
Abstract
Background:
Using mRNA expression-derived signatures as predictors of individual patient outcome has been a goal ever since the introduction of microarrays. Here, we addressed whether analyses of tumour mRNA at the exon level can improve on the predictive power and classification accuracy of gene-based expression profiles using neuroblastoma as a model.
Methods:
In a patient cohort comprising 113 primary neuroblastoma specimens expression profiling using exon-level analyses was performed to define predictive signatures using various machine-learning techniques. Alternative transcript use was calculated from relative exon expression. Validation of alternative transcripts was achieved using qPCR- and cell-based approaches.
Results:
Both predictors derived from the gene or the exon levels resulted in prediction accuracies >80% for both event-free and overall survival and proved as independent prognostic markers in multivariate analyses. Alternative transcript use was most prominently linked to the amplification status of the MYCN oncogene, expression of the TrkA/NTRK1 neurotrophin receptor and survival.
Conclusion:
As exon level-based prediction yields comparable, but not significantly better, prediction accuracy than gene expression-based predictors, gene-based assays seem to be sufficiently precise for predicting outcome of neuroblastoma patients. However, exon-level analyses provide added knowledge by identifying alternative transcript use, which should deepen the understanding of neuroblastoma biology.
Similar content being viewed by others


Main
Neuroblastoma is a childhood tumour with a wide range of clinical courses, spanning from spontaneous differentiation to rapid progression and fatal outcome. A few genetic alterations have been described to be causative for neuroblastoma including mutations of the genes encoding ALK (Chen et al, 2008b; George et al, 2008; Mosse et al, 2008) or PHOX2B (reviewed in Janoueix-Lerosey et al (2010); Maris (2010)). Genomic sequencing of primary neuroblastoma recently revealed an enrichment for mutations in genes involved in neuritogenesis (Molenaar et al, 2012). However, the origin of the majority of spontaneous neuroblastomas remains unclear (Park et al, 2008). One subgroup of aggressive neuroblastomas is characterised by amplification of the MYCN oncogene, which occurs in about 20–25% of all primary neuroblastoma (Seeger et al, 1985). In the absence of the MYCN amplification, it remains difficult to define the individual risk of relapse and assign adequate therapy (Maris et al, 2007). Expression profiling has been performed in recent years to identify signatures predictive of outcome, and these signatures are still being refined for the use in clinically applicable settings (Wei et al, 2004; Ohira et al, 2005; Schramm et al, 2005b; Oberthuer et al, 2006, 2010). A comparison of published microarray studies has revealed little overlap of genes within signatures identified to predict neuroblastoma outcome, although integration of data seemed possible (Chen et al, 2008a). Several explanations for the diversity of genes in these signatures have been suggested such as the use of different technical platforms for expression profiling, the choice of prediction algorithms and inter-laboratory variation (Irizarry et al, 2005). It has also been argued that genes in the same pathways are co-regulated implying a similar predictive value and redundancy of information. Still, the fact that genes are used by one predictor and are absent in others may be a function of the probes being derived from different parts of the same gene for the different technical platforms. Methods to overcome this limitation of gene-based expression profiling include RNA sequencing or exon-level mRNA expression analyses. Here, we chose Affymetrix HuEx1.0 arrays that were designed to interrogate every known and predicted exon in the human genome. Reporter sequences that extend beyond protein coding regions to other areas of the transcribed genome are also included on these arrays. In principle, the combination of coverage and precise localisation information allows for analyses of regulation on the sub-gene level. This is important as a substantial proportion of protein coding genes are predicted to be alternatively spliced. In neuroblastomas, alternative splicing of the NTRK1/TrkA receptor has already been reported to have an impact on tumour aggressiveness (Tacconelli et al, 2004), suggesting that the phenomenon of differential transcripts use has a major impact on neuroblastoma biology. In this study, we wanted to explore whether analyses of mRNA expression resolved to the exon level was capable of improving prognostic prediction of neuroblastoma patient outcome above the level currently achieved using gene expression-based predictors. We also aimed to identify factors associated with alternative exon use in neuroblastoma and sought to gain novel biological insights into neuroblastoma biology by exon-level gene expression analyses.
Materials and methods
Patient cohort
The 113 primary neuroblastoma specimens were from tumour banks in Cologne and Essen, Germany, Ghent, Belgium and Valencia, Spain. All patients were diagnosed between 1998 and 2007 and treated according to the German neuroblastoma trials NB97, NB 2004 or the respective SIOPEN protocol. Written informed consent was obtained from patients or their parents. As many patients were identified within general neuroblastoma screening in Germany (Schilling et al, 2002), patients were selected to recreate a representative distribution of tumours stages. Of the 113 NB patients enclosed in our study, 57 presented with localised disease (stage 1: n=31; stage 2: n=11; and stage 3: n=15) and 56 with metastatic disease at the time of diagnosis. The mean and median ages of our patient cohort at diagnosis were 934 and 472 days, respectively. Approximately two third of patients were >1 year of age at the time of diagnosis (n=72) and the remainder (n=41) were <1 year. Amplification of the MYCN oncogene occurred in 21 patients. Median and mean follow-up were 2066 and 1976 days, respectively. In total, 26 patients died of their disease. Patient characteristics are summarised in Table 1.
RNA preparation
Representative areas of histologically confirmed, snap-frozen neuroblastomas were cut on dry ice. No preselection for tumour cell fraction or microenvironment was performed. Total RNA extraction of NB tumour samples was performed in by silica gel-based membrane purification methods (RNeasy Mini kit or MicroRNeasy kit, Qiagen, Hilden, Germany) according to the manufacturers’ instructions. RNA quality was controlled by capillary gel electrophoresis using either the Experion system (high-sensitivity chips, Bio-Rad, Munich, Germany) or the Bioanalyser 2100 (RNA nano kits, Agilent, Palo Alto, CA, USA).
Array hybridisations
Fragmentation of cRNA and hybridisation to HuEx1.0 microarrays (Affymetrix, Santa Clara, CA, USA), washing, staining and scanning of the arrays in a GeneArray scanner (Agilent) were basically performed as described previously (Schramm et al, 2005a, 2005b). Quality control of hybridisations and RMA normalisation was performed using Genomics Suite (Partek Inc., St. Louis, MO, USA) using the default settings. Data were stored in the R2 microarray analysis and visualisation platform (AMC Amsterdam, http://r2.amc.nl, Amsterdam, The Netherlands) for exploration and visualisation. Microarray data have been submitted to the GEO database (acc. no. GSE32664).
Relapse prediction using shrunken centroids
A shrunken centroids (prediction analyses for microarrays (PAMs); Tibshirani et al, 2002) model was used to predict event-free survival (EFS) or relapse as well as overall survival (OS) or death using exon- and gene expression values. Two filter steps were applied before running the classification algorithms. First, all features of the core probe set having a 0.3-quantile score >5.0 were discarded. Then features that were significantly differentially expressed between two classes were identified using an F-test (significance level 0.05) to check for equal variances in each class for every exon or gene. Student’s t-test was performed for exons or genes with equal class variance and Welch's t-test was used for exons or genes with non-equal class variance to calculate P-values. These steps were repeated 20 times on bootstrap samples (drawing with replacement) from the data. Only exons and genes having P-values ⩽0.05 in at least 95% of the rounds were kept as significantly associated to the outcome or label and these data were used to build the shrunken centroids model, which further reduced the number of features by shrinking the centroids to the relevant features. Cross-validation delivered a shrinkage parameter of 1.2 as a sensible trade-off between prediction accuracy classifier size for both gene and exon data.
Shrunken centroids analysis
Implementations of support vector machines (SVMs) and PAMs in Rapid Miner 5.1 (Rapid-I, Dortmund, Germany) were used to predict patient outcome, and patients were divided into a training (n=73) and a test set (n=40). RapidMiner was applied in conjunction with the RapidMiner Feature Selection Extension. Analyses of maximum divergent courses (MDCs) were carried out on a subset of 63 patients. MDC was defined by patients with localised disease without MYCN amplification, which had no relapse on the one hand and patients with metastatic (INSS stage 4) disease who died of their disease on the other hand. Test set data were not used in any of the validation steps until the predictor was applied.
Detection of differential gene or exon expression
The ‘apt-probeset-summarise’ tool (Affymetrix Power Tools 1.10.0) was run to compute exon and gene expression values using the following analysis parameters on the core probe set: rma-bg (background correction as described in Irizarry et al (2003)), quant-norm (quantile normalisation), pm-only (no mismatch probe adjustments) and med-polish (median polish as described in Irizarry et al, (2003)). Genes and exons were discarded if their normalised log2 expression count was <5 for at least 1/3 of the patients. Relative exon expression was computed for each gene with at least two exons represented, and utilised all exons representing a given gene as r=(e−g)/g, where e is the normalised log2 exon expression and g is the normalised log2 expression of the gene. Differential expression between classes was determined by Welch's t-test, followed by false discovery correction according to Benjamini–Hochberg. Differential exon use in transcripts from the same coding gene was determined on relative exon expression levels. Genes or exons with a false discovery rate <0.05 were reported as significantly differentially expressed. To estimate differential exon use, significantly differentially expressed genes were discarded and differential exon expression between groups was then redetermined.
Cell culture and in vitro analyses
Cultivation of the human neuroblastoma cell lines IMR32, NB69, SH-EP, WAC2 and SH-EP MYC-ER was performed as described previously (Schramm et al, 2005c; Schulte et al, 2008). Induction of MYCN in SH-EP MYC-ER was achieved using 4-hydroxy tamoxifen as described (Schulte et al, 2008). Propodium iodide-based FACS analyses of DNA content was carried out as in (Eggert et al, 2001). Cell viability assays using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide were performed as described in (Schulte et al, 2005). RNA was harvested from subconfluently growing cells using the RNeasy Mini Kit (Qiagen) and cDNA was subsequently prepared as described (Schulte et al, 2005). Semiquantitative qPCR was performed using standard protocols on a ABI StepOnePlus Real-Time PCR system (Life Technologies, Carlsbad, CA, USA) using primers spanning INCENP exons 1–2 and 10–11, respectively (Life Technologies).
Results
Association of known risk factors with outcome in our study cohort
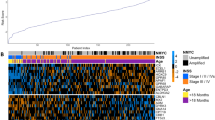
We first evaluated the distribution of known risk factors (Table 1) and their association with clinical outcome in the cohort of 113 neuroblastomas that were analysed by expression profiling in this study. The relative number of patients with MYCN amplification and metastatic disease at time of diagnosis resembled the situation found in the overall patient population. As expected, MYCN amplification, metastatic disease, age at diagnosis >1 year and low NTRK1 expression were all associated with a significantly worse outcome in univariate analyses (Figure 1).

Significant association of clinical and biological features with outcome (OS) as visualised by Kaplan–Meier analyses. (A) MYCN status (MYCN sc=MYCN single copy; MNA=MYCN-amplified); (B) metastatic spread (local vs metastatic); (C) age at diagnosis (age < or >1 year at diagnosis); and (D) TrkA expression (TrkA expression < or > median).
Comparison of the predictive power of gene-level or exon level-based predictors
In contrast to other studies focussing on only gene expression, we evaluated both gene- and exon-level expressions as the basis for a reliable predictor of neuroblastoma outcome. Tumour expression profiles from the 113 neuroblastoma patients (Table 1) were obtained using Affymetrix HuExST1.0 arrays. Expression data for both the gene and exon levels were processed and normalised as described in the Materials and methods section. Several methods were evaluated for prediction, including linear SVM and random trees/forests (data not shown), but PAM-based prediction was the most stable and reliable method. After filtering and enrichment for significantly differentially expressed genes, SVM- or PAM-based methods were used to build a predictor for EFS or OS using gene or exon expression data. Support vector machine-based predictors were less stable when data were split into a training set (n=73) and a test set (n=40, see also Materials and methods), which resulted in decrease in accuracy and specificity in the test set (data not shown). The PAM-based signature predicted OS with an accuracy of 83.1% (sensitivity 84.7%, specificity 82.6%, Table 2). When a published PAM-based gene set was used as input for prediction (Vermeulen et al, 2009a), accuracy was comparable (86.1%) but this was accompanied by a decrease in sensitivity, which dropped to 79.6%, whereas specificity was higher (89.4%). Prediction analyses for microarray utilising only the patients with MDCs only (n=63) produced the highest prediction accuracy (89.2%, sensitivity=85.2%, specificity=91.4%). Interestingly, both prediction accuracy and the individual classification were highly similar using either expression from the exon or gene levels and the same prediction method and data sets (Table 2).
Gene- and exon-derived PAM signatures are independent prognostic markers of survival in multivariate analyses
As we wanted to test the robustness of our predictors and to correlate them with known prognostic markers, backward logistic regression was performed. As the gene- and exon-derived signatures functioned similarly in predicting the individual patient outcome, we analysed their performance separately when building the regression models. Both gene- and exon-based predictors were the only significant independent predictors of OS in our cohort in multivariate analyses (Table 3, P=0.007, odds ratio=9.7, for the gene-based predictor; P=0.005, odds ratio=10.7, for the exon-based predictor).
Identification of factors associated with differential exon usage in neuroblastoma
We analysed the impact of clinical and biological features on both differential gene and exon expression with the aim to identify master regulators governing alternative transcript use. Using the filtering steps described in the Materials and Methods section, we first determined the number of differentially expressed genes associated with either INSS stages, age at diagnosis, MYCN amplification, survival or NTRK1 expression (Table 4). Survival, MYCN amplification and NTRK1 expression were associated with a massive shift of the transcriptome, reflected by significantly differential expression of >1000 genes each. This also held true when analyses were performed using expression at the exon level, which resulted in >10 000 significantly differentially used exons for each group. Interestingly, when differential exon use was assessed for genes, whose expression was not significantly altered between groups, NTRK1 expression and MYCN amplification status were strongly associated with alternative exon use. When tumours with high NTRK1 expression were compared with tumours with low NTRK1 expression, 471 exons were found to be differentially expressed, whereas MYCN amplification correlated with 306 differentially used exons. This finding suggests that both, MYCN and NTRK1, could be involved in governing alternative transcript use.
Validation of known and novel transcript variants in primary NB
We first validated a variant of the neurotrophin receptor NTRK1, designated TrkA-III, which has been previously associated with aggressive tumour behaviour in NB (Tacconelli et al, 2004), using our expression data. This variant lacks exons six, seven and nine resulting in altered NTRK1 signalling independent of ligand binding (Tacconelli et al, 2004). Differential expression of exons six and seven was already visually detectable from the individual tumour profiles plotted as a heatmap (Figure 2A). NTRK1 gene expression was highly correlated with survival, as expected, whereas expression of exons six or seven was not (Figures 2B and C). Notably, expression of NTRK1 is not only correlated to prognosis, but also to patient age: younger patients have higher NTRK1 levels than older patients. Thus, we could validate that differential NTRK1 variants are expressed in NB and that these variants correlate with different patient outcome for the first time in a large patient cohort.

Association of alternative exon usage in NTRK1 with outcome. (A) The heat map shows NTRK1 exon-level expression (rows, exon 1 on top) in our patient cohort. Exon 6/7 depicts the exons, for which the respective probes on the array are located. Red indicates exon upregulation and green indicates downregulation of an exon as compared with the median expression of the respective exon across all patients in our study. High NTRK1 expression was also correlated with young (<1 year) patient age. Age=patient age at diagnosis; green >1 year; red <1 year. (B) Differential association of NTRK1 exon expression with EFS or relapse. Left: NTRK1 exon 2 expression is high in EFS and low in patients with relapse (P=9 × 10−15). Right: correlation between NTRK1 exon 6 expression in NB patients with EFS is less pronounced although still significant (P=0.002). (C) Real-time PCR of NTRK1 exons 5–8 shows preferential expression in patients with EFS when compared with patients who experienced relapse (M=marker, − and + depict the negative and positive controls, respectively).
We also wanted to gain first insights into transcripts and novel transcript variants associated with unfavourable clinical features as these could serve as novel therapeutic targets. For this reason, we focussed on genes and exons, whose expression was linked to survival and the amplification status of MYCN (Table 4). Patterns of alternative exon use included different transcriptional start and stop sites as well as differential expression of isoforms from the same coding gene (Supplementary Figure 1). To validate the MYCN-induced regulation of one of these transcript isoforms, we selected the gene coding for INCENP, a member of the chromosomal passenger complex involved in mitotic progression. Statistical analyses suggested a significant upregulation of INCENP exons 1–5 corresponding to the INCENP transcript variant, NM_020238, in patients with MYCN-amplified tumours (Figure 3A). This is exemplified for exon 5, whose expression was significantly associated with MYCN amplification (P<0.001, Figure 3B), whereas no difference in INCENP exon 15 expression was observed between tumours with and without MYCN amplification (Figure 3C). INCENP was also induced upon MYCN activation or stable overexpression in vitro in two cell model systems based on the SH-EP NB cell line, which does not express detectable levels of MYCN. We used SH-EP derivates engineered to stably or inducibly overexpress MYCN (designated WACII and SH-EP MYCN-ER, respectively, Figure 3D). Although PCR-based assays validated INCENP as a MYCN-inducible target in vitro, specific isoforms were not differentially induced in assays specifically measuring both INCENP transcript variants compared with the longer INCENP transcript variant (NM_020238_2, Figure 3D, right). Thus, INCENP could be validated as a MYCN-inducible target in vitro, confirming the data obtained from the primary tumours, but an isoform-specific induction of INCENP variants by MYCN remains to be confirmed.

Expression pattern of the INCENP gene in primary NB and in in vitro NB models. (A) A shorter INCENP isoform comprising only exons 1–5 (NM_020238) was preferentially expressed in MYCN-amplified patients, whereas expression differences were less pronounced for the longer INCENP transcript NM_020238_2. Data are presented as log2 of mean expression in each group ±s.e.m. (B) Expression of INCENP exon 5 was significantly elevated in MYCN-amplified patients (MNA) when compared with patients with normal MYCN (MYCNsc), whereas expression of exon 15 was not (C). (D) Semiquantitative real-time PCR revealed that expression of INCENP exons 1–2 and 10–11 was upregulated in vitro in a MYCN-negative NB cell lines, SH-EP, upon MYCN overexpression (WAC2) or MYCN induction (SH-EP MYCN+).
Interestingly, alternative exon usage of the first exons of the cell cycle regulating genes CDKN2B and CCNB1 was significantly different between aggressive, deadly or MYCN-amplified tumours and patients with EFS or normal MYCN status (Figures 4A and B). The most 5′ probe sets of CDKN2B are expressed at very low levels, so a functional role for the observed expression difference remains to be determined. The expression of a shorter CCNB1 isoform was verified in neuroblastoma cell lines (Figure 4C) and inhibition of Cdk1/CCNB1 interaction using the RO3306 small molecule inhibitor resulted in an increase in the fraction of apoptotic cells (Figure 4 D). Taken together, differential expression of transcript variants suggested from the analyses of primary tumours could be validated in the in vitro models.

Differential transcript expression of the cell-cycle-associated genes, p15INK4B (CDKN2B) and cyclin B1 (CCNB1). (A) CDKN2B isoforms are differentially regulated between tumours with and without MYCN amplification, but not between tumours from patients with EFS and non-survivors (DoD). (B) A short isoform of CCNB1 (red line) is preferentially expressed in tumours from non-survivors (DoD). For both, A and B, data are presented as log2 of mean expression in each group ±s.e.m. (C) Higher expression of this shorter CCNB1 isoform was also detected in NB cell lines. (D) Inhibition of the CCNB1/Cdk1 interaction by a small molecule inhibitor induced apoptosis in these cell lines.
Discussion
Prediction of the individual risk and consequent adaptation of treatment is one promise of personalised cancer medicine. Using gene expression profiling for individual treatment decisions has been investigated for a decade now for many cancer types and also for neuroblastoma. Although early studies published in this field suffered from data overfitting and lack of general applicability, large studies have meanwhile provided evidence for the high sensitivity and accuracy of gene expression-based classifiers using different platforms (Vermeulen et al, 2009a, 2009b; Oberthuer et al, 2010). International consortia such as MAQC have defined standard operating procedures for validating microarray data (Shi et al, 2010). Although advances have been achieved for both technical improvements as well as standardisation, concerns still remain on the clinical usability of classifiers derived from expression profiling. Here, we addressed whether the higher information density of expression profiling at the exon level rather than the gene-level expression could increase the power of tumour expression-based outcome prediction. For this purpose, we used a cohort of 113 neuroblastoma patients, which were profiled using Affymetrix Exon Arrays (HuEx1.0ST). Using exon-level expression achieved comparable prediction accuracy, sensitivity and specificity for both OS and EFS compared with using gene-level expression. These findings also held true, when a published PAM-derived signature of 59 genes (Vermeulen et al, 2009a) and the exons making up these genes were used to predict outcome in this cohort. Prediction accuracy increased when patient cohorts with maximum diversity (MDCs) in clinical courses were used. For the best clinical courses, we only included patients with localised disease without MYCN amplification, which had never experienced a relapse. Patients with the worst clinical course had metastatic spread at time of diagnosis (INSS stage 4) and were eventually succumbing to their disease. As previously noticed, survival prediction is possible with near perfect accuracy whereas prediction of relapse is more difficult. One reason for this phenomenon could be that MYCN amplification is a major risk factor for relapsing neuroblastoma and results in massive reprogramming of the tumour transcriptome to create this aggressive tumour phenotype. However, MYCN amplification alone cannot account for all aggressive, relapsing courses of the disease. Given the heterogeneity of transcriptomes from tumours with and without MYCN amplification, the definition of a perfect relapse signature is still an unresolved task. Nevertheless, the gene- and exon-derived signatures in our study were the only independent markers of survival in multivariate analyses, corroborating their potential usefulness for patient stratification and, in the long run, individually adapted risk assessment.
Exon-level profiling promises to yield unprecedented insights into the fine tuning of transcript use associated with aggressive or benign tumours. MYCN amplification has long been linked to aggressiveness in NB (Schwab et al, 1983; Seeger et al, 1985), but has only recently been associated with alternative exon use in primary NB (Guo et al, 2011). First mechanistic insights into MYC-driven splice site usage were provided by linking alternative splicing of the Raf kinase to differential MYC expression (Rauch et al, 2011). Here, we identified NTRK1 as a second biological parameter governing gene composition, when patients with tumours strongly expressing NTRK1 were compared with patients whose tumours weakly expressed NTRK1. High NTRK1 expression, which has previously been shown to be associated with outcome in NB (Nakagawara et al, 1993), was also correlated with excellent outcome in our data set (Figure 2A). Of note, expression of NTRK1 exons 6 and 7 was even lower in advanced stage tumours (Figure 2C), confirming previous results (Tacconelli et al, 2004).
We identified differential expression of >200 predicted alternative transcripts between neuroblastomas with and without MYCN amplification or with high and low NTRK1 expression. Thus, we are still at the beginning to understand the global changes induced by MYCN and NTRK1 in neuroblastoma. The interplay of these factors is complicated by the fact that MYCN can directly bind to the NTRK1 promoter and negatively regulate NTRK1 expression (Iraci et al, 2011). Although first attempts have been made to catalogue alternative splice site usage in oncogenic kinases (Druillennec et al, 2012), we are still far from having an overview of the functional relevance of all splicing changes intrinsic to aggressive cancer cells. Moreover, it remains to be determined whether alternative transcript use rather than alternative splicing has a role in determining the expression profiles underlying tumour aggressiveness. In neuronal development, alternative transcript use was identified as a major cause for transcriptome diversity (Pal et al, 2011), and similar patterns were observed in medulloblastomas (Menghi et al, 2011). It is evident that the altered tumour transcriptome results from aberrant transcripts due to mutations, aberrant transcript amounts from changes in regulatory networks and also from alternative transcript use, but how the latter is produced must be analysed in more tumour entities to evaluate its contribution to cancer biology.
In neuroblastoma, there are hints linking functional modules such as cell cycle regulation (Murphy et al, 2011) and protein biosynthesis (Boon et al, 2001) to MYCN and aggressive tumour behaviour. Moreover, gene expression of members of the chromosomal passenger complex, most prominently survivin, has been previously linked to NB. Survivin upregulation was linked to neuroblastoma aggressiveness (Islam et al, 2000), but this could be attributed to an unspecific gain of chromosome 17q, which occurs than half of all neuroblastomas (Bown et al, 1999). Here, we provide first evidence that a second member of the chromosomal passenger complex, INCENP, is a potential MYCN target in neuroblastoma as INCENP mRNA was upregulated upon MYCN induction in vitro as well as in patients having tumours with amplified MYCN. In addition, exon-level analyses of the INCENP coding region suggested a shorter hypothetical INCENP isoform differentially expressed between tumours with and without MYCN amplification. However, the existence of such an isoform, for which only a cDNA clone has previously been described, remains to be proven experimentally. In vitro studies corroborated MYCN-induced upregulation of INCENP exons 1–2 and 11–12. Nevertheless, it is intriguing to speculate that transcriptional reprogramming of aggressive tumour cells may not only cause differential gene expression but also alternative transcript use, which could here be exemplified for a CCNB1 variant.
In summary, expression signatures based on the gene or exon level proved to be equally powerful in predicting the clinical outcome of neuroblastoma patients, rendering gene-based assays as sufficiently precise for potential diagnostic purposes. The power of exon-based expression analyses lies in the identification of alternatively used transcripts in tumours, which can provide new insights into the aberrant circuits of the most aggressive tumour fraction, harbouring a plethora of options for therapeutic interventions.
Change history
04 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voute PA, Schwab M, Versteeg R (2001) N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J 20 (6): 1383–1393
Bown N, Cotterill S, Lastowska M, O'Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, Nicholson J, Bernheim A, Betts DR, Vandesompele J, Van Roy N (1999) Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med 340 (25): 1954–1961
Chen QR, Song YK, Wei JS, Bilke S, Asgharzadeh S, Seeger RC, Khan J (2008a) An integrated cross-platform prognosis study on neuroblastoma patients. Genomics 92: 195–203
Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S (2008b) Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455 (7215): 971–974
Druillennec S, Dorard C, Eychene A (2012) Alternative splicing in oncogenic kinases: from physiological functions to cancer. J Nucleic Acids 2012: 639062
Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM (2001) Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res 61 (4): 1314–1319
George RE, Sanda T, Hanna M, Frohling S, Luther W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT (2008) Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455 (7215): 975–978
Guo X, Chen QR, Song YK, Wei JS, Khan J (2011) Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and MYCN amplification status. BMC Med Genomics 4: 35
Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, Herold S, Eilers M, Bernardoni R, Della Valle G, Perini G (2011) A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res 71 (2): 404–412
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4 (2): 249–264
Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, Griffin C, Hilmer SC, Hoffman E, Jedlicka AE, Kawasaki E, Martinez-Murillo F, Morsberger L, Lee H, Petersen D, Quackenbush J, Scott A, Wilson M, Yang Y, Ye SQ, Yu W (2005) Multiple-laboratory comparison of microarray platforms. Nat Methods 2 (5): 345–350
Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y, Nakagawara A (2000) High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 19 (5): 617–623
Janoueix-Lerosey I, Schleiermacher G, Delattre O (2010) Molecular pathogenesis of peripheral neuroblastic tumors. Oncogene 29 (11): 1566–1579
Maris JM (2010) Recent advances in neuroblastoma. N Engl J Med 362 (23): 2202–2211
Maris JM, Hogarty MD, Bagatell R, Cohn SL (2007) Neuroblastoma. Lancet 369 (9579): 2106–2120
Menghi F, Jacques TS, Barenco M, Schwalbe EC, Clifford SC, Hubank M, Ham J (2011) Genome-wide analysis of alternative splicing in medulloblastoma identifies splicing patterns characteristic of normal cerebellar development. Cancer Res 71 (6): 2045–2055
Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R (2012) Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483: 589–593
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 (7215): 930–935
Murphy DM, Buckley PG, Bryan K, Watters KM, Koster J, van Sluis P, Molenaar J, Versteeg R, Stallings RL (2011) Dissection of the oncogenic MYCN transcriptional network reveals a large set of clinically relevant cell cycle genes as drivers of neuroblastoma tumorigenesis. Mol Carcinog 50 (6): 403–411
Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM (1993) Association between high levels of expression of the Trk gene and favorable outcome in human neuroblastomas. N Engl J Med 328: 847–854
Oberthuer A, Berthold F, Warnat P, Hero B, Kahlert Y, Spitz R, Ernestus K, Konig R, Haas S, Eils R, Schwab M, Brors B, Westermann F, Fischer M (2006) Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol 24 (31): 5070–5078
Oberthuer A, Hero B, Berthold F, Juraeva D, Faldum A, Kahlert Y, Asgharzadeh S, Seeger R, Scaruffi P, Tonini GP, Janoueix-Lerosey I, Delattre O, Schleiermacher G, Vandesompele J, Vermeulen J, Speleman F, Noquera R, Piqueras M, Benard J, Valent A, Avigad S, Yaniv I, Weber A, Christiansen H, Grundy RG, Schardt K, Schwab M, Eils R, Warnat P, Kaderali L, Simon T, Decarolis B, Theissen J, Westermann F, Brors B, Fischer M (2010) Prognostic impact of gene expression-based classification for neuroblastoma. J Clin Oncol 28 (21): 3506–3515
Ohira M, Oba S, Nakamura Y, Isogai E, Kaneko S, Nakagawa A, Hirata T, Kubo H, Goto T, Yamada S, Yoshida Y, Fuchioka M, Ishii S, Nakagawara A (2005) Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell 7 (4): 337–350
Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV (2011) Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res 21 (8): 1260–1272
Park JR, Eggert A, Caron H (2008) Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am 55 (1): 97–120
Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, Kolch W (2011) c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res 71 (13): 4664–4674
Schilling FH, Spix C, Berthold F, Erttmann R, Fehse N, Hero B, Klein G, Sander J, Schwarz K, Treuner J, Zorn U, Michaelis J (2002) Neuroblastoma screening at one year of age. N Engl J Med 346 (14): 1047–1053
Schramm A, Schulte JH, Astrahantseff K, Apostolov O, Limpt V, Sieverts H, Kuhfittig-Kulle S, Pfeiffer P, Versteeg R, Eggert A (2005a) Biological effects of TrkA and TrkB receptor signaling in neuroblastoma. Cancer Lett 228 (1-2): 143–153
Schramm A, Schulte JH, Klein-Hitpass L, Havers W, Sieverts H, Berwanger B, Christiansen H, Warnat P, Brors B, Eils J, Eils R, Eggert A (2005b) Prediction of clinical outcome and biological characterization of neuroblastoma by expression profiling. Oncogene 24 (53): 7902–7912
Schramm A, von Schuetz V, Christiansen H, Havers W, Papoutsi M, Wilting J, Schweigerer L (2005c) High activin A-expression in human neuroblastoma: suppression of malignant potential and correlation with favourable clinical outcome. Oncogene 24 (4): 680–687
Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, Schramm A, Christiansen H, Eilers M, Eggert A, Berwanger B (2008) MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer 122 (3): 699–704
Schulte JH, Schramm A, Klein-Hitpass L, Klenk M, Wessels H, Hauffa BP, Eils J, Eils R, Brodeur GM, Schweigerer L, Havers W, Eggert A (2005) Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA- and TrkB-expressing neuroblastomas. Oncogene 24 (1): 165–177
Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Godstein M, Trent J (1983) Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and neuroblastoma tumors. Nature 305: 245–248
Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D (1985) Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313 (18): 1111–1116
Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, Shaughnessy JD, Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M, Chen M, Cheng J, Cheng J, Chou J, Davison TS, Delorenzi M, Deng Y, Devanarayan V, Dix DJ, Dopazo J, Dorff KC, Elloumi F, Fan J, Fan S, Fan X, Fang H, Gonzaludo N, Hess KR, Hong H, Huan J, Irizarry RA, Judson R, Juraeva D, Lababidi S, Lambert CG, Li L, Li Y, Li Z, Lin SM, Liu G, Lobenhofer EK, Luo J, Luo W, McCall MN, Nikolsky Y, Pennello GA, Perkins RG, Philip R, Popovici V, Price ND, Qian F, Scherer A, Shi T, Shi W, Sung J, Thierry-Mieg D, Thierry-Mieg J, Thodima V, Trygg J, Vishnuvajjala L, Wang SJ, Wu J, Wu Y, Xie Q, Yousef WA, Zhang L, Zhang X, Zhong S, Zhou Y, Zhu S, Arasappan D, Bao W, Lucas AB, Berthold F, Brennan RJ, Buness A, Catalano JG, Chang C, Chen R, Cheng Y, Cui J, Czika W, Demichelis F, Deng X, Dosymbekov D, Eils R, Feng Y, Fostel J, Fulmer-Smentek S, Fuscoe JC, Gatto L, Ge W, Goldstein DR, Guo L, Halbert DN, Han J, Harris SC, Hatzis C, Herman D, Huang J, Jensen RV, Jiang R, Johnson CD, Jurman G, Kahlert Y, Khuder SA, Kohl M, Li J, Li L, Li M, Li QZ, Li S, Li Z, Liu J, Liu Y, Liu Z, Meng L, Madera M, Martinez-Murillo F, Medina I, Meehan J, Miclaus K, Moffitt RA, Montaner D, Mukherjee P, Mulligan GJ, Neville P, Nikolskaya T, Ning B, Page GP, Parker J, Parry RM, Peng X, Peterson RL, Phan JH, Quanz B, Ren Y, Riccadonna S, Roter AH, Samuelson FW, Schumacher MM, Shambaugh JD, Shi Q, Shippy R, Si S, Smalter A, Sotiriou C, Soukup M, Staedtler F, Steiner G, Stokes TH, Sun Q, Tan PY, Tang R, Tezak Z, Thorn B, Tsyganova M, Turpaz Y, Vega SC, Visintainer R, von Frese J, Wang C, Wang E, Wang J, Wang W, Westermann F, Willey JC, Woods M, Wu S, Xiao N, Xu J, Xu L, Yang L, Zeng X, Zhang J, Zhang L, Zhang M, Zhao C, Puri RK, Scherf U, Tong W, Wolfinger RD MAQC Consortium (2010) The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol 28 (8): 827–838
Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, Sferra R, Rucci N, Argenti B, Screpanti I, Gulino A, Mackay AR (2004) TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 6 (4): 347–360
Tibshirani R, Hastie T, Narasimhan B, Chu G (2002) Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA 99 (10): 6567–6572
Vermeulen J, De Preter K, Laureys G, Speleman F, Vandesompele J (2009a) 59-gene prognostic signature sub-stratifies high-risk neuroblastoma patients. Lancet Oncol 10 (11): 1030
Vermeulen J, De Preter K, Naranjo A, Vercruysse L, Van Roy N, Hellemans J, Swerts K, Bravo S, Scaruffi P, Tonini GP, De Bernardi B, Noguera R, Piqueras M, Cañete A, Castel V, Janoueix-Lerosey I, Delattre O, Schleiermacher G, Michon J, Combaret V, Fischer M, Oberthuer A, Ambros PF, Beiske K, Bénard J, Marques B, Rubie H, Kohler J, Pötschger U, Ladenstein R, Hogarty MD, McGrady P, London WB, Laureys G, Speleman F, Vandesompele J (2009b) Predicting outcomes for children with neuroblastoma using a multigene-expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol 10 (7): 663–671
Wei JS, Greer BT, Westermann F, Steinberg SM, Son CG, Chen QR, Whiteford CC, Bilke S, Krasnoselsky AL, Cenacchi N, Catchpoole D, Berthold F, Schwab M, Khan J (2004) Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res 64 (19): 6883–6891
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft, Collaborative Research Effort SFB876, subprojects C1‘DimRed’ (to AS and KM) and B1 ‘Spectra’ (to SR) and PI10/00015 and RD06/0020/0102 (RTICC, ISCIII and ERDF) Spain (to RN). We thank Ellen Mahlow and Sabine Dreesmann for their excellent technical assistance, Harald Stephan for his help in preparing the figures and Kathy Astrahantseff for proof reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Schramm, A., Schowe, B., Fielitz, K. et al. Exon-level expression analyses identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict primary neuroblastoma outcome. Br J Cancer 107, 1409–1417 (2012). https://doi.org/10.1038/bjc.2012.391
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.391
Keywords
This article is cited by
-
The ETS transcription factor ETV5 is a target of activated ALK in neuroblastoma contributing to increased tumour aggressiveness
Scientific Reports (2020)
-
The oncogenic neurotrophin receptor tropomyosin-related kinase variant, TrkAIII
Journal of Experimental & Clinical Cancer Research (2018)
-
The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines
Archives of Toxicology (2018)
-
RIT2: responsible and susceptible gene for neurological and psychiatric disorders
Molecular Genetics and Genomics (2018)
-
Radiation-induced alternative transcription and splicing events and their applicability to practical biodosimetry
Scientific Reports (2016)
