
Abstract
Background:
We investigated the association between skin rash and plasma creatine kinase (CK) levels in oncology phase I trials.
Methods:
We analysed data from 295 patients treated at our institution within 25 phase I trials which included CK measurements in the protocol. Trials involved drugs targeting EGFR/HER2, m-TOR, VEGFR, SRC/ABL, aurora kinase, BRAF/MEK, PARP, CDK, A5B1 integrin, as well as oncolytic viruses and vascular disrupting agents.
Results:
Creatine kinase measurements were available for 278 patients. The highest levels of plasma CK during the trial were seen among patients with Grade (G) 2/3 rash (median 249 U l−1) compared with G1 (median 81 U l−1) and no rash (median 55 U l−1) (P<0.001). There was a significant reduction in CK after the rash resolved (mean 264.2 vs 100.1; P=0.012) in 25 patients, where serial CK values were available. In vitro exposure of human keratinocytes to EGFR, MEK and a PI3Kinase/m-TOR inhibitor led to the increased expression of CK-brain and not CK-muscle or mitochondrial-CK.
Conclusion:
Plasma CK elevation is associated with development of skin rash caused by novel anticancer agents. This should be studied further to characterise different isoforms as this will change the way we report adverse events in oncology phase I clinical trials.
Similar content being viewed by others
Main
The patterns of toxicities caused by anticancer agents have changed with the advent of the development of targeted agents, and myelotoxicity is no longer the predominant limiting toxicity. Skin rash is a frequent side-effect of targeted anticancer drugs including drugs inhibiting the EGFR–RAS–RAF–MEK and PI3 kinase–AKT–m-TOR pathways (Figure 1) (Moore et al, 2007; O’Donnell et al, 2008; Banerji et al, 2010; Flaherty et al, 2010; Yap et al, 2010; Moreno Garcia et al, 2011).

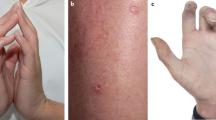
Types of skin rash caused by molecularly targeted agents. Skin rash often causes dose-limiting toxicities in phase I trials of molecularly targeted agents. (A) Acneiform skin rash typical of EGFR or MEK inhibitors. (B) Maculopapular skin rash typical of PI3K, AKT or m-TOR inhibitors.
Creatine (Cr) and creatine kinase (CK) have an important role in human soft tissue. In the cell, CK catalyses the reversible phosphorylation between ATP and Cr to produce phosphocreatine (PCr) and ADP, acting as a cytosolic energy buffer. Three predominant isoforms of CK exist with different subcellular locations: two cytosolic isoforms (CK-muscle (CK-M) and CK-brain (CK-B)) and one mitochondrial isoform (mitochondrial-CK (mt-CK)) (Wallimann et al, 2011). Cytosolic isotypes form homo- or hetero-dimers with tissue specificity: CK-MM (muscle), CK-MB (heart) and CK-BB (brain and skin) (Schlattner et al, 2002). During wound healing, keratinocytes overexpress CK-BB to cope with the increased metabolism caused by nucleotide biosynthesis (Schlattner et al, 2002). We hypothesised that skin toxicity caused by novel anticancer agents was associated with keratinocyte stress, thus leading to increased levels of CK in the skin and possibly circulating CK levels in the plasma. We thus aimed to study the association, if any, of plasma CK in patients who developed skin rash in phase I trials.
Methods
Trial and patient inclusion
This was a retrospective study including 295 consecutive patients treated within phase I clinical trials at the Drug Development Unit, Royal Marsden Hospital, Sutton, UK. We reviewed 104 trial protocols and found 25 that mandated regular plasma CK measurements. All patients consented to individual phase I clinical trials that were approved by the institutional research ethics committee, and this project was reviewed by the Royal Marsden Hospital Audit Committee.
Data collection
Compliance to CK measurement during the trial was assessed. To assess the dynamics of CK, we included several data points: baseline CK, first elevation (level and day) and highest level (and day of highest level). A complete list of concomitant medications (such as use of statins) was not available. The baseline plasma CK elevation was defined as levels above 140 U l−1 by local laboratory reference ranges. To avoid potential bias due to baseline elevation or significant changes below the threshold of CK elevation, we also calculated the ratio: higher CK-on trial/baseline CK and considered ‘increased CK’ if the ratio was ⩾1.5. The 1.5-fold cutoff was arbitrary. If rash occurred, we attempted to collect CK measurements at time points when the rash resolved, if documented. Representative photographs of skin rash caused by MEK and AKT inhibitors were taken after informed consent from patients and have been anonymised.
Please see Supplementary Data for tissue culture, chemical reagents and western blotting.
Statistical analysis
To compare the difference between changes in plasma CK, and because of the limited number of patients with skin rash, we performed non-parametric tests. Mann–Whitney U and Kruskal–Wallis one-way analysis of variance tests were used to compare CK levels with rash when two or three categories were included, respectively. The Wilcoxon test was used to analyse paired CK values at diagnosis and resolution of rash. For categorical variables, χ2 and Fisher’s exact test were used as required. A P-value of ⩽0.05 was considered statistically significant and all tests were two-tailed. SPSS v13.0.1 (IBM, Portsmouth, UK) was used for all the analyses.
Results
Data from 295 patients were considered in the analysis. CK measurements were available for 278 patients (94%), indicating compliance to protocol. For further analysis of the association of CK and skin rash, only the 278 patients who had CK measurement on file were used. Sixty-four patients (23%) developed a high CK during the study, including 42 (15%) patients who had an 1.5 × elevation compared with baseline during the study. In 25 patients who developed rash, it was possible to find documented paired CK at development and resolution of rash. Fifty patients (18%) developed drug-induced skin rash during the trial (Figure 1).
There was an association between development of rash and CK rise; 17% (39 out of 228) of patients who did not develop skin rash had a CK rise during the trial compared with 35% (9 out of 26) of patients who developed G1 and 67% (16 out of 24) of patients who developed G2/3 rash (P<0.001). The levels of CK during the trial were significantly elevated among patients with G2/3 rash (mean 406 U l−1; median 249 U l−1) compared with G1 (mean 127 U l−1; median 81 U l−1; P=0.01) and no rash (mean 92 U l−1; median 55; P<0.001; Figure 2. In addition, it was possible to find documented CK when rash developed and when it resolved in 25 cases. There was significant difference in the CK during rash and CK at resolution (mean 264.2 vs 100.1; P=0.012).

Serum creatine kinase levels and rash grade. Two hundred and seventy-eight patients with available CK measurements were included in the analysis. The highest CK level on study was selected for analysis.
In vitro analysis
Adult human keratinocytes were exposed to increasing concentrations of an EGFR (gefitinib), PI3K/m-TOR (PI-103) and MEK inhibitor (PD0325901) at increasing doses for 24 h. The expression of CK-B, CK-M and mt-CK was studied in the lysates. There was an increase in the expression of CK-B (Figure 3). CK-M and mt-CK were not detected in the lysates.

Expression of CK isoforms in human keratinocytes. Expression of the three different isoforms of CK in adult human keratinocytes after exposure to vehicle control and two concentrations (100 nM and 1000 nM) of gefitinib (EGFR inhibitor) and PDO325901 (MEK inhibitor). An increase in CK-B expression was seen after exposure to the three drugs. Mt-CK and CK-M were not detectable in the samples studied. Abbreviation: GAPDH; glyceraldehyde 3-phosphate dehydrogenase.
Discussion
This is the first time that an association between skin rash and elevated plasma CK in patients treated with different novel anticancer agents has been reported. Further pointers that the association is real are the fact that there was a temporal relationship between the CK levels dropping when the rash resolved in a subset of patients where this data was available and the fact that the relationship between skin rash and CK seem to spread across a wide range of tumours and anticancer agents (Supplementary Table 1). However, this study has the limitations of a retrospective analysis and only total CK and not its specific isoforms were measured. CK is known to be elevated in patients treated with a range of drugs such as statins (Stolcpart et al, 2010) and anticancer agents, trabectedin (Stoyianni et al, 2011) and imatinib (Gordon et al, 2010). In most instances these elevations are thought to be due to inflammation in muscles and relate to CK-MM. CK-BB subtype is usually undetectable in serum but can be responsible for 1–18% of the total CK activity (Skogseid et al, 1992; Kovesdi et al, 2010). It has also been found to be overexpressed in different tumour cells, such as breast (Zarghami et al, 1996) and lung (Niklinski et al, 1991) cancers. CK elevation has also been reported in patients with toxic epidermal necrosis. However, it was found that CK elevation of CK-BB fraction represented only 3% of the serum total CK whereas 94% was CK-MM (Caux et al, 1994). The present analysis includes total plasma CK measurement and does not differentiate between isotypes.
We conducted preclinical experiments to explore this further. We detected a rise in expression of CK-B in keratinocytes after in vitro exposure to EGFR, PI3K/m-TOR and MEK inhibitors. This was consistent with previous reports that there was an increased expression of CK-BB in keratinocytes, when they were under stress (Schlattner et al, 2002), or in exudates within blisters (Paavonen et al, 1988; Kiistala et al, 1989). It is therefore possible that the rise in plasma CK associated with skin toxicity is due to a rise in CK-BB, and this has not been documented before. Future studies should analyse different circulating isoforms of CK to find the source of the rise of plasma CK associated with skin rash.
These findings have several clinical implications. First, at present, a raised CK would be reported as a musculoskeletal adverse or serious adverse event in phase I trials. However, it is possible that these are manifestations of skin toxicity. Second, these findings may give an insight into the treatment of skin rash. It has previously been shown that repair mechanisms in keratinocytes require ATP. The free energy of ATP is stored and transported in the form of PCr, where CK activity can replenish cellular ATP in situ and exogenous Cre protected against free radical stress (Lenz et al, 2005). On the basis of the observations that skin rash is associated with a raised CK and keratinocytes show increased expression of CK (CK-B) after exposure to different signal transduction agents, it could be hypothesised that topical Cre supplementation (to increase the PCr cytosolic energy buffer in keratinocytes) may be useful to prevent or treat rash caused by novel anticancer agents. Indeed, topical Cre has been investigated for the treatment of skin damage associated with UV light (Lenz et al, 2005; Knott et al, 2008). In conclusion, this is the first report of an association of skin rash and total circulating CK in the setting of phase I trials of novel anticancer agents. This has a number of clinical implications that include adverse event reporting and the clinical management of rash. These findings merit further prospective evaluation in clinical trials.
Change history
09 November 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, Desar IM, Timmer-Bonte JN, Eckhardt SG, Lewis KD, Brown KH, Cantarini MV, Morris C, George SM, Smith PD, van Herpen CM (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16 (5): 1613–1623
Caux F, Chosidow O, Philippon C, Roujeau JC, Revuz J (1994) Increased serum and blister fluid levels of creatine kinase in patients with toxic epidermal necrolysis. Br J Dermatol 130 (3): 337–341
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363 (9): 809–819
Gordon JK, Magid SK, Maki RG, Fleisher M, Berman E (2010) Elevations of creatine kinase in patients treated with imatinib mesylate (Gleevec). Leuk Res 34 (6): 827–829
Kiistala U, Paavonen T, Saarelainen I, Aronen H, Asko-Seljavaara S, Kariniemi AL, Ingervo L, Niemitalo S (1989) Epidermis is the origin of high creatine kinase levels in skin blister fluid. Acta Derm Venereol 69 (4): 284–287
Knott A, Koop U, Mielke H, Reuschlein K, Peters N, Muhr GM, Lenz H, Wensorra U, Jaspers S, Kolbe L, Raschke T, Stab F, Wenck H, Gallinat S (2008) A novel treatment option for photoaged skin. J Cosmet Dermatol 7 (1): 15–22
Kovesdi E, Luckl J, Bukovics P, Farkas O, Pal J, Czeiter E, Szellar D, Doczi T, Komoly S, Buki A (2010) Update on protein biomarkers in traumatic brain injury with emphasis on clinical use in adults and pediatrics. Acta Neurochir (Wien) 152 (1): 1–17
Lenz H, Schmidt M, Welge V, Schlattner U, Wallimann T, Elsasser HP, Wittern KP, Wenck H, Stab F, Blatt T (2005) The creatine kinase system in human skin: protective effects of creatine against oxidative and UV damage in vitro and in vivo. J Invest Dermatol 124 (2): 443–452
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25 (15): 1960–1966
Moreno Garcia V, Baird RD, Shah KJ, Basu B, Tunariu N, Blanco M, Cassier PA, Pedersen JV, Puglisi M, Sarker D, Papadatos-Pastos D, Omlin AG, Biondo A, Ware JA, Koeppen H, Levy GG, Mazina KE, De Bono JS (2011) A phase I study evaluating GDC-0941, an oral phosphoinositide-3 kinase (PI3K) inhibitor, in patients with advanced solid tumors or multiple myeloma. J Clin Oncol 29 (15s): 199s (abstract no. 3021)
Niklinski J, Furman M, Palynyczko Z, Laudanski J, Bulatowicz J (1991) Carcinoembryonic antigen, neuron-specific enolase and creatine kinase-BB as tumor markers for carcinoma of the lung. Neoplasma 38 (6): 645–651
O'Donnell A, Faivre S, Burris HA, Rea D, Papadimitrakopoulou V, Shand N, Lane HA, Hazell K, Zoellner U, Kovarik JM, Brock C, Jones S, Raymond E, Judson I (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 26 (10): 1588–1595
Paavonen T, Aronen H, Saarelainen I, Neittaanmaki H, Hjelm I, Kiistala U (1988) The BB-isoenzyme is a major component of creatine kinase in skin blister fluid. Br J Dermatol 118 (6): 753–757
Schlattner U, Mockli N, Speer O, Werner S, Wallimann T (2002) Creatine kinase and creatine transporter in normal, wounded, and diseased skin. J Invest Dermatol 118 (3): 416–423
Skogseid IM, Nordby HK, Urdal P, Paus E, Lilleaas F (1992) Increased serum creatine kinase BB and neuron specific enolase following head injury indicates brain damage. Acta Neurochir (Wien) 115 (3–4): 106–111
Stolcpart RS, Olson KL, Delate T, Rasmussen J, Rehring TF, Merenich JA (2010) The risk for significant creatine kinase elevation with statins. Am J Cardiovasc Drugs 10 (3): 187–192
Stoyianni A, Kapodistrias N, Kampletsas E, Pentheroudakis G, Pavlidis N (2011) Trabectedin-related rhabdomyolysis: an uncommon but fatal toxicity. Tumori 97 (2): 252–255
Wallimann T, Tokarska-Schlattner M, Schlattner U (2011) The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40 (5): 1271–1296
Yap TA, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Tunariu N, Sullivan D, Yan L, De Bono JS, Tolcher AW (2010) First-in-class phase I trial of a selective Akt inhibitor, MK2206 (MK), evaluating alternate day (QOD) and once weekly (QW) doses in advanced cancer patients (pts) with evidence of target modulation and antitumor activity. J Clin Oncol 28 (15s): 235s (abstract no. 3009)
Zarghami N, Giai M, Yu H, Roagna R, Ponzone R, Katsaros D, Sismondi P, Diamandis EP (1996) Creatine kinase BB isoenzyme levels in tumour cytosols and survival of breast cancer patients. Br J Cancer 73 (3): 386–390
Acknowledgements
This work was supported by Cancer Research UK (grant numbers: C309/A8274; A309/A11566; C51/A6883); an ECMC grant (C51/A7401; C12540/A15573); and by the Fundacion Para la Investigacion del Hospital Universitario La Paz (FIBHULP): Grant REX-09.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Presented in part at the 22nd EORTC-NCI-AACR symposium on ‘Molecular targets and Cancer Therapeutics’, Berlin, Germany, 2010.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Moreno Garcia, V., Thavasu, P., Blanco Codesido, M. et al. Association of creatine kinase and skin toxicity in phase I trials of anticancer agents. Br J Cancer 107, 1797–1800 (2012). https://doi.org/10.1038/bjc.2012.482
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.482
Keywords
This article is cited by
-
Phase I and pharmacokinetics/pharmacodynamics study of the MEK inhibitor RO4987655 in Japanese patients with advanced solid tumors
Investigational New Drugs (2015)
-
Lung, CNS and musculo-skeletal targeted therapy-induced toxicity: imaging features
Cancer Imaging (2014)
-
Im Fokus: die Blockade mit Kinaseinhibitoren
Im Focus Onkologie (2014)
