
Abstract
Background:
Preclinical studies in prostate cancer (PC) models demonstrated the anti-tumour activity of the first fully synthetic epothilone, sagopilone. This is the first study to investigate the activity and safety of sagopilone in patients with metastatic castration-resistant PC (CRPC).
Methods:
Chemotherapy-naïve patients with metastatic CRPC received sagopilone (one cycle: 16 mg m−2 intravenously over 3 h q3w) plus prednisone (5 mg twice daily). The primary efficacy evaluation was prostate-specific antigen (PSA) response rate (⩾50% PSA reduction confirmed ⩾28 days apart). According to the Simon two-stage design, ⩾3 PSA responders were necessary within the first 13 evaluable patients for recruitment to continue until 46 evaluable patients were available.
Results:
In all, 53 patients received ⩾2 study medication cycles, with high compliance. Mean individual dose was 15.1±1.4 mg m−2 during initial six cycles, mean dose intensity 94±9%. The confirmed PSA response rate was 37%. Median overall progression-free survival was 6.4 months. The most commonly reported adverse events (>10% of patients) were peripheral neuropathy (94.3%), fatigue (54.7%) and pain in the extremities (47.2%). Sagopilone was associated with very little haematological toxicity.
Conclusion:
This study shows that first-line sagopilone has noteworthy anti-tumour activity and a clinically significant level of neuropathy for patients with metastatic chemotherapy-naïve CRPC.
Similar content being viewed by others
Main
In the United States, the most common cancer for men is prostate cancer (PC), which accounts for 9% of all cancer-related deaths in men (Vishnu and Tan, 2010). In the United Kingdom, the life-time risk of being diagnosed with PC is 1 in 10 (Cancer Research UK, 2010). Although PC has a favourable prognosis if detected at an early stage, it is generally incurable in patients with metastatic disease. Hormone therapy reduces androgen levels, providing rapid disease control, but patients with metastatic PC often become resistant to this treatment and develop castration-resistant PC (CRPC). Several treatment options are available to these patients, with docetaxel being the current standard of care, but in general these treatments provide only temporary disease control (Petrylak et al, 2004; Tannock et al, 2004; DeVita et al, 2008; National Comprehensive Cancer Network, 2011).
Epothilones are a novel alternative class of tubule-binding compounds with a similar mode of action to that of taxanes (Bollag et al, 1995; Altaha et al, 2002). However, unlike taxanes, epothilones can retain activity in multidrug-resistant cancer cells that overexpress the P-glycoprotein efflux pump (Bollag et al, 1995). Ixabepilone, a half-synthetic analogue of epothilone B, has demonstrated encouraging prostate-specific antigen (PSA) response rates in chemotherapy-naïve patients with advanced PC (Galsky et al, 2005; Hussain et al, 2005). Neutropenia and neuropathy were reported as grade 3 or 4 adverse events (AEs) in both studies.
Sagopilone (ZK-EPO; Bayer HealthCare Pharmaceuticals, Berlin, Germany) is the first fully synthetic member of the novel epothilone class of microtubule stabilisers and is not associated with multidrug resistance (Klar et al, 2006). Preclinical studies demonstrated the anticancer activity of sagopilone in many human cancer xenograft models, including PC models and taxane-resistant models (Hoffmann et al, 2008). Compared with taxanes, sagopilone had greater cellular uptake, with no recognition by efflux mechanisms, and enhanced intrinsic activity in preclinical systems (Hoffmann et al, 2008). In phase I and II clinical studies, sagopilone (as a monotherapy or in combination with carboplatin or cisplatin) was well tolerated and demonstrated clinical benefit in several solid tumour types (Rustin et al, 2007; Gauler et al, 2008; McMeekin et al, 2008; Wenk et al, 2008; Schmid et al, 2010).
The single-arm, multicentre, phase II study reported here investigated the efficacy and safety of sagopilone in chemotherapy-naïve patients with CRPC.
Methods
Study design and objectives
This was a prospective, single-arm, multicentre, phase II study conducted at 18 trial sites in the USA and Argentina. A Simon two-stage (optimal) design was used. At least 3 out of the first 13 patients needed to achieve a PSA 50% response (⩾50% reduction compared with study baseline and maintained for ⩾28 days) in stage 1 for recruitment to continue until a target of 46 fully evaluable patients had been reached. Based on standards available at the time of study design, the study outcome would be considered positive if a PSA response was observed in at least 28% of patients (13 out of 46).
The primary efficacy end point was the proportion of patients with a PSA response. Secondary efficacy end points were: objective response and duration of objective response according to modified Response Evaluation Criteria in Solid Tumours (modRECIST; only in patients with measurable disease); time to progression (TTP); time to PSA response; duration of PSA response; progression-free survival (PFS); overall survival; safety; and tolerability.
Patients received one dose of 16 mg m−2 sagopilone intravenously over 3 h once every 3 weeks, plus 5 mg oral prednisone twice daily, for two to six cycles. Patients subsequently demonstrating a sustained clinical benefit could receive extended therapy. Study medication was permanently discontinued if patients developed grade 3 or 4 neurotoxicity or grade 3 or 4 drug-related toxicities that did not resolve within 5 weeks of the last infusion, or any grade 4 non-haematological toxicity.
This study met all local legal and regulatory requirements, followed the Declaration of Helsinki and conformed to Good Publication Practice guidelines. The protocol was approved by an independent ethics committee at each study centre. All patients provided written, informed consent before enrolment into the study.
Patients
All patients (aged ⩾18 years) had histologically or cytologically confirmed metastatic adenocarcinoma of the prostate and had received no prior chemotherapy. All patients had progressive disease as demonstrated by a PSA increase on two or more consecutive measurements taken at least 7 days apart. The last PSA measurement had to be ⩾50% more than the lowest value achieved under the most recent prior treatment, with a minimal value of ⩾5 ng ml−1. For patients undergoing 4–6 weeks of washout from anti-androgen treatment, the post-washout PSA level had to remain elevated. Other inclusion criteria included: a World Health Organization performance status of 0–2; adequate function of the major organs; and serum testosterone levels <50 ng ml−1. In addition, all patients had to have recovered adequately from previous surgery and/or radiation.
Exclusion criteria were: any prior chemotherapy for PC; use of any investigational drug within 4 weeks or five half-lives before the start of study medication; previous symptomatic brain metastases requiring whole-brain irradiation; active infection; and any concurrent malignancy other than non-melanoma skin cancer.
Patients were withdrawn from the study if they had: disease progression; unacceptable toxicity; a delay in treatment of >2 weeks; dose postponement more than twice during the study owing to toxicities; or concurrent conditions that precluded continuing study medication.
Assessments
The primary end point of PSA response was defined as a decrease of ⩾50% compared with study baseline, with the response maintained for at least 28 days. Measurable lesions were defined by a diameter of ⩾20 mm using conventional techniques or ⩾10 mm with spiral computed tomography scan. Patients with measurable disease were evaluated by magnetic resonance imaging or computed tomography scan every two cycles and responses were assessed according to modRECIST (modified from Therasse et al, 2000) to include the category of ‘unknown’ as a further possible outcome of tumour evaluation: ‘unknown’ indicates that the target or non-target lesion response is unknown and no new lesions have been documented). Bone metastases were assessed at baseline, with further evaluation based on investigator-determined need and not mandated by the protocol.
The secondary end points were: duration of PSA response, defined as the time from initial PSA response to first PSA progression; time to PSA progression, defined as the time from enrolment into the study to first PSA progression; TTP, defined as the time from enrolment to progression (according to PSA criteria or modRECIST) or death from tumour; overall PFS, defined as the time from enrolment to first progression (by PSA criteria or modRECIST) or death; and overall survival, defined as the time from enrolment until death from any cause or date of last follow-up.
Adverse events and serious AEs (SAEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v 3.0. Standard laboratory assessments, such as serum chemistry, haematology, coagulation and urinalysis, were performed at the individual study centres.
Statistical analysis
The primary efficacy analysis was designed to investigate the null hypothesis that the PSA response rate was ⩽0.20, vs an alternative hypothesis that the PSA response rate was >0.20 (with one-sided type 1 error rate of 0.10). A Simon two-stage design minimised the number of exposed patients in case of insufficient efficacy; based on this design, the study had a planned sample size of 46 evaluable patients.
An interim analysis was planned based on the results of the first 13 evaluable patients. If there were fewer than three PSA responders, the study would be discontinued. Otherwise, the final analysis would be based on the first 46 evaluable patients. The null hypothesis would be rejected only if ⩾13 PSA responders were identified.
The primary end point was assessed using the primary analysis set. For stage 1 this included the first 13 per protocol (PP) patients (i.e., all patients assigned to study treatment with no major protocol deviation) for whom the primary efficacy variable was assessable, and for stage 2 this included the next 33 PP patients for whom the primary efficacy variable was assessable. Additionally, the primary efficacy variable was assessed descriptively for the PP set, which included all full analysis set patients assigned to study treatment without major protocol deviation. All secondary efficacy variables were analysed in the PP set. Safety analyses were investigated using the safety analysis set, which included all patients in the full analysis set with at least one intake of the study medication.
Demographic and baseline characteristics, safety end points, and clinical laboratory evaluations were analysed by descriptive statistics and/or frequency tables where appropriate. Secondary time-to-event efficacy end points were analysed using the Kaplan–Meier estimates and corresponding graphs, and other secondary efficacy end points were analysed descriptively.
Results
Between August 2006 and May 2008, 70 patients were screened for inclusion in this study; 53 patients were enroled and received at least two cycles of study medication. Patient numbers were not equally distributed across sites (competitive accrual; 1–11 patients were accrued per site). Patient demographics and baseline disease characteristics are shown in Table 1. All 53 patients were included in the safety analyses, while 5 patients were excluded from the efficacy analysis: 2 did not meet all eligibility criteria and 3 were missing a baseline PSA assessment.
A median of five treatment cycles per patient were administered, with an individual median dose of 16 mg m−2 (range 11.8–16.0 mg m−2). The individual mean dose during the first six cycles was 15.1±1.4 mg m−2 (range 11.8–16.0 mg m−2) and the mean dose intensity (individual mean dose divided by planned dose according to the assigned treatment group) was 94±9%.
The median duration for which patients remained on the study, from start of study to end of any follow-up, was 190 days (interquartile range 122–247 days). The median follow-up time, from last study medication to last clinic visit, was 85 days (range 38–107 days).
Fifteen patients (15/53, 28.3%) discontinued the study medication before cycle 6: thirteen patients owing to AEs, mainly neuropathy (eleven patients); one patient owing to non-compliance; and one patient withdrew consent.
One patient continued to receive treatment after the cut-off date for analysis and received 27 treatment cycles. The data from the additional cycles were collected but not included in these analyses.
Efficacy
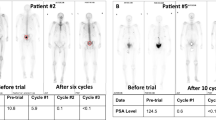
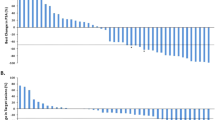
Of the 46 patients in the primary analysis set, 17 (37%) had a confirmed ⩾50% PSA reduction from baseline, demonstrating that this study met its primary end point (Figure 1).

Maximum relative percentage change in prostate-specific antigen within 3 months of sagopilone treatment (primary analysis set).
In the full analysis set, 40 patients had measurable disease. A total of 36 patients in the PP set had measurable disease, of whom 12 (33.3%) responded (Table 2). One patient had complete response; eleven patients had partial response. Median duration of objective response could not be calculated as only three of these responders experienced either a recurrence of the disease or disease progression during the observation period.
The duration of PSA response was analysed using the data from the 17 initial PSA responders and was estimated to be 289 days (95% confidence interval (CI): lower limit 166 – upper limit could not be calculated) (Figure 2). Median time to PSA progression was 232 days (95% CI: lower limit 198 – upper limit could not be calculated).

Duration of prostate-specific antigen response in the per protocol set.
Median TTP was 168 days (95% CI: 131–232) and median PFS was 195 days (95% CI: 131–232) (Figure 3). Progression-free survival in patients with PSA response and those with objective response was 232 days (95% CI: 208–308) and 211 days (95% CI: 140–308), respectively (Figure 4). Median overall survival could not be calculated as only three patients died during the observation period.

Kaplan–Meier plot of overall progression-free survival in the per protocol set.

Kaplan–Meier plot of progression-free survival in (A) patients with prostate-specific antigen response and (B) patients with objective response (per protocol set).
Safety
All 53 patients received at least one dose of 16 mg m−2 sagopilone via a 3-h infusion and were therefore included in the safety analysis set. All patients reported at least one AE during the study treatment. Almost all (98.1%) of the observed AEs were drug-related.
The incidence of AEs indicating neurotoxicity was high, with 94.3% of patients (50 out of 53) reporting peripheral neuropathy during study treatment (Table 3) and >50% (30 out of 53) still experiencing this AE at follow-up. The highest incidences of grade 3 or 4 AEs were reported for peripheral neuropathy (12 out of 53, 22.6%), pain in extremities (4 out of 53, 7.5%) and fatigue (4 out of 53, 7.5%). Grade 4 AEs were observed in single cases only (1 out of 53, 1.9%), with the exception of pulmonary embolism (2 out of 53, 3.8%). One patient developed a fatal grade 5 AE (gastrointestinal haemorrhage).
As a result of AEs, treatment was discontinued in 20 patients, 16 owing to peripheral neuropathy. At least one treatment was postponed in 17 patients (because of AEs in 15 patients). The majority of dose postponements were associated with subsequent dose reduction. Twenty patients required a dose reduction from 16 mg m−2 to 12 mg m−2, three of whom required a further dose reduction to 9 mg m−2. Toxicity management by dose reduction and/or dose delay facilitated drug compliance.
A total of 19 SAEs were experienced by 18.9% (10 out of 53) of patients during study treatment and/or follow-up, with pulmonary embolism the only SAE to be reported in more than one patient (two patients). In 4 of these 10 patients, at least one SAE was identified as drug-related. Three patients discontinued sagopilone owing to an SAE.
Four patients died during this study, three from progressive disease and one due to gastrointestinal haemorrhage (grade 5 AE), which was not drug-related.
Most changes in the CTCAE grade of quantitative laboratory parameters were from grade 0 to grade 1 or 2. Fifteen patients had a change to CTCAE grade 4, with the highest incidence rate of changes to CTCAE grade 4 in creatinine and low calcium levels. Creatinine levels for the three affected patients returned to normal by week 12 of the follow-up period. All other changes to CTCAE grade 4 were reported in a maximum of two patients. No clinically relevant changes were observed in heart rate or blood pressure.
Discussion
The results of this trial clearly demonstrate that sagopilone has clinical activity in patients with CRPC. In our study, PSA reduction, measurable disease response and TTP are generally comparable to those observed in chemotherapy-naïve patients following docetaxel treatment (Montgomery et al, 2007; Buch-Hansen et al, 2010). Progression-free survival was similar for both the PSA responders and the objective responders, suggesting concordance, with respect to this measurement, across these two groups.
Recognising the limitations of this small, single-arm, phase II study, measurable disease response in 1 out of 3 patients (33%) represents a relatively high level of activity, although this could be a chance finding.
In the absence of docetaxel, these results would support phase III evaluation. However, while positive, our efficacy and safety results are probably insufficiently compelling to support a head-to-head challenge against docetaxel, in the context of currently available treatment options.
The toxicity profile was notable in our study. Peripheral neuropathy was common and frequently resulted in the discontinuation of study medication. Diarrhoea, however, was relatively uncommon and haematological, and hepatic and renal toxicity were nearly absent. Given the relatively encouraging level of anti-tumour activity observed, strategies to prevent, reduce or mitigate the neuropathy were considered. Other than conventional, symptomatic management of neuropathy, which masks some of the symptoms but does not address the underlying pathological process, no additional viable methods were found to adequately address this toxicity. In this study, detailed data on the treatment administered to manage neuropathy and its outcomes were not collected.
The measurable disease response rate in the study is encouraging and all other measures are competitive. However, the potential development of neuropathy may limit the ability to deliver sagopilone for at least nine cycles, as with docetaxel in the phase III setting (Tannock et al, 2004). Therefore, overall results do not support further phase III development of sagopilone in patients with CRPC.
The recent success of second-line cabazitaxel is a reminder that successful strategies to target microtubules in PC do not start and end with docetaxel (de Bono et al, 2010). In view of the promising level of activity observed in our trial, investigation of sagopilone in docetaxel-treated patients would be of interest. Sagopilone may provide a clinically meaningful level of activity for these patients as it was developed to be unaffected by multidrug-resistance-mediated mechanisms and has demonstrated activity in taxane-resistant preclinical models (Klar et al, 2006; Hoffmann et al, 2008).
Although the landscape for treating advanced PC is changing, there remains a significant need to identify and develop new cytotoxic agents for treating CRPC. Only cabazitaxel has been added to the chemotherapy armamentarium since the approval of docetaxel in 2004. Efforts have been directed towards evaluating the role of epothilones in metastatic CRPC, and to date two epothilones, ixabepilone and patupilone, have been investigated for the treatment of this disease (Galsky et al, 2005; Hussain et al, 2005, 2009; Chi et al, 2012). In a phase II study, weekly patupilone (2.5 mg m−2) showed minimal activity in patients with PC (Hussain et al, 2009), although response improved with 3-weekly patupilone (8 mg m−2) in a second phase II study (Chi et al, 2012). More robust activity was seen with ixabepilone, although this was not significantly different from that observed with docetaxel (Galsky et al, 2005; Hussain et al, 2005).
The PSA response rate and PFS observed in our sagopilone study are comparable to those reported for ixabepilone and patupilone (Galsky et al, 2005; Hussain et al, 2005; Chi et al, 2012). Differences between these compounds are more evident in the toxicity profiles: neutropenia and neuropathy associated with ixabepilone (Galsky et al, 2005; Hussain et al, 2005), diarrhoea associated with patupilone (Chi et al, 2012), and neuropathy associated with sagopilone. Patupilone may present an alternative to cabazitaxel, but perhaps only for patients with significant residual haematological toxicity following docetaxel. Our study adds to the limited experience with epothilone treatment for PC and, when taken in context with patupilone and ixabepilone, suggests that although this class of drugs has clinical activity, when applied to the broad CRPC population, epothilones are unlikely to significantly influence the outcomes of patients with advanced PC. Overall, it is difficult to envisage a place for any of these treatments in the current treatment landscape for PC, in which there has been considerable change recently.
However, the relatively high measurable disease response rate seen in our trial is interesting and merits further investigation. As our understanding of the heterogeneity of advanced PC increases at both molecular and clinical levels, we may learn that various subsets of PC respond differently to microtubule-targeted agents. Following the observation of a higher than expected response rate in measurable disease, clinical research efforts are currently underway to examine taxane–platinum combinations in the relatively ill-defined entity of ‘anaplastic’ CRPC, a subset of patients with a proclivity to develop soft-tissue metastases. Further investigations may be justified to evaluate whether sagopilone could be a more suitable microtubule disassembly inhibitor in this subset. In the study reported in this manuscript, a high proportion of patients had a dose reduction from 16 mg m−2, suggesting that a lower starting dose of sagopilone should be considered.
In conclusion, we have demonstrated with PSA and measurable disease response rates a significant level of anti-tumour activity for sagopilone in patients with CRPC. The frequency of neuropathy was relatively high, distinctive and dose-limiting, and discourages further development. Moreover, the fact that the overall activity is comparable to that of docetaxel suggests that a head-to-head comparison with docetaxel in chemotherapy-naïve CRPC is probably not justified. On the other hand, a drug with this level of activity deserves further evaluation in the second-line setting and for different patient subsets to acquire a better understanding of which patients may derive greater benefit from either taxanes or epothilones.
Change history
08 August 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Altaha R, Fojo T, Reed E, Abraham J (2002) Epothilones: a novel class of non-taxane microtubule-stabilizing agents. Curr Pharm Des 8: 1707–1712
Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM (1995) Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res 55: 2325–2333
Buch-Hansen TZ, Bentzen L, Hansen S, Hoeyer M, Jensen NV, Saxe C, Sengeloev L (2010) Phase I/II study on docetaxel, gemcitabine and prednisone in castrate refractory metastatic prostate cancer. Cancer Chemother Pharmacol 66: 295–301
Cancer Research UK (2010) Prostate cancer risks and causes. Available at: http://www.cancerhelp.org.uk/type/prostate-cancer/about/prostate-cancer-risks-and-causes Accessed 20 July 2011
Chi KN, Beardsley E, Eigl BJ, Venner P, Hotte SJ, Winquist E, Ko YJ, Sridhar SS, Weber D, Saad F (2012) A phase 2 study of patupilone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel: Canadian Urologic Oncology Group study P07a. Ann Oncol 23: 53–58
de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376: 1147–1154
DeVita VT, Lawrence TS, Rosenberg SA (2008) Cancer: Principles and Practice of Oncology 8th edn Lippincott Williams & Wilkins: Philadelphia
Galsky MD, Small EJ, Oh WK, Chen I, Smith DC, Colevas AD, Martone L, Curley T, DeLaCruz A, Scher HI, Kelly WK (2005) Multi-institutional randomized phase II trial of the epothilone B analog ixabepilone (BMS-247550) with or without estramustine phosphate in patients with progressive castrate metastatic prostate cancer. J Clin Oncol 23: 1439–1446
Gauler TC, Christoph DC, Gamarra F, Fuhr H, Gonschorek C, Giurescu M, Frickhofen N, Huber RM, Schuler M, Eberhardt WEE (2008) Phase I trial of the novel epothilone sagopilone (ZK-EPO) in combination with cisplatin as first-line therapy in patients with extensive-disease small-cell lung cancer (ED-SCLC). J Clin Oncol 26: 19081
Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, Skuballa W, Vivet S, Lichtner RB, Vicencio JM, Panaretakis T, Siemeister G, Lage H, Nanty L, Hammer S, Mittelstaedt K, Winsel S, Eschenbrenner J, Castedo M, Demarche C, Klar U, Kroemer G (2008) Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res 68: 5301–5308
Hussain A, DiPaola RS, Baron AD, Higano CS, Tchekmedyian NS, Johri AR (2009) Phase II trial of weekly patupilone in patients with castration-resistant prostate cancer. Ann Oncol 20: 492–497
Hussain M, Tangen CM, Lara PN, Vaishampayan UN, Petrylak DP, Colevas AD, Sakr WA, Crawford ED (2005) Ixabepilone (epothilone B analogue BMS-247550) is active in chemotherapy-naive patients with hormone-refractory prostate cancer: a Southwest Oncology Group trial S0111. J Clin Oncol 23: 8724–8729
Klar U, Buchmann B, Schwede W, Skuballa W, Hoffmann J, Lichtner RB (2006) Total synthesis and antitumor activity of ZK-EPO: the first fully synthetic epothilone in clinical development. Angew Chem Int Ed Engl 45: 7942–7948
McMeekin S, Patel R, Verschraegen C, Celano P, Burke J, Plaxe S, Ghatage P, Giurescu M, Stredder C, Wang Y (2008) Phase I/II study of sagopilone (ZK-EPO) plus carboplatin in women with recurrent platinum-sensitive ovarian cancer. Ann Oncol 19 (Suppl 8): Abstract 665O, pviii211
Montgomery RB, Nelson PS, Lin D, Ryan CW, Garzotto M, Beer TM (2007) Diethylstilbestrol and docetaxel: a phase II study of tubulin active agents in patients with metastatic, androgen-independent prostate cancer. Cancer 110: 996–1002
National Comprehensive Cancer Network (2011) NCCN Clinical Practice Guidelines in Oncology. Prostate Cancer V.4.2011. Available at: http://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf Accessed 20 July 2011
Petrylak DP, Tangen CM, Hussain MHA, Lara PN, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED (2004) Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 351: 1513–1520
Rustin GJ, Reed NS, Jayson G, Ledermann JA, Adams M, Stredder C, Wagner A, Giurescu M, The ZK-EPO Study Group in Platinum-Resistant Ovarian Cancer (2007) Phase II trial of the novel epothilone ZK-EPO in patients with platinum resistant ovarian cancer. J Clin Oncol 25: 5527
Schmid P, Kiewe P, Possinger K, Korfel A, Lindemann S, Giurescu M, Reif S, Wiesinger H, Thiel E, Kühnhardt D (2010) Phase I study of the novel, fully synthetic epothilone sagopilone (ZK-EPO) in patients with solid tumors. Ann Oncol 21: 633–639
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA, for the TAX327 Investigators (2004) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351: 1502–1512
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, Van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92: 205–216
Vishnu P, Tan WW (2010) Update on options for treatment of metastatic castration-resistant prostate cancer. Onco Targets Ther 3: 39–51
Wenk D, DeConti RC, Urbas P, Andrews S, Sondak VK, Maker N, Weber JS, Daud AI (2008) Phase II trial of sagopilone (ZK-EPO), a novel epothilone, in patients with metastatic melanoma. J Clin Oncol 26: 9046
Acknowledgements
We take full responsibility for the scope, direction, and content of the manuscript and have approved the submitted manuscript. We thank all study investigators: Shabbir Ahmad, Blair Medical Associates, Altoona, PA, USA; Norberto Brocato, Policlínica Bancaria, Buenos Aires, Argentina; Mario Felix Bruno, Hospital General de Agudos ‘Dr Teodoro Alvarez’, Buenos Aires, Argentina; James Burke, Billings Clinic, Billings, MT, USA; Nashat Gabrail, Gabrail Cancer Center, Canton, OH, USA; Ralph Hauke, University of Nebraska Medical Center, Omaha, NE, USA; Glen Justice, Pacific Coast Haematology/Oncology Medical Group Inc., Fountain Valley, CA, USA; Robert Montgomery, VA Puget Sound Health Care System, Seattle, WA, USA; Latha Neerukonda, JPS Centre For Cancer Care, Fort Worth, TX, USA; Debra Prow, JPS Centre For Cancer Care, Fort Worth, TX, USA; Avi Samson Retter, National Cancer Institute/National Institutes of Health, Bethesda, MD, USA; William Tingle, Florida Urology Specialists, Sarasota, FL, USA. We thank Katherine Wilson, PhD, at Complete HealthVizion for her assistance in the preparation and revision of the draft manuscript, based on detailed discussion and feedback from all the authors. Editorial assistance was funded by Bayer HealthCare Pharmaceuticals. This work was funded by Bayer HealthCare Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Martin Giurescu and Katrin Roth are employees of Bayer HealthCare Pharmaceuticals. Yao Wang was an employee of Bayer HealthCare Pharmaceuticals when the phase II clinical trial of first-line sagopilone was conducted. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Beer, T., Smith, D., Hussain, A. et al. Phase II study of first-line sagopilone plus prednisone in patients with castration-resistant prostate cancer: a phase II study of the Department of Defense Prostate Cancer Clinical Trials Consortium. Br J Cancer 107, 808–813 (2012). https://doi.org/10.1038/bjc.2012.339
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.339
Keywords
This article is cited by
-
Update on the Role of Epothilones in Metastatic Breast Cancer
Current Breast Cancer Reports (2013)
