growth of both liver cancer cell lines. This result was reconfirmed by the study carried out using CM: the CM collected from the BMSC/IFN-β, but not BMSCs, significantly attenuated growth of the two HCC cell lines. In the second study, the mechanism by which IFN-β caused growth attenuation of the cancer cells was determined by studying cell cycle using flow cytometric determination, BrdU incorporation, and western blot analysis. Both studies clearly indicate that IFN-β-dependent cell growth attenuation is mainly because of the arrest of G1/S check point. Then, the in vivo effect of BMSC/IFN-β was evaluated using Huh7 cell xenografts in NOD/SCID mouse. Multiple systemic administrations of BMSC/IFN-β markedly attenuated tumour formation. These results strongly suggest that BMSC/IFN-β therapy is a viable option for the treatment of HCC. To the best of our knowledge, this is the first demonstration of the efficacy of IFN-β gene-transfected stem cell therapy on HCC. Although a number of hurdles remain, this success opens the possibility of BMSC/IFN-β therapy for HCC.
As IFN-β treatment has been shown to downregulate PI3K/Akt pathway in neuroblastoma cells (Dedoni et al, 2010), it is conceivable that IFN-β-induced growth inhibition in HCC cells is mediated through PI3K/Akt pathway. When BMSC/IFN-β reached the tumour site, they produced the cytokine IFN-β, which ultimately caused inhibition of cell growth in the tumour cells. This mechanism was evidenced by the decrease of the tumour growth in BMSC/IFN-β-treated NOD/SCID mouse. As un-engineered BMSCs consistently exhibit a minor growth attenuation of liver cancer cells (Figure 4B–E), they may have some intrinsic ability to attenuate liver cancer cells. Our data also suggest a possible regulation of FOXO3a, a member of the forkhead box class O transcription factor family, activity by BMSC/IFN-β. In consistence with previously identified significance of the Akt oncogenic kinase in HCC, our data indicate that FOXO3a phosphorylation inhibited by BMSC/IFN-β is associated with reducing Akt activation. It is noteworthy that FOXO3a and Akt-regulated transcription factor have been found to be functionally important in the development of human HCC (Saxena et al, 2007; Kim, 2008; Lu et al, 2009). These new data not only provide a line of evidence for the regulatory effect of BMSC/IFN-β on Akt phosphorylation, but also link the BMSC/IFN-β-triggered Akt signalling cascade inhibition with the lowly proliferative phenotype of HCC cells. Nevertheless, how BMSC/IFN-β inhibits the phosphorylation of Akt kinase remains to be investigated.
In terms of a potential application of BMSC/ IFN-β to human patients, the low immunogenicity of BMSCs has significant merit. However, many issues must be clarified before human trials: whether this therapy is also effective in developed HCC, what is the minimal effective dose, whether frequent applications promote tumour development, and so on. Nevertheless, the preliminary findings of the present study support further investigation of BMSC/IFN-β therapy as a novel strategy for the treatment of liver cancer or other solid malignant tumours.
Change history
03 September 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Angelucci C, Iacopino F, Ferracuti S, Urbano R, Sica G. (2007) Recombinant human IFN-β affects androgen receptor level, neuroendocrine differentiation, cell adhesion, and motility in prostate cancer cells. J Interferon Cytokine Res 27: 643–652.
Aravalli RN, Steer CJ, Cressman EN (2008) Molecular mechanisms of hepatocellular carcinoma. Hepatology 48: 2047–2063.
Buchwalder PA, Buclin T, Trinchard I, Munafo A, Biollaz J (2000) Pharmacokinetics and pharmacodynamics of IFN-beta 1a in healthy volunteers. J Interferon Cytokine Res 20: 857–866.
Chan X, Shi-Bin X, Dong-Ying X, Liang P, Shao-Quan Z, Jun-Qiang X, Zhi-Liang G (2010) Bone marrow mesenchymal stem cell has poor proliferation but non-tumorigenicity in cancer environment. Lab Medicine 41: 551–556.
Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C (2009) p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ 16: 1203–1214.
Dedoni S, Olianas MC, Onali P (2010) Interferon-β induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J Neurochem 115: 1421–1433.
Einhorn S, Grander D (1996) Why do so many cancer patients fail to respond to interferon therapy? J Interferon Cytokine Res 16: 275–281.
Elzaouk L, Moelling K, Pavlovic J (2006) Anti-tumor activity of mesenchymal stem cells producing IL-12 in a mouse melanoma model. Exp Dermatol 15: 865–874.
Ho KK, Myatt SS, Lam EW (2008) Many forks in the path: cycling with FoxO. Oncogene 27: 2300–2311.
Huang H, Tindall DJ (2007) FoxO transcription factors. J Cell Sci. 120: 2479–2487.
Izawa JI, Sweeney P, Perrotte P, Kedar D, Dong Z, Slaton JW, Karashima T, Inoue K, Benedict WF, Dinney CP (2002) Inhibition of tumorigenicity and metastasis of human bladder cancer growing in athymic mice by interferon-beta gene therapy results partially from various antiangiogenic effects including endothelial cell apoptosis. Clin Cancer Res 8: 1258–1270.
Kim BC (2008) FoxO3a mediates transforming growth factor-beta1-induced apoptosis in FaO rat hepatoma cells. BMB Rep 41: 728–732.
Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ, Lam EW, Burgering BM (2002) Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol 22: 2025–2036.
Lu M, Ma J, Xue W, Cheng C, Wang Y, Zhao Y, Ke Q, Liu H, Liu Y, Li P, Cui X, He S, Shen A (2009) The expression and prognosis of FOXO3a and Skp2 in human hepatocellular carcinoma. Pathol Oncol Res 15: 679–687.
Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19: 2783–2810.
Matrone A, Grossi V, Chiacchiera F, Fina E, Cappellari M, Caringella AM, Di Naro E, Loverro G, Simone C (2010) p38α is required for ovarian cancer cell metabolism and survival. Int J Gynecol Cancer 20: 203–211.
Mohr A, Lyons M, Deedigan L, Harte T, Shaw G, Howard L, Barry F, O'Brien T, Zwacka R (2008) Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med 12: 2628–2643.
Nakahara N, Pollack IF, Storkus WJ, Wakabayashi T, Yoshida J, Okada H (2003) Effective induction of antiglioma cytotoxic T cells by coadministration of interferon-beta gene vector and dendritic cells. Cancer Gene Ther 10: 549–558.
Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, Andreeff M, Lang FF (2005) Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 65: 3307–3318.
Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H, Bizen A, Honmou O, Niitsu Y, Hamada H (2004) Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther 11: 1155–1164.
Natsume A, Mizuno M, Ryuke Y, Yoshida J (1999) Antitumor effect and cellular immunity activation by murine interferon-beta gene transfer against intracerebral glioma in mouse. Gene Ther 6: 1626–1633.
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108.
Peng L, Xie DY, Lin BL, Liu J, Zhu HP, Xie C, Zheng YB, Gao ZL (2011) Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: short-term and long-term outcomes. Hepatology 54: 820–828.
Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, Ponnazhagan S (2008) Cancer gene therapy using mesenchymal stem cells expressing interferon-β in a mouse prostate cancer lung metastasis model. Gene Ther 15: 1446–1453.
Salmon P, Le Cotonnec JY, Galazka A, Abdul-Ahad A, Darragh A (1996) Pharmacokinetics and pharmacodynamics of recombinant human interferon-beta in healthy male volunteers. J Interferon Cytokine Res 16: 759–764.
Sasaki S, Ishida T, Toyota M, Ota A, Suzuki H, Takaoka A, Yasui H, Yamamoto H, Takagi H, Maeda M, Seito T, Tsujisaki M, Shinomura Y, Imai K (2011) Interferon-α/β and anti-fibroblast growth factor receptor 1 monoclonal antibody suppress hepatic cancer cells in vitro and in vivo. PLoS One 6: e19618.
Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA (2007) Concomitant activation of the JAK/STAT, PI3K/Akt, and ERK signaling is involved in leptin-ediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res 67: 2497–2507.
Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH (2002) Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol 22: 7842–7852.
Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M (2002) Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res 62: 3603–3608.
Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE, Andreeff M (2004) Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst 96: 1593–1603.
Tang ED, Nuñez G, Barr FG, Guan KL (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274: 16741–16746.
Wilderman MJ, Sun J, Jassar AS, Kapoor V, Khan M, Vachani A, Suzuki E, Kinniry PA, Sterman DH, Kaiser LR, Albelda SM (2005) Intrapulmonary IFN-beta gene therapy using an adenoviral vector is highly effective in a murine orthotopic model of bronchogenic adenocarcinoma of the lung. Cancer Res 65: 8379–8387.
Wong VL, Rieman DJ, Aronson L, Dalton BJ, Greig R, Anzano MA (1989) Growth-inhibitory activity of interferon-beta against human colorectal carcinoma cell lines. Int J Cancer 43: 526–530.
Xu L, Xie K, Fidler IJ (1998) Therapy of human ovarian cancer by transfection with the murine interferon beta gene: role of macrophage-inducible nitric oxide synthase. Hum Gene Ther 9: 2699–2708.
You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. (2006) FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med 203: 1657–1663.
Zhang F, Lu W, Dong Z (2002) Tumor-infiltrating macrophages are involved in suppressing growth and metastasis of human prostate cancer cells by INF-β gene therapy in nude mice. Clin Cancer Res 8: 2942–2951.
Acknowledgements
This work was supported by The Natural Science Foundation of China (no. 81101864)and the National Program on twelfth five-year project (no. 2012ZX10002007-002-008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Xie, C., Xie, DY., Lin, BL. et al. Interferon-β gene-modified human bone marrow mesenchymal stem cells attenuate hepatocellular carcinoma through inhibiting AKT/FOXO3a pathway. Br J Cancer 109, 1198–1205 (2013). https://doi.org/10.1038/bjc.2013.422
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.422
