
Abstract
Background:
Mutations in genes for hereditary non-polyposis colorectal cancer (HNPCC) in ovarian cancer patients remains poorly defined. We sought to estimate the frequency and characteristics of HNPCC gene mutations in a population-based sample of women with epithelial ovarian cancer.
Methods:
The analysis included 1893 women with epithelial ovarian cancer ascertained from three population-based studies. Full-germline DNA sequencing of the coding regions was performed on three HNPCC genes, MLH1, MSH2 and MSH6. Collection of demographic, clinical and family history information was attempted in all women.
Results:
Nine clearly pathogenic mutations were identified, including five in MSH6, two each in MLH1 and MSH2. In addition, 28 unique predicted pathogenic missense variants were identified in 55 patients. Pathogenic mutation carriers had an earlier mean age at diagnosis of ovarian cancer, overrepresentation of cancers with non-serous histologies and a higher number of relatives with HNPCC-related cancers.
Conclusions:
Our findings suggest that fewer than 1% of women with ovarian cancer harbour a germline mutation in the HNPCC genes, with overrepresentation of MSH6 mutations. This represents a lower-range estimate due to the large number of predicted pathogenic variants in which pathogenicity could not definitively be determined. Identification of mismatch repair gene mutations has the potential to impact screening and treatment decisions in these women.
Similar content being viewed by others

Main
It is estimated that 13% of all invasive ovarian cancer patients carry mutations in BRCA1 or BRCA2 (Whittemore et al, 1992; Taylor and Schwartz, 1994), and an additional proportion of patients carry mutations in genes that increase risk of hereditary non-polyposis colorectal cancer (HNPCC). Hereditary non-polyposis colorectal cancer is an autosomal dominant syndrome of cancer predisposition of the colorectum, endometrium, stomach or ovaries associated with DNA mismatch repair (MMR) gene mutations. Other less frequent sites include cancers of the renal pelvis, ureter, small bowel, pancreas and brain (Lynch et al, 2009). Patients may develop multiple primary cancers, and colorectal cancers are characterised by young age of onset (Vasen, 2005). Hereditary non-polyposis colorectal cancer is believed to account for 2–4% (Hampel et al, 2005, 2008) of unselected cases of colorectal cancer, but the fraction of ovarian cancer due to HNPCC is not well established. The lifetime risk of ovarian carcinoma in females carrying HNPCC mutations has been estimated to be up to 12%, with a recent report suggesting cumulative risks of 20% or higher for the MLH1 and MSH2 genes; however, risk may vary with the specific gene involved (Bonadona et al, 2011).
In most studies of HNPCC families to-date, MLH1 and MSH2 account for 80–90% of observed mutations in the MMR genes (Papadopoulos et al, 1994; Peltomaki and Vasen, 1997). However, most studies were clinic-based surveys of individuals tested based on clinical criteria, such as the Amsterdam criteria (Vasen et al, 1999), and few included MSH6 testing. Mutations in MSH6 (Miyaki et al, 1997; Sjursen et al, 2010) are infrequent in classical HNPCC families, and thus may have been underestimated in prior studies.
Identification of germline MMR genes enables individuals to benefit from up-to-date cancer risk management options as outlined in the NCCN guidelines (Lynch, 2006). In fact, frequent colonoscopy has been shown to improve outcome (Syngal et al, 1998; Vasen et al, 1998). Furthermore, endometrial and ovarian cancer risk management options include both screening and prophylactic surgery, once childbearing is complete. In addition, preliminary studies in colorectal cancer suggest that knowledge of germline MMR gene mutations may provide an opportunity to refine cancer treatment (Elsaleh et al, 2000, 2001; Hemminki et al, 2000).
The objective of the current study was to estimate the frequency of mutations in the three HNPCC genes in a population-based sample of women with epithelial ovarian cancer. Secondary objectives included evaluation of demographic, clinical, histopathologic and family history characteristics of germline mutation carriers.
Materials and methods
Participants
Data for this study were drawn from three population-based studies of epithelial ovarian cancer: the Familial Ovarian Tumour Study (FOTS) in Toronto (Risch et al, 2001), the Tampa Bay Ovarian Cancer Study (TBOCS) at the Moffitt Cancer Center (Pal et al, 2005) and the North Carolina Ovarian Cancer Study (NCOCS) at the Duke University (Wenham et al, 2003), with details about study design, populations and data collection methods published previously. Briefly, FOTS cases were identified through monitoring of pathology reports submitted to the population-based Ontario Cancer Registry for province-wide recruitment. The TBOCS cases were recruited through a rapid case ascertainment mechanism in the two most populous counties in the Tampa Bay region. The NCOCS cases were identified through a rapid case ascertainment mechanism in a 48-county region located in the central portion of North Carolina (Wenham et al, 2003). The study protocol was approved by the institutional review board at each centre, and written informed consent was obtained from all participants. There was no mechanism of rapid ascertainment in place to recruit the FOTS cases, in contrast to the TBOCS and NCOCS cases, which resulted in a longer time from diagnosis to ascertainment in the Toronto centre compared with the American centres.
Eligibility criteria for study enrolment included diagnosis of incident, pathologically confirmed primary epithelial ovarian cancer, either borderline or invasive, and age 20 years or above. Each study collected questionnaire data concerning demographic, clinical and family history information, and reviewed medical and pathology records for determining tumour histopathology. Specimen collection included blood for DNA extraction and analysis.
Gene sequencing
All 45 coding exons of the MLH1 (NM_000249.3), MSH2 (NM_000251.1) and MSH6 (NM_000179.2) genes were amplified and sequenced in 44 fragments in germline DNA. Primers were designed using Primer 3 software (Rozen and Skaletsky, 2000) to cover at least 20 bp at each 5′- and 3′-side of the exons. The amplified DNA fragments were sequenced by using the BigDye Terminator Cycle Sequencing kit on an ABI 3730xl DNA Analyzer (Applied Biosystems Co., Foster City, CA, USA). Sequencing chromatograms generated by the analyser were examined for variant detection using Mutation Surveyor software (SoftGenetics LLC., State College, PA, USA). All sequences were compared with the related NCBI (http://www.ncbi.nlm.nih.gov) reference sequences for variant detection. The chromatograms of all the computationally determined variants were confirmed manually.
All insertions and deletions in the gene-coding regions, nonsense variants and variants located at the essential splice site sequences were considered as potentially pathogenic, and their DNA fragments were also sequenced in the reverse direction for confirmation. Insertions and deletions for which no functionality data were available were classified as ‘unknown pathogenicity’. Missense variants first were searched in the colon cancer gene variant database (http://chromium.liacs.nl/LOVD2/colon_cancer/home.php) for functional effects. If functional effect was not known, we used Align-GVGD (Tavtigian et al, 2006) to predict the likely functional effect based on library alignments from human to pufferfish for MLH1 and MSH2. As no alignment data were available for MSH6 in the Align-GVGD website, a protein alignment was constructed for predicting the functional effect of MSH6 missense variants from MSH6 homologues in eight species (human, chimpanzee, dog, cow, pig, mouse, chicken, frog and zebrafish). Missense variants with class C45 and higher defined by Align-GVGD were classified as ‘predicted pathogenic’. The class C45 was chosen as a cut-off point for Align-GVGD based on receiver-operating characteristic curve analysis using BRCA1 and BRCA2 missense variants with known functional effects reported in the Breast Cancer Information Core database (http://research.nhgri.nih.gov/projects/bic/) or Myriad’s published data (Easton et al, 2007). The rationale for using BRCA1 and BRCA2 mutations for the receiver-operating characteristic curve analysis was the limited sample size of known pathogenic missense mutations in the MLH1, MSH2 and MSH6 genes. The chosen cut-off point corresponds to 90% specificity and 50% sensitivity. We also used PolyPhen2 (Adzhubei et al, 2010) and SIFT (Ng and Henikoff, 2003) for predicting the functional effect of missense variants. We considered a missense variant as a predicted pathogenic if two of the three in silico tools (Align-GVGD, PolyPhen-2 and SIFT) predicted it as pathogenic and the carrier frequency of that mutation was less than 1% among 6481 study subjects of the NHLBI Exome Sequencing Project (ESP6500) (http://evs.gs.washington.edu/EVS/).
Data collection
All participants completed questionnaires by which demographic and family history information was obtained. Family history data included types and ages of cancer diagnoses in first-, second- and third-degree relatives. In addition, the FOTS data set included ages of relatives (current, or if deceased, age at death). Medical records were retrieved on all participants to abstract information on tumour histology. Information on date of diagnosis (based on pathology report) and date of study enrolment (based on date consent form was signed) were collected, to determine time between diagnosis and enrolment (calculated in days).
Statistical analyses
Site differences in descriptive statistics of clinical factors across the three sites were evaluated using analysis of variance and Kruskal–Wallis tests for continuous variables (e.g., time to diagnosis and attained age) and Pearson’s χ2-tests for categorical variables. Similarly, clinical variables were compared across mutation subtypes (i.e., pathogenic, predicted pathogenic, predicted non-pathogenic, no mutation detected). Known benign polymorphic variants were combined with the ‘no mutation’ group. All reported P-values are two-sided. All analyses were carried out with SAS version 9.1.3 (SAS Institute, Inc., Cary, NC, USA).
To compare the cumulative incidence of cancer among the first-degree relatives of proband carriers of different mutation subtypes, a survival analysis approach among the FOTS study subjects for whom we had detailed family history information was used. Each first-degree relative was considered as an observation and followed from birth until the occurrence of any type of cancer, death from another cause, or the date of the study interview, using the Cox proportional hazard model. Separate analyses were carried out for all cancer outcomes, all HNPCC cancer outcomes, colorectal cancer outcome and ovarian cancer outcome. Covariates for each observation included the proband’s mutation status. Hazard ratios of mutation subtypes and 95% confidence interval (CI) were estimated for each model.
Results
In total, 1893 ovarian cancer patients were included, with 1521 from FOTS, 126 from TBOCS and 246 from NCOCS, as shown in Table 1. Overall, mean age at diagnosis and racial distribution were similar between sites, other than a higher proportion of Black subjects in the Duke sample. The median time between diagnosis and study enrolment was similar between the Duke and Moffitt cases, although longer for the Toronto cases because of the difference in participant ascertainment procedure. At all three sites, the majority of cancers were of serous histology. However, the proportion of non-serous cases was higher in the Ontario case than in the other two sites. Family histories of HNPCC-associated cancers were similar across the three sites.
Germline genetic testing of coding regions in the MLH1, MSH2 and MSH6 genes revealed sequence changes at 161 different nucleotides. As shown in Table 2, nine pathogenic mutations were found, five in MSH6, two in MLH1 and two in MSH2. Four mutations were insertions or deletions and five were nonsense mutations. Seven of the nine pathologic mutations have been previously reported and each of the nine was seen in a single individual. We detected two additional changes of ‘unknown pathogenicity’ in MSH6: an in-frame deletion of two amino acids (MSH6 c.936_941delGAAAAG), not known to belong to any known functional protein domain, and a frame-shift insertion (MSH6 c.4065_4066insGTGA) at the C terminus that truncates two terminal amino acids. In addition, there were 101 missense variants (Supplementary Table 1), including 5 benign polymorphisms, and 96 unclassified variants (in 128 participants) and 49 silent variants (Supplementary Table 2). Overall, the frequency of pathogenic mutations was 0.5% (95% CI 0.2–0.8; Table 2). However, this frequency was 1.6% (95% CI 0.4–2.7) when considering only invasive cancers of the endometrioid and clear cell subtypes.
Through the Align-GVGD, PolyPhen and SIFT algorithms, pathogenicity was evaluated in the 96 unclassified variants, by which 28 unclassified variants (in 55 participants) were classified as ‘predicted pathogenic’ and 68 (in 99 participants) as ‘predicted non-pathogenic’. To further evaluate our predictions of pathogenicity, we compared the penetrance among 9015 first-degree relatives of 1521 FOTS participants by mutation subtype. Results showed that cumulative risk to age 80 years of any cancer in those with pathogenic mutations was 56.7% compared with 44.2, 21.0 and 27.4% for those in the ‘predicted pathogenic’, predicted ‘non-pathogenic’, and no mutations groups, respectively. A similar result was seen for the risk of developing HNPCC-related cancer, but the difference between the groups did not reach customary statistical significance (Table 3). Cumulative risk of ovarian cancer among relatives of mutation carriers compared with relatives of non-carriers was not elevated, likely due to the limited number of relatives with ovarian cancer.
The average age of onset of ovarian cancer was 47.1 years in the ‘pathogenic group’ (Table 4) and 53.2 years in the ‘predicted pathogenic group’, compared with 56.1 years in those with no mutations. Non-serous cancers comprised 77.8% in the ‘pathogenic group’ and 66% in the ‘predicted pathogenic group’, compared with 41.9% in those with no mutations. The number of relatives with HNPCC-related cancers was highest in the ‘pathogenic’ group; however, findings in the other three groups indicated similar proportions. Of note, the proportion of mutations in the four mutation subgroups was similar across study sites (P=0.2). Specifically, the proportion of mutation carriers in the Moffitt, Duke and Toronto samples was 0.8%, 0.4% and 0.5%, respectively, despite the longer time from diagnosis to enrolment in the Toronto cases.
Finally, review of family histories of those with pathogenic and predicted pathogenic mutations indicated the two families that met clinical criteria for HNPCC (Vasen et al, 1999) carried pathogenic MLH1 mutations. Furthermore, the two families with pathogenic MSH2 mutations had striking family histories, despite not meeting clinical criteria. All remaining families did not meet clinical criteria.
Discussion
We determined the frequency of germline mutations in three MMR genes in a population-based sample of 1893 women with ovarian cancer. Our findings suggest that pathogenic germline MMR mutations are found in less than 1% of unselected ovarian cancer cases, with a higher frequency in those with invasive cancers of the endometrioid and clear cell subtypes. Of the nine pathogenic mutations clearly identified, the majority (55%) were detected in the MSH6 gene.
The pathogenic mutations clearly identified in our study were classified based on protein truncation or on previous epidemiologic or functional studies. Our findings likely represent an underestimate of the true proportion, as there were several missense variants for which pathogenicity could not be definitively determined. Furthermore, we did not test for additional less common MMR genes (PMS2, EPCAM) or for large gene rearrangements, which may have detected additional pathogenic mutations (de Jong et al, 2004; Grabowski et al, 2005; van der Klift et al, 2005; Gylling et al, 2009; Kovacs et al, 2009).
For the many missense variants detected in the study, databases such as UniProt (www.uniprot.org) and Human Genome Mutation Database (www.hgmd.org) were explored to determine pathogenicity. Of note, UniProt, a protein database, had many errors in functional classification of missense variants and, thus, is unreliable for definitively determining the functional effect of individual variants. Similarly, the Human Genome Mutation Database, a manually curated database, classifies many missense mutations as disease-causing without providing compelling evidence from the literature to support the assignments. Consequently, we focused on bioinformatic tools to establish the pathogenicity of missense variants (Tavtigian et al, 2008). Specifically, we used the Align-GVGD (Tavtigian et al, 2006) algorithm by means of a manually curated protein alignment across species of broad phylogenic scope in combination with a stringent cut-off point to predict the dysfunctional effect of a variant. This method outperforms other bioinformatic tools (Akbari et al, 2011) such as SIFT (Ng and Henikoff, 2003) and PolyPhen2 (Adzhubei et al, 2010). However, we used the combination of these three tools for predicting the functional effect of the missense variants. We also looked at the carrier frequency of these variants in ESP6500 database and considered the frequently reported ones as predicted ‘non-pathogenic’. We detected 28 missense mutations (in 55 participants) classified as ‘predicted pathogenic’ using these tools. Thus, if all of these are in fact pathogenic, the prevalence of MMR gene mutation carriers among ovarian cancer patients would be 3.6% or higher. Of note, we chose not use the MAPP-MMR tool (Chao et al, 2008; which is another bioinformatics tool with no clear advantage to other tools), because it only predicts the functional effects in MLH1 and MSH2 genes, whereas over 40% of the missense variants identified in our sample were in MSH6. Furthermore, because of conflicting reports in the literature regarding pathogenicity of certain variants (e.g., MLH1 p.Lys618A1a, observed in 24 participants in our study, was previously reported as both non-pathogenic (Raevaara et al, 2005) and pathogenic; Pastrello et al, 2011), we reported predictions of pathogenicity according to aforementioned criteria rather than based on prior published reports, which are in essence based on predictions.
Prior estimates of the frequency of germline MMR mutations in ovarian cancer patients have been less than 5% (Rubin et al, 1998; Stratton et al, 1999), based on studies of 116 patients or fewer and limited to the MLH1 and MSH2 genes only. Moreover, one of the studies was restricted to women aged 30 years or less at diagnosis (Stratton et al, 1999). Thus, although our prevalence estimates are generally lower than prior reports, the validity and generalisability of our data is enhanced by the much larger sample size, performance of testing in all three genes (i.e., including MSH6) and inclusion criteria that encompassed a wider age group of participants.
Although most studies have focused on MLH1 and MSH2, several have reported ovarian cancer in the MSH6 tumour spectrum (Miyaki et al, 1997; Wu et al, 1999; Wagner et al, 2001; Bonadona et al, 2011). However, in contrast to prior reports suggesting only 10% of HNPCC families harbour MSH6 mutations (Wijnen et al, 1999; Berends et al, 2002), such mutations accounted for over half of the clearly pathogenic mutations in our study. The low percentage of MSH6 mutations may be due to inadequate clinical criteria to accurately identify these families (Sjursen et al, 2010), who typically present with later age of colorectal cancer, and higher risk but later age of endometrial cancer in females (Wijnen et al, 1999; Wagner et al, 2001; Devlin et al, 2008; Ramsoekh et al, 2009). Thus, although infrequent in classical HNPCC families, our results suggest MSH6 mutations may be better represented in families with ovarian cancer. In fact, a recent study of 67 MLH1, MSH2 and MSH6 mutation carriers ascertained through a family cancer clinic included 10 ovarian cancers, of which the majority were seen in MSH6 carriers (Ramsoekh et al, 2009), consistent with our study findings. Interestingly, none of the families with pathogenic MSH6 mutations in our study met clinical criteria for HNPCC (Vasen et al, 1999). In contrast, both MLH1 families met clinical criteria, and both MSH2 families had striking family histories, despite not meeting clinical criteria. Of note, clinical criteria do not include cancers of the ovary and stomach, yet these cancers are much more common in HNPCC compared with those of the small bowel, ureter and renal pelvis, which are included. Ultimately, our study underscores the limitations of the use of HNPCC clinical diagnostic criteria based only on personal and family cancer history to identify MSH6 families.
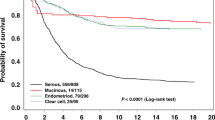
Our results were consistent with prior reports, suggesting an earlier age at diagnosis of ovarian cancer in carriers (mean ranging from 41 to 49 years) compared with sporadic cases (mean ranging from 60 to 65 years) (Watson and Lynch, 2001; Crijnen et al, 2005; Malander et al, 2006). Moreover, our age of diagnosis ranged from 40 to 59 years, which suggests that prophylactic surgery offered before age 40 years is likely to prevent the majority of HNPCC-associated ovarian cancers. Furthermore, the overrepresentation of non-serous tumours, particularly of endometrioid and clear cell subtypes (Pal et al, 2008a, 2008b; Ketabi et al, 2011), reported in HNPCC is consistent with our findings. Finally, as in our prior studies (Akbari et al, 2011), we used a cohort analysis to evaluate the Align-GVGD predictions. For example, if the predicted pathogenic mutations have the same penetrance, on average as the clearly pathogenic mutations, then we would expect to see cancer risks in first-degree relatives of patients in the two subgroups to be similar. In our study, although the penetrance of predicted pathogenic variants was slightly lower than the pathogenic variants, it was still consistently higher than the ‘predicted non-pathogenic’ and ‘no mutation’ groups, thus suggesting that many in this class are likely pathogenic. A deviation from this trend was seen in the lifetime risk of ovarian cancer illustrated in Table 3, which was lowest in the ‘pathogenic mutation’ group; however, this may be a spurious finding due to the limited number of relatives with ovarian cancer. Overall, the combined frequency of pathogenic and predicted pathogenic mutations was 3.6% (95% CI 2.9–4.6; Table 2). However, this frequency was 6.3% (95% CI 4.3–9.2) when considering only invasive cancers of the endometrioid and clear cell subtypes.
A number of strengths support the current study, including the large sample size, the population-based study design and comprehensive collection of clinical and demographic data. Despite these strengths, there remain some limitations, including our testing strategy that may have led to underestimating mutation frequency. Specifically, because of limited resources, large rearrangement testing was not performed, which may account for 10–20% of mutations in the MMR genes (Grabowski et al, 2005; van der Klift et al, 2005; Gylling et al, 2009). Similarly, testing for the two additional MMR genes, which has recently become available (i.e., PMS2 (de Jong et al, 2004) and EPCAM (Kovacs et al, 2009)) was also not performed in the current study. In addition, differences in demographic and clinical variables across sites (as reported in Table 1) could potentially affect our results. Notably, the time to diagnosis in FOTS cases was significantly longer than that for TBOCS or NCOCS, leading to overrepresentation of cases with longer survival in the FOTS data set. However, the main concern here is whether a longer time to diagnosis led to overrepresentation of mutation-positive cases in the FOTS data set, thereby biasing our mutation prevalence estimate. If this were true, the proportion of mutation-positive cases in the FOTS sample would be higher, which was not the case (as illustrated in Table 4). Therefore, it appears the longer time to diagnosis in the FOTS data set did not appear to substantially affect the mutation prevalence estimate. Nevertheless, the presence of survival bias within the FOTS data may have skewed the clinical characteristics within the overall data set as suggested by the higher proportions of non-serous cases observed. However, there remained an association between non-serous histologies and HNPCC mutations, which was likely underestimated because of the lower numbers of serous cases within the data set. Finally, we were limited in our ability to predict pathogenicity in many of the missense mutations because of sparse data available in the literature and mutation databases. To address this, we used an in silico approach to predict the dysfunctional effect of the detected missense variants, recognising that inherent inaccuracy of bioinformatics tools could result in some misclassification of the variants. However, our findings suggest that many are likely to be pathogenic based on similarities in the clinical and demographic characteristics with the pathogenic group (as shown in Tables 3 and 4).
In summary, we estimate that less than 1% of unselected ovarian cancer patients may have mutations in the MMR genes, with overrepresentation of MSH6 mutations in those with invasive cancers of the endometrioid and clear cell subtypes. Ovarian tumours with MMR-deficiency are characterised by early age of onset and overrepresentation of non-serous cancers. Our results suggest that current clinical criteria for HNPCC are insufficient to identify the majority of ovarian cancer patients with mutations. These findings highlight the need to consider HNPCC testing in women with ovarian cancer, particularly those with invasive cancers of the endometrioid and clear cell subtypes, even if they do not meet strict clinical criteria for the condition.
Change history
26 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249
Akbari MR, Zhang S, Fan I, Royer R, Li S, Risch H, McLaughlin J, Rosen B, Sun P, Narod SA (2011) Clinical impact of unclassified variants of the BRCA1 and BRCA2 genes. J Med Genet 48: 783–786
Berends MJ, Wu Y, Sijmons RH, Mensink RG, van der Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, van der Zee AG, Hofstra RM, Kleibeuker JH (2002) Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 70: 26–37
Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Nogues C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frebourg T, Sobol H, Lasset C, Bonaiti-Pellie C (2011) Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305: 2304–2310
Chao EC, Velasquez JL, Witherspoon MS, Rozek LS, Peel D, Ng P, Gruber SB, Watson P, Rennert G, Anton-Culver H, Lynch H, Lipkin SM (2008) Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Hum Mutat 29: 852–860
Crijnen TE, Janssen-Heijnen ML, Gelderblom H, Morreau J, Nooij MA, Kenter GG, Vasen HF (2005) Survival of patients with ovarian cancer due to a mismatch repair defect. Fam Cancer 4: 301–305
de Jong AE, van Puijenbroek M, Hendriks Y, Tops C, Wijnen J, Ausems MG, Meijers-Heijboer H, Wagner A, van Os TA, Brocker-Vriends AH, Vasen HF, Morreau H (2004) Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res 10: 972–980
Devlin LA, Graham CA, Price JH, Morrison PJ (2008) Germline MSH6 mutations are more prevalent in endometrial cancer patient cohorts than hereditary non polyposis colorectal cancer cohorts. Ulster Med J 77: 25–30
Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ, Goldgar DE (2007) A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 81: 873–883
Elsaleh H, Joseph D, Grieu F, Zeps N, Spry N, Iacopetta B (2000) Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet 355: 1745–1750
Elsaleh H, Powell B, McCaul K, Grieu F, Grant R, Joseph D, Iacopetta B (2001) P53 alteration and microsatellite instability have predictive value for survival benefit from chemotherapy in stage III colorectal carcinoma. Clin Cancer Res 7: 1343–1349
Grabowski M, Mueller-Koch Y, Grasbon-Frodl E, Koehler U, Keller G, Vogelsang H, Dietmaier W, Kopp R, Siebers U, Schmitt W, Neitzel B, Gruber M, Doerner C, Kerker B, Ruemmele P, Henke G, Holinski-Feder E (2005) Deletions account for 17% of pathogenic germline alterations in MLH1 and MSH2 in hereditary nonpolyposis colorectal cancer (HNPCC) families. Genet Test 9: 138–146
Gylling A, Ridanpaa M, Vierimaa O, Aittomaki K, Avela K, Kaariainen H, Laivuori H, Poyhonen M, Sallinen SL, Wallgren-Pettersson C, Jarvinen HJ, Mecklin JP, Peltomaki P (2009) Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer 124: 2333–2340
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, Panescu J, Fix D, Lockman J, LaJeunesse J, Comeras I, de la Chapelle A (2008) Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 26: 5783–5788
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la Chapelle A (2005) Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352: 1851–1860
Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H (2000) Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology 119: 921–928
Ketabi Z, Bartuma K, Bernstein I, Malander S, Gronberg H, Bjorck E, Holck S, Nilbert M (2011) Ovarian cancer linked to Lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol Oncol 121: 462–465
Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E (2009) Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 30: 197–203
Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR (2009) Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet 76: 1–18
Lynch PM (2006) Current approaches in familial colorectal cancer: a clinical perspective. J Natl Compr Cancer Netw 4: 421–430
Malander S, Rambech E, Kristoffersson U, Halvarsson B, Ridderheim M, Borg A, Nilbert M (2006) The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol Oncol 101: 238–243
Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T (1997) Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17: 271–272
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814
Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005) BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104: 2807–2816
Pal T, Permuth-Wey J, Kumar A, Sellers TA (2008a) Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin Cancer Res 14: 6847–6854
Pal T, Permuth-Wey J, Sellers TA (2008b) A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 113: 733–742
Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomaki P, Mecklin JP, de la Chapelle A, Kinzler KW, Vogelstein B (1994) Mutation of a mutL homolog in hereditary colon cancer. Science 263: 1625–1629
Pastrello C, Pin E, Marroni F, Bedin C, Fornasarig M, Tibiletti MG, Oliani C, Ponz de Leon M, Urso ED, Della Puppa L, Agostini M, Viel A (2011) Integrated analysis of unclassified variants in mismatch repair genes. Genet Med 13: 115–124
Peltomaki P, Vasen HF (1997) Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 113: 1146–1158
Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomaki P, Kohonen-Ccorish M, Mangold E, Macrae F, Greenblatt M, de la Chapelle A, Nystrom M (2005) Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology 129: 537–549
Ramsoekh D, Wagner A, van Leerdam ME, Dooijes D, Tops CM, Steyerberg EW, Kuipers EJ (2009) Cancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical management. Hered Cancer Clin Pract 7: 17
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B, Narod SA (2001) Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68: 700–710
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users for biologist programmers. In Methods Mol Biol Misener S, Krawetz SA, (eds) pp 365–386. Humana Press: Totowa, NJ
Rubin SC, Blackwood MA, Bandera C, Behbakht K, Benjamin I, Rebbeck TR, Boyd J (1998) BRCA1, BRCA2 and hereditary nonpolyposis colorectal cancer gene mutations in an unselected ovarian cancer population: relationship to family history and implications for genetic testing. Am J Obstet Gynecol 178: 670–677
Sjursen W, Haukanes BI, Grindedal EM, Aarset H, Stormorken A, Engebretsen LF, Jonsrud C, Bjornevoll I, Andresen PA, Ariansen S, Lavik LA, Gilde B, Bowitz-Lothe IM, Maehle L, Moller P (2010) Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet 47: 579–585
Stratton JF, Thompson D, Bobrow L, Dalal N, Gore M, Bishop DT, Scott I, Evans G, Daly P, Easton DF, Ponder BA (1999) The genetic epidemiology of early-onset epithelial ovarian cancer: a population-based study. Am J Hum Genet 65: 1725–1732
Syngal S, Weeks JC, Schrag D, Garber JE, Kuntz KM (1998) Benefits of colonoscopic surveillance and prophylactic colectomy in patients with hereditary nonpolyposis colorectal cancer mutations. Ann Intern Med 129: 787–796
Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A (2006) Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet 43: 295–305
Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB (2008) In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat 29: 1327–1336
Taylor KJ, Schwartz PE (1994) Screening for early ovarian cancer. Radiology 192: 1–10
van der Klift H, Wijnen J, Wagner A, Verkuilen P, Tops C, Otway R, Kohonen-Corish M, Vasen H, Oliani C, Barana D, Moller P, Delozier-Blanchet C, Hutter P, Foulkes W, Lynch H, Burn J, Moslein G, Fodde R (2005) Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 44: 123–138
Vasen HF (2005) Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 4: 219–225
Vasen HF, van Ballegooijen M, Buskens E, Kleibeuker JK, Taal BG, Griffioen G, Nagengast FM, Menko FH, Meera Khan P (1998) A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer 82: 1632–1637
Vasen HF, Watson P, Mecklin JP, Lynch HT (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116: 1453–1456
Wagner A, Hendriks Y, Meijers-Heijboer EJ, de Leeuw WJ, Morreau H, Hofstra R, Tops C, Bik E, Brocker-Vriends AH, van Der Meer C, Lindhout D, Vasen HF, Breuning MH, Cornelisse CJ, van Krimpen C, Niermeijer MF, Zwinderman AH, Wijnen J, Fodde R (2001) Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet 38: 318–322
Watson P, Lynch HT (2001) Cancer risk in mismatch repair gene mutation carriers. Fam Cancer 1: 57–60
Wenham RM, Schildkraut JM, McLean K, Calingaert B, RC Bentley, Marks J, Berchuck A (2003) Polymorphisms in BRCA1 and BRCA2 and risk of epithelial ovarian cancer. Clin Cancer Res 9: 4396–4403
Whittemore AS, Harris R, Itnyre J (1992) Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. II. Invasive epithelial ovarian cancers in white women. Collaborative Ovarian Cancer Group. Am J Epidemiol 136: 1184–1203
Wijnen J, de Leeuw W, Vasen H, van der Klift H, Moller P, Stormorken A, Meijers-Heijboer H, Lindhout D, Menko F, Vossen S, Moslein G, Tops C, Brocker-Vriends A, Wu Y, Hofstra R, Sijmons R, Cornelisse C, Morreau H, Fodde R (1999) Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet 23: 142–144
Wu Y, Berends MJ, Mensink RG, Kempinga C, Sijmons RH, van Der Zee AG, Hollema H, Kleibeuker JH, Buys CH, Hofstra RM (1999) Association of hereditary nonpolyposis colorectal cancer-related tumours displaying low microsatellite instability with MSH6 germline mutations. Am J Hum Genet 65: 1291–1298
Acknowledgements
This study was supported by grants R01 CA111914 (TP), K07 CA108987 (TP), R01 CA063682 (HAR), R01 CA063678 (SN) and R01 CA080978 (SN) from the National Cancer Institute. The sequencing was done by the Genome Quebec Innovation Centre at McGill University.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Pal, T., Akbari, M., Sun, P. et al. Frequency of mutations in mismatch repair genes in a population-based study of women with ovarian cancer. Br J Cancer 107, 1783–1790 (2012). https://doi.org/10.1038/bjc.2012.452
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.452
Keywords
This article is cited by
-
An epigenetic hypothesis for ovarian cancer prevention by oral contraceptive pill use
Clinical Epigenetics (2023)
-
Impact of germline mutations in cancer-predisposing genes on long-term survival in patients with epithelial ovarian cancer
British Journal of Cancer (2022)
-
Comparison of universal screening in major lynch-associated tumors: a systematic review of literature
Familial Cancer (2022)
-
Identifying sequence variants contributing to hereditary breast and ovarian cancer in BRCA1 and BRCA2 negative breast and ovarian cancer patients
Scientific Reports (2019)
-
Predictors of next-generation sequencing panel selection using a shared decision-making approach
npj Genomic Medicine (2018)
