
Abstract
Background:
PM00104 binds guanines at DNA minor grooves, impacting DNA replication and transcription. A phase I study was undertaken to investigate safety, dose-limiting toxicities (DLTs), recommended phase II dose (RP2D), pharmacokinetics (PKs) and preliminary antitumour activity of PM00104 as a 1- or 3-h infusion three-weekly.
Methods:
Patients with advanced solid tumours received PM00104 in a dose escalation trial, as guided by toxicity and PK data.
Results:
A total of 47 patients were treated; 27 patients on the 1-h schedule (0.23–3.6 mg m−2) and 20 patients on the 3-h schedule (1.8–3.5 mg m−2). Dose-limiting toxicities comprised reversible nausea, vomiting, fatigue, elevated transaminases and thrombocytopenia, establishing the 1-h schedule RP2D at 3.0 mg m−2. With the 3-h schedule, DLTs of reversible hypotension and neutropenia established the RP2D at 2.8 mg m−2. Common PM00104-related adverse events at the RP2D comprised grade 1–2 nausea, fatigue and myelosuppression. In both schedules, PKs increased linearly, but doses over the 1-h schedule RP2D resulted in higher than proportional increases in exposure. A patient with advanced urothelial carcinoma had RECIST shrinkage by 49%, and three patients had RECIST stable disease ⩾6 months.
Conclusion:
PM00104 is well tolerated, with preliminary evidence of antitumour activity observed. The 1-h 3-weekly schedule is being assessed in phase II clinical trials.
Similar content being viewed by others

Main
PM00104 (Zalypsis, PharmaMar, Madrid, Spain) is a novel marine-derived synthetic tetrahydroisoquinoline alkaloid, which is structurally related to jorumycins (isolated from the Pacific nudibranch Jorunna funebris; Rinehart et al, 1990; Fontana et al, 2000) and renieramycins (from marine sponges; Figure 1; Oku et al, 2003). PM00104 showed potent cytotoxic activity against a wide range of human tumour cell lines, with IC50 values in the low nanomolar range (Leal et al, 2009). It has also demonstrated antitumour effects in multiple tumour xenograft mouse models, including prostate, gastric, pancreatic, bladder, breast, gastric and hepatocellular cancers (Elices et al, 2005a; Greiner et al, 2007; LePage et al, 2007a; Guillen et al, 2009), as well as multiple myeloma (Ocio et al, 2009).

Structural formula of PM00104 (Zalypsis; C37H38F3N3O8).
PM00104 exerts its antitumour effects through the covalent modification of guanines in the DNA minor groove, thereby affecting DNA replication and transcription (Guirouilh-Barbat et al, 2009; Leal et al, 2009). DNA adducts are transformed into DNA double-strand breaks at nanomolar concentrations of PM00104 (Herrero et al, 2007), as detected by an increased proportion of cells with abundant γH2AX foci formation in solid tumour and multiple myeloma cell lines (Guirouilh-Barbat et al, 2009; Leal et al, 2009; Ocio et al, 2009). DNA damage induced by PM00104 results in S-phase cell cycle arrest in various solid tumour cell lines (Magro et al, 2007; Leal et al, 2009) and activates DNA replication checkpoints. PM00104 caused tumour cells to accumulate in S-phase and induced apoptotic cell death, which was dependent on caspase activity, resulting in cleavage of poly (ADP-ribose) polymerase-1 and cytokeratin 18 (Leal et al, 2009; Ocio et al, 2009).
In preclinical studies, PM00104 given by intravenous injection produced toxicological effects primarily affecting the bone marrow, reticuloendothelial system and gastrointestinal tract in dogs and rats, as well as the liver and injection site. The gastrointestinal, bone marrow and lymphoid system alterations were completely reversible 2–3 weeks after the last drug administration (Yin et al, 2006).
The elimination kinetics of intravenous PM00104 in animals followed a biexponential profile, with calculated terminal half-lives of ∼7 and 10 h for mice and dogs, respectively, indicating a relatively slow plasma clearance (Yin et al, 2005a). The volume of distribution at steady state (Vss) was greater than plasma volume for all animal species tested, suggesting extensive extravascular distribution (Yin et al, 2006). In vitro metabolism studies of PM00104 demonstrated extensive hepatic metabolism; PM00104 mainly undergoes microsomal metabolism, with plasma protein binding >99%.
Given its preclinical antitumour activity and acceptable pharmacokinetic (PK) and toxicity profile in preclinical studies, a first-in-man, open-label, phase I study of PM00104 was undertaken. The primary objectives were to assess the safety and tolerability of PM00104, determine dose-limiting toxicities (DLTs) and establish the recommended phase II dose (RP2D) for PM00104 as a 1- or 3-h 3-weekly intravenous infusion. The secondary objectives were to characterise the PK and preliminary anticancer activity for these two 3-weekly schedules of PM00104.
Materials and methods
This phase I study was conducted at the Royal Marsden NHS Foundation Trust (Sutton, UK), Hospital Universitario 12 de Octubre (Madrid, Spain) and Vall d'Hebron University Hospital (Barcelona, Spain) in accordance with the Declaration of Helsinki and the ICH Harmonised Tripartite Guideline for Good Clinical Practice and approved by the relevant regulatory authorities and independent ethics committees.
Inclusion criteria
The study inclusion criteria were as follows: ⩾18 years with histopathological or cytological confirmation of cancer not amenable to conventional treatments; written informed consent; the Eastern Cooperative Oncology Group performance status ⩽2; life expectancy ⩾3 months; no residual toxicities from previous therapies (excluding alopecia and ⩽grade 1 (G1) peripheral neuropathy); adequate bone marrow, renal and hepatic function. Left ventricular ejection fraction (LVEF) ⩾50% was required. Exclusion criteria are detailed in the Supplementary Methods.
Exclusion criteria
Patients were excluded if they had evidence of progressive central nervous system metastases or any symptomatic brain or leptomeningeal metastases; increased cardiac risk or other relevant clinical conditions such as active infection or uncontrolled endocrine diseases. Radiotherapy, chemotherapy, hormonal or biological therapies within 4 weeks before administration of PM00104 (8 weeks for extensive radiotherapy and 6 weeks for mitomycin C or nitrosurea therapy) were not permitted. Patients were also excluded if they were pregnant or breast-feeding, or if appropriate contraceptive measures were not being used.
Drug administration
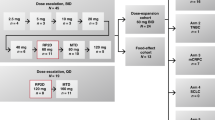
PM00104 was provided as a powder concentrate in 2.5 mg vials and reconstituted in 50–100 ml of 5% dextrose or saline solution for injection. PM00104 was initially administered as a 1-h intravenous infusion 3 weekly. A second schedule in which the duration of infusion was increased from 1 to 3 h was also planned a priori as an alternative strategy if toxicity due to PM00104-related maximum plasma concentration (Cmax), or if a more than proportional increase in Cmax was observed during dose escalation with the 1-h schedule. The starting dose for the 1-h 3-weekly schedule was 0.23 mg m−2, that is, one-tenth of the maximum tolerated dose found for the most sensitive rodent species (mouse) in animal toxicology studies. The starting dose for the 3-h 3-weekly schedule was 1.8 mg m−2 (one dose level below the RP2D for the 1-h 3-weekly regimen). In both schedules, treatment was continued until discontinuation criteria were met, including disease progression, unacceptable toxicity or withdrawal of consent. Further details on DLT definitions and dose escalation design can be found in the Supplementary Methods.
Study assessments
Assessments before, during and after study treatment included a medical history, physical examination, electrocardiogram and echocardiogram, haematology and chemistry profile, clotting studies and relevant scanning to evaluate tumour status. Toxicity was assessed at baseline and during treatment. All adverse events (AEs) and laboratory variables were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v.3.0 (National Cancer Institute, 2006). As preclinical repeat-dose studies in rats had shown mortality related to cardiac degeneration, haemorrhage and mineralisation in some animals treated at the maximum tolerated doses in repeated cycles, a central independent cardiological assessment (including measurement of troponin I, standard ECG and evaluation of left ventricular ejection fraction by echocardiography) was carried out in phase I trials. The objective antitumour response was graded according to RECIST v.1.0 (Therasse et al, 2000) based on tumour size measurements made every two cycles.
Dose-limiting toxicities and dose escalation
Dose-limiting toxicities were determined in the first treatment cycle and defined as follows: grade 4 neutropenia >5 days; grade 4 neutropenia associated with fever (⩾38 °C) or infection; grade 4 thrombocytopenia, and any other grade 3/4 PM00104-related non-haematological toxicity, except for nausea and/or vomiting unless they occurred despite optimal medical management; delay exceeding 2 consecutive weeks due to persistent drug-related toxicity; troponin I increase ⩾0.1 ng ml−1 with evidence of cardiac damage by echocardiogram or electrocardiogram, and decrease in LVEF >20% from baseline and/or LVEF <50%.
In the 1-h schedule, the dose of PM00104 was escalated by increments of 100% (50% if grade 2 toxicity occurred, and 25% if grade 3 or 4 toxicity occurred), while in the 3-h schedule, in view of the higher starting dosing, doses were escalated by increments of 25%. Dose escalation followed a 3+3 design; cohort expansion to six patients was required if one DLT was reported, and dose escalation stopped if two DLTs were observed. The RP2D was defined as the highest dose in which no >1 of a cohort of six patients had a DLT in the first cycle.
Pharmacokinetics
Blood samples for PK analyses were collected on day 1 of cycles 1 and 2 at the following time-points: pre-dose, 10 min before the end of the infusion, and 0.25, 0.5, 1, 1.5, 2, 3, 4, 5 and 7 h post infusion. Additional samples were collected on days 2, 3, 4, 6, 8, 11 and 15. Blood samples were centrifuged at 2500 g for 15 min at 4 °C, and plasma was separated, placed in polypropylene tubes and stored at −20 °C until analysis. The concentrations of PM00104 in plasma samples were measured using a validated high-performance liquid chromatograph system coupled with electrospray ionisation tandem mass spectrometry (LC/MS-MS method; Yin et al, 2005b). Percentage coefficient of variation at the lower limit of quantification (LLOQ) varied from 2.26–14.69. Lower limit of quantification was ∼15 pg ml−1 in plasma. Non-compartmental PK parameters were calculated using WinNonLin version 5.2 (Pharsight, Mountain View, CA, USA).
Results
Patient characteristics
A total of 47 patients were enroled between December 2004 and January 2010, with 27 and 20 patients treated with the 1- and 3-h schedules of PM00104, respectively (Table 1).
Recommended phase II dose
PM00104 1-h schedule
The dose was increased between cohorts until the 3.6 mg m−2 cohort, where two of six patients experienced DLTs in the first cycle (Table 2). One patient with suprarenal carcinoma developed G3 fatigue, nausea and vomiting on the first day of treatment despite maximal medical management. Nausea and vomiting improved to G1 within 3 days, while fatigue improved to G1 within 15 days of onset. A second patient with metastatic colorectal carcinoma had a transient G3 transaminase increase and G4 thrombocytopenia on days 2 and 7 of treatment, respectively. The transaminases returned to baseline levels after 7 days, while thrombocytopenia resolved within 14 days. As the previous dose level (1.8 mg m−2) was not associated with any significant toxicity, an intermediate dose level (3.0 mg m−2) was explored. No DLTs occurred in an expanded cohort of nine patients, and hence 3.0 mg m−2 was established as the RP2D of PM00104 on the 1-h 3-weekly schedule.
PM00104 3-h schedule
One DLT occurred at the first dose level (1.8 mg m−2) and two at the fifth dose level of 3.5 mg m−2 (Table 2). The patient with the DLT at 1.8 mg m−2 was a patient with metastatic NSCLC, who developed G3 hypotension on day 2 of PM00104. All cardiac assessments were normal and the DLT resolved within 2 days with intravenous fluids. The patient developed G2 vomiting after the end of his PM00104 infusion on day 1 and was also on regular antihypertensive medications, which could have contributed to the hypotension. No further DLTs were observed in the 1.8 mg m−2 cohort, and dose escalation continued with no DLTs until 3.5 mg m−2.
At this dose of 3.5 mg m−2 of PM00104, in the initial cohort of three patients, one patient with metastatic urothelial carcinoma developed G3 hypotension on day 2 of treatment associated with nausea, vomiting and diarrhoea, which resolved within 1 day. Another patient with melanoma developed G3 transaminase elevation and G2 hypotension due to Group A Streptococcus toxic shock syndrome, which resolved within 5 days of onset. As the relationship of these AEs to PM00104 in both patients was unclear, the 3.5 mg m−2 cohort was expanded to include three additional patients. Two of these subsequent three patients experienced DLTs; a patient with metastatic colorectal carcinoma had an asymptomatic and reversible G3 hypotension on day 2 of PM00104. He was treated with intravenous fluids with resolution of the DLT within 3 days without sequelae. A second patient with urothelial carcinoma developed G4 neutropenia on day 11 of treatment, which resolved after 7 days.
In view of the similar toxicities and the absence of an advantage in the PK exposure of PM00104 with the 3-h infusion, it was concluded that the 3-h infusion did not have any added benefit over the 1-h infusion. Furthermore, prolongation of infusion duration did not allow a higher PM00104 dose to be reached. Hence, the RP2D for this schedule was declared at 2.8 mg m−2.
Toxicity profile at the RP2D
Most drug-related AEs or laboratory abnormalities (Table 3) experienced by patients receiving PM00104 at the RP2D of 3.0 mg m−2 for the 1-h schedule and 2.8 mg m−2 for the 3-h schedule were mild or moderate.
PM00104 1-h schedule
The most common AEs related to PM00104 treatment were nausea (n=6), fatigue (n=5) and injection site phlebitis (n=4). No treatment-related AE reached G4. Frequent haematological abnormalities irrespective of the grade or relationship with treatment included anaemia (n=8), neutropenia (n=6) and leucopenia (n=4). Grade 3/4 neutropenia appeared in four patients at a median of 15 days after dosing, reaching the nadir on the same day and lasting a median of 7 days. Frequent biochemical abnormalities included raised ALP (n=4) and transaminase levels (n=3). Only one patient had grade 3 AST increase, with the onset and peak of the event on day 3 of cycle 2. No data on AST recovery were available, as this patient died due to disease progression.
PM00104 3-h schedule
The most common AEs related to PM00104 treatment were constipation, nausea and vomiting (n=3). No treatment-related AE reached G3 or G4, or had consequences on treatment. The most frequent haematological abnormalities irrespective of grade or relationship with treatment were anaemia and neutropenia. Only one patient had severe neutropenia, reaching grade 4, with the onset and nadir of the event on day 18, and lasting 3 days. Frequent biochemical abnormalities were ALP and ALT increases (n=2). No severe biochemical abnormalities occurred at the RP2D.
PM00104 administration and tolerability
PM00104 1-h schedule
A total of 73 cycles (median of 2 cycles per patient; range, 1–7) were administered from 0.23 to 3.6 mg m−2. About 10.9% of infusions were delayed due to PM00104-toxicities (median delay of 7 days; range, 3–11 days), with transient G1–G3 neutropenia and G2–G3 fatigue most commonly observed. At the 1-h schedule RP2D of 3.0 mg m−2, the median dose intensity was 1 mg m−2 per week and the median relative dose intensity was 100% (range, 54.2–100.5%).
PM00104 3-h schedule
In all, 58 cycles (median of 2 cycles per patient; range, 1–8) were administered from 1.8 to 3.5 mg m−2. About 13.2% of infusions were delayed due to PM00104-toxicities (median delay of 7 days; range, 5–8 days), with transient G2–G3 neutropenia and G2 fatigue most commonly observed. At the 3-h schedule RP2D of 2.8 mg m−2, the median dose intensity was 0.93 mg m−2 per week and the median relative dose intensity was 100% (range, 85.8–100%).
Cardiological assessment
Non-clinically relevant increases in cardiac troponin I >ULN were observed in 5 of 27 patients treated with the 1-h schedule and in 6 of 20 patients treated with the 3-h 3-weekly schedule. There was no clear pattern for the onset and duration of raised troponin I levels following PM00104 treatment. These increases in cardiac troponin I levels were not associated with any concomitant symptoms or alterations in electrocardiograms and echocardiograms. A review of electrocardiograms and echocardiograms by an independent board-certified cardiologist found no evidence of cardiac toxicity. Cardiac MRI scans undertaken in two patients treated within the 3-h 3-weekly schedule confirmed no evidence of drug-induced cardiac damage.
In one patient in the 1-h infusion cohort, cardiac abnormalities comprising G2 T-wave inversion on electrocardiogram was considered dose-limiting. Full cardiac workup was undertaken, including a coronary angiogram and echocardiogram, none of which revealed significant findings that could account for the electrocardiogram changes.
Pharmacokinetics
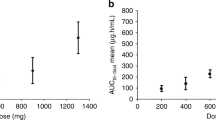
Relevant PK data for PM00104 administered in 1- and 3-h schedules are shown in Table 4. With both schedules, area under the curve (AUC) and Cmax increased dose proportionally up to the RP2D (Figure 2).

Pharmacokinetics: Cmaxvs dose level. (A) 1-h 3-weekly schedule; (B) 3-h 3-weekly schedule and (C) dose level vs AUC by infusion length.
Weight and BSA were related to the Vss, but not to PM00104 clearance (Supplementary Figure 1). Gender and performance status did not have an impact on PK parameters. An increase in AST was directly related with longer half-life, while renal function did not affect PK parameters. The plasma proteins were inversely related with Vss: an increase in plasma proteins was related to a decrease in Vss. The analysis of the effect of PM00104 PK exposure on biochemical and haematological parameters (measured as the ratio of the worst level in cycle 1 to the baseline level of each parameter) showed that a longer half-life and increases in AUC and Cmax were related with higher transaminases and bilirubin, and lower counts of neutrophils, platelets and white blood cells, whereas haemoglobin and renal function were not affected.
Antitumour activity
A total of 39 of 47 patients treated with both schedules of PM00104 were evaluable for antitumour efficacy assessments. Tumour shrinkage was observed in a patient with metastatic urothelial cancer (previous history of prostate adenocarcinoma) treated with 3.5 mg m−2 of PM00104 in a 3-h schedule. This heavily pretreated patient achieved RECIST tumour shrinkage of 49.0% after two cycles of PM00104 (Figure 3). However, the patient developed a symptomatic brain metastasis 7 days after his post-cycle 2 CT evaluation. The patient temporarily discontinued PM00104 to receive whole-brain radiotherapy, but after acquiring appropriate Ethics Committee permission, proceeded to receive further cycles of PM100104. He eventually developed disease progression in his brain metastasis after cycle 4, leading to drug discontinuation after 4.2 months. Three other patients had RECIST disease stabilisation >6 months, including patients with advanced urothelial carcinoma (3.0 mg m−2; 1-h schedule), head and neck squamous cell carcinoma (3.0 mg m−2; 1-h schedule) and NSCLC (1.8 mg m−2; 3-h schedule), lasting 9.8, 8.6 and 6.1 months, respectively (Supplementary Table 1). All three patients had radiological disease progression before enrolment on study.

Pre- and post-treatment CT scans of the patient treated with four cycles of PM00104 3.5 mg m−2 3-h 3-weekly and with partial response according to Response Evaluation Criteria in Solid Tumors (RECIST). These are CT scans from a 64-year-old male with metastatic urothelial cancer who had previously received cisplatin/gemcitabine, radiotherapy to the pelvis and carboplatin/paclitaxel. The maximum tumour shrinkage in the target lesion (right iliac lymph node) was 49.0% by RECIST measurements. However, disease progression was found in the brain after cycle 4, leading to drug discontinuation (time to progression of 4.2 months).
Discussion
The primary objectives of this phase I study, to assess the safety and establish the RP2D of two schedules of PM00104 in patients with advanced solid tumours, were met. All DLTs observed were fully reversible. Dose-limiting toxicities observed with the 1-h schedule of PM00104 included G3 transaminitis, fatigue, nausea and vomiting and G4 thrombocytopenia (n=1 each). Dose-limiting toxicities with the 3-h schedule of PM00104 included G3 hypotension (n=2) and G4 neutropenia >5 days (n=1). These findings are consistent with preliminary results from other phase I trials evaluating other schedules of PM00104, in which myelosuppression, nausea and vomiting have also been observed (Soria et al, 2008; Capdevila et al, 2009), in keeping with this agent’s mechanism of action. For the 1-h 3-weekly schedule, the RP2D was established at 3.0 mg m−2. Prolongation of 3-weekly infusion duration from 1 to 3 h did not result in higher exposures of PM00104, with DLTs observed at a dose of 3.5 mg m−2. As a result, the dose declared as the RP2D for the 3-h 3-weekly schedule was 2.8 mg m−2.
Both schedules were generally well tolerated and associated with manageable and reversible toxicity. PM00104-related AEs at the RP2D for both schedules were mostly G1–G2, with the most frequently observed toxicities being transient nausea, vomiting and fatigue. As some infusion site reactions occurred with the 1-h 3-weekly schedule, the administration of PM00104 via central intravenous access during this study became mandatory in all clinical trials of PM00104. Furthermore, as PM00104 has a moderate emetogenic risk, primary prophylaxis utilising anti-emetics according to the American Society of Clinical Oncology guidelines (Kris et al, 2006) with dexamethasone and 5-HT3 antagonists was pursued. The most common G3–G4 haematological abnormalities with both schedules of PM00104 at the respective RP2Ds were transient and comprised neutropenia and leucopenia (1-h), and neutropenia alone (3-h).
In this phase I trial, isolated increments in cardiac troponin I levels were observed in a minority of patients (n=5, 1-h 3-weekly; n=6, 3-h 3-weekly), with no associated symptoms or alterations in electrocardiograms or echocardiograms. Data from this and other phase I studies of PM00104 (Soria et al, 2008; Capdevila et al, 2009) suggest that there is no evidence of acute cardiac damage in patients treated with PM00104. Troponin I was tested from the outset of this phase I trial because preclinical data in rats identified haemorrhage, necrosis and mineralisation of cardiac muscle. In other phase I studies of PM00104 (Soria et al, 2008; Capdevila et al, 2009), increases in troponin I were also observed, but no other cardiac studies were found to be abnormal. Serial ECGs, echocardiograms, cardiac MRI and coronary angiogram studies in patients remained normal, while LVEF was maintained within normal ranges during the trials. The long-term effects of such a rise in troponin I levels is currently unknown, but may suggest that elevations in troponin I levels observed in this small subset of patients receiving PM00104 may simply reflect a biochemical finding, rather than clinical sequelae of cardiac damage. A phase II study of PM00104 with a sub-analysis of troponin I using a 1-h 3-weekly schedule is currently ongoing.
Hypotension observed on this trial was transient, and no underlying drug-related pathophysiological mechanism has been established. In patients with symptoms including emesis and diarrhoea, dehydration may have had a role as all episodes reversed with intravenous rehydration. In a pooled analysis of 144 patients treated in four phase I single-agent trials of PM00104, 5% (n=8) had hypotension, although this was G3 in only three patients (2%). No other relevant cardiovascular events were reported concomitant with hypotension.
Non-compartmental analysis of the PK profile of PM00104 demonstrated dose proportionality of the main parameters (AUC and Cmax) up to the RP2D. A population PK analysis including data from this and other phase I studies (Perez-Ruixo et al, 2012) confirmed that PM00104 has linear PKs.
Three patients with advanced solid tumours had RECIST stable disease ⩾6 months. Furthermore, a patient with advanced urothelial carcinoma had a RECIST partial response, with tumour shrinkage by 49% after 2 months of treatment, indicating that PM00104 has antitumour activity. PM00104 is a large molecule that does not cross the blood brain barrier, which may explain the progression of a brain metastasis in this patient.
In conclusion, the primary objective of this phase I trial was met, and the RP2D was established as 3.0 mg m−2 for the 1-h PM00104 schedule and as 2.8 mg m−2 for the 3-h PM00104 schedule. In view of the well-tolerated toxicities, active PK profile and preliminary evidence of antitumour activity observed for PM00104 in this first-in-human study, multiple phase II clinical trials have now been initiated. Currently, PM00104 is being assessed in phase II studies undertaken in patients with advanced endometrial cancer, cervical cancer, multiple myeloma, advanced Ewing family of tumours and advanced urothelial cancer (http://www.clinicaltrials.gov).
Change history
23 January 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Capdevila J, Clive S, Tabernero J, Lardelli P, Soto-Matos A, Baselga J, Piera A, Pardos I, Rye R, Smyth JF (2009) Phase I study of the novel anticancer drug PM00104 as a 24-hour IV infusion every 3 weeks (q3w) in patients (pts) with advanced solid tumors or lymphoma. J Clin Oncol 27(15 (suppl)): Abstract 2568
Elices M, LePage D, Grant W, Sasak H, Caylor T, Martin J, Guillén MJ, Cuevas Marchante C, Avilés P, Faircloth G (2005a) Profile of Zalypsis™ (PM00104) against various human solid tumors. 17th AACR-NCI-EORTC International Conference on ‘Molecular Targets and Cancer Therapeutics: Discovery, Biology and Clinical Applications’ Clin Cancer Res (11(24 Part 2): 9085s–9086s (Abstract B216)
Elices MJ, Grant W, Guillen MJ, Cuevas Marchante C, Aviles P, Faircloth GT (2005b) The novel compound PM00104 exhibits significant in vivo activity againts breast tumors. AACR American Association for Cancer Research; 96th Annual Meeting. Abstract 623
Fontana A, Cavaliere P, Wahidulla S, Naik CG, Cimino G (2000) A new antitumor isoquinoline alkaloid from the marine nudibranch Jorunna funebris. Tetrahedron 56: 7305–7308
Greiner T, Maier A, Bausch N, Fiebig HH, LePage D, Guillén MJ, Cuevas C, Avilés P (2007) Preclinical evaluation of PM00104 to support the selection of tumor indications for clinical studies. 19th AACR-NCI-EORTC International Conference on ‘Molecular Targets and Cancer Therapeutics: Discovery, Biology and Clinical Applications’: Abstract C60
Guillen MJ, Lepage D, Grant W, Cuevas C, Aviles P (2009) Abstract #2018: Antitumor activity of PM00104 in hepatocellular carcinoma (HCC) experimental models. AACR Meeting Abstracts 2009 (2_Annual_Meeting): 2018
Guirouilh-Barbat J, Antony S, Pommier Y (2009) Zalypsis (PM00104) is a potent inducer of gamma-H2AX foci and reveals the importance of the C ring of trabectedin for transcription-coupled repair inhibition. Molecular Cancer Therapeutics 8(7): 2007–2014
Herrero A, Botet J, Cuevas C, Gago F, Moreno S (2007) Genome-wide screen reveals antitumor Zalypsis(R) as a strong inducer of DNA double strand breaks. AACR Meeting Abstracts 2007 (1_Annual_Meeting): 5733
Kris MG, Hesketh PJ, Somerfield MR, Feyer P, Clark-Snow R, Koeller JM, Morrow GR, Chinnery LW, Chesney MJ, Gralla RJ, Grunberg SM (2006) American society of clinical oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol 24(18): 2932–2947
Leal JF, Garcia-Hernandez V, Moneo V, Domingo A, Bueren-Calabuig JA, Negri A, Gago F, Guillen-Navarro MJ, Aviles P, Cuevas C, Garcia-Fernandez LF, Galmarini CM (2009) Molecular pharmacology and antitumor activity of Zalypsis in several human cancer cell lines. Biochem Pharmacol 78(2): 162–170
LePage D, Sasak H, Cheney L, Oyama S, Cuevas C, Avilés P, Maxuitenko Y, Faircloth G (2007a) Antitumor activity of Zalypsis® in human pancreas tumors. AACR American Association for Cancer Research; Annual Meeting: Abstract 1519
LePage D, Sasak H, Guillen MJ, Grant W, Cuevas C, Aviles P (2007b) Antitumor activity of Zalypsis® (PM00104) in experimental models of bladder, gastric and liver cancer. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics: Abstract C62
Magro P, Alvarez E, Kolb E . Mandola M, Scotto K (2007) Characterization of tumor cell response to the novel anti-tumor agent Zalypsis (R). AACR Meeting Abstracts 2007 (1_Annual_Meeting): 1529
National Cancer Institute (2006) Common Terminology Criteria for Adverse Events (CTCAE) v.3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf, 1–72
Ocio EM, Maiso P, Chen X, Garayoa M, Alvarez-Fernandez S, San-Segundo L, Vilanova D, Lopez-Corral L, Montero JC, Hernandez-Iglesias T, de Alava E, Galmarini C, Aviles P, Cuevas C, San-Miguel JF, Pandiella A (2009) Zalypsis: a novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 113(16): 3781–3791
Oku N, Matsunaga S, van Soest RW, Fusetani N (2003) Renieramycin J, a highly cytotoxic tetrahydroisoquinoline alkaloid, from a marine sponge Neopetrosia sp. J Nat Prod 66(8): 1136–1139
Perez-Ruixo C, Valenzuela B, Fernandez Teruel C, Gonzalez-Sales M, Miguel-Lillo B, Soto-Matos A, Perez-Ruixo JJ (2012) Population pharmacokinetics of PM00104 (Zalypsis((R))) in cancer patients. Cancer Chemother Pharmacol 69(1): 15–24
Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH, Martin DG (1990) Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J Org Chem 55(15): 4512–4515
Soria JC, Plummer R, Soto A, Massard C, Calvert H, Prados R, Angevin E, Jones C, de las Heras B (2008) Phase I study of the novel anti-cancer drug PM00104 as a 1-hour weekly infusion resting every fourth week in patients with advanced solid tumors or lymphoma. Eur J Cancer 6(Suppl 12): 57 (abstract no.179)
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Nat Cancer Inst 92(3): 205–216
Yin J, Avilés P, Guillén MJ, Ly C, Lee W, Munt S, Cuevas C, Faircloth GT (2005a) Pharmacokinetic evaluation of a novel anti-tumor agent, zalypsis (PM00104), in mice and dogs. 17th AACR-NCI-EORTC International Conference on ‘Molecular Targets and Cancer Therapeutics: Discovery, Biology and Clinical Applications’ Filadelfia, EEUU, 14–18 Noviembre, 2005 Clin Cancer Res 11(24 Part 2): 9072s (Abstract B163)
Yin J, Aviles P, Lee W, Ly C, Guillen MJ, Munt S, Cuevas C, Faircloth G (2005b) Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM00104, a novel antineoplastic agent, in mouse, rat, dog, and human plasma. Rapid Commun Mass Spectrom 19(5): 689–695
Yin J, Aviles P, Wright T, Guillen MJ, Ly C, Lee W, Munt S, Cuevas C, Faircloth GT (2006) Toxicokinetic evaluation of a novel anti-tumour agent, Zalypsis(R) (PM00104), in dogs. AACR Meeting Abstracts 2006(1): 728–72
Acknowledgements
This study was supported by PharmaMar, SA. The Drug Development Unit of the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research is supported in part by a programme grant from Cancer Research UK. Support was also provided by the Experimental Cancer Medicine Centre (to The Institute of Cancer Research) and the National Institute for Health Research Biomedical Research Centre (jointly to the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JS de Bono and J Tabernero served as paid consultants for PharmaMar. JA Lopez-Martin was a former employee of PharmaMar.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yap, T., Cortes-Funes, H., Shaw, H. et al. First-in-man phase I trial of two schedules of the novel synthetic tetrahydroisoquinoline alkaloid PM00104 (Zalypsis) in patients with advanced solid tumours. Br J Cancer 106, 1379–1385 (2012). https://doi.org/10.1038/bjc.2012.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.99
Keywords
This article is cited by
-
Phase II clinical trial of PM00104 (Zalypsis®) in urothelial carcinoma patients progressing after first-line platinum-based regimen
Cancer Chemotherapy and Pharmacology (2014)
-
Phase I study of carboplatin in combination with PM00104 (Zalypsis®) in patients with advanced solid tumors
Investigational New Drugs (2014)
-
A phase I pharmacokinetic study of PM00104 (Zalypsis®) administered as a 24-h intravenous infusion every 3 weeks in patients with advanced solid tumors
Cancer Chemotherapy and Pharmacology (2013)
-
Phase I study of PM00104 (Zalypsis®) administered as a 1-hour weekly infusion resting every fourth week in patients with advanced solid tumors
Investigational New Drugs (2013)
-
Phase II study of weekly PM00104 (ZALYPSIS®) in patients with pretreated advanced/metastatic endometrial or cervical cancer
Medical Oncology (2013)
