
Abstract
Background:
Inter-patient pharmacokinetic variability can lead to suboptimal drug exposure, and therefore might impact the efficacy of sorafenib. This study reports long-term pharmacokinetic monitoring of patients treated with sorafenib and a retrospective pharmacodynamic/pharmacokinetic analysis in melanoma patients.
Patients and methods:
Heavily pretreated patients with stage IV melanoma were started on sorafenib 400 mg twice daily (bid). In the absence of limiting toxicity, dose escalation of 200 mg bid levels was done every 2 weeks. Plasma sorafenib measurement was performed at each visit, allowing a retrospective pharmacodynamic/pharmacokinetic analysis for safety and efficacy.
Results:
In all, 19 of 30 patients underwent dose escalation over 400 mg bid, and 28 were evaluable for response. The overall disease control rate was 61% (95% confidence interval (CI): 42.6–78.8), including three confirmed responses (12%). Disease control rate and progression-free survival (PFS) were improved in patients with high vs low exposure (80% vs 32%, P=0.02, and 5.25 vs 2.5 months, P=0.005, hazard ratio (HR)=0.28 (95% CI: 0.11–0.73)). In contrast, drug dosing had no effect on PFS. In multivariate analysis, drug exposure was the only factor associated with PFS (HR=0.36 (95% CI: 0.13–0.99)). Diarrhoea and anorexia were correlated with drug dosing, while hypertension and hand–foot skin reaction were correlated with drug exposure.
Conclusions:
Although sorafenib had modest efficacy in melanoma, these results suggest a correlation between exposure and efficacy of sorafenib. Therefore, dose optimisation in patients with low exposure at standard doses should be evaluated in validated indications.
Similar content being viewed by others


Main
Sorafenib is an oral agent that inhibits a large spectrum of cellular targets (VEGFR-2, PDGFR, c-KIT, FLT-3, CRAF, wild-type BRAF or BRAFV600E; Wilhelm et al, 2004). The recommended dose of sorafenib in patients with hepatocellular carcinoma and advanced renal cell cancer is 400 mg twice daily (bid) (Strumberg et al, 2007). In preclinical studies, sorafenib efficiently inhibited BRAF activity in BRAF-mutated melanomas, leading to growth retardation in preclinical studies (Sharma et al, 2005; Wilhelm et al, 2008). A phase II trial of sorafenib in 37 metastatic melanoma patients reported a modest activity, with only three partial response (8%; Min et al, 2008). Another phase II randomized discontinuation trial confirmed these results, with no confirmed objective response, and only 19% of stable disease (Eisen et al, 2006). Unfortunately, BRAF mutations were not predictive of clinical outcome in several trials involving sorafenib in melanoma patients (Eisen et al, 2006; Flaherty et al, 2008; Amaravadi et al, 2009; Ott et al, 2010). Recently, the BRAFV600E inhibitor vemurafenib has shown significant clinical activity in patients with advanced melanoma (Chapman et al, 2011). Hence, it is unclear whether sorafenib exerts anti-tumour activity in melanoma through the inhibition of BRAF or other targets, such as c-Kit. For instance, imatinib, another c-Kit inhibitor, is active in KIT-mutated melanomas (Guo et al, 2011). NRAS, GNAQ and GNA11 are other potential molecular targets, particularly in uveal melanoma (Alsina et al, 2003; Van Raamsdonk et al, 2010).
Sorafenib dose-limiting toxicities (DLTs) included diarrhoea, hypertension and hand–foot skin reaction (HFSR). Notably, doses increases from 400 to 800 mg bid did not substantially increase sorafenib area under the curve (AUC) in phase I trials (Strumberg et al, 2007). However, intra-patient dose escalation has not been evaluated by pharmacokinetics. Owing to a large inter-patient variability (∼50%) of sorafenib area under the plasma concentration–time curve over 12 h (AUC; Strumberg et al, 2007; Hornecker et al, 2011), a suboptimal exposure to sorafenib could result in a lack of anti-tumour activity in some patients. To date, this hypothesis could not be ruled out, as sorafenib exposure was not assessed in previous phase II and III trials. Otherwise, dose adjustment of sorafenib based on plasma exposure is not currently recommended. In addition, two clinical trials suggest potential benefit for sorafenib dose-escalation strategies in RCC, even after failure of sorafenib 400 mg bid dosing (Amato et al, 2008; Escudier et al, 2009).
In this context, we hypothesised that optimisation of sorafenib exposure might improve its efficacy in patients with metastatic melanoma, and that sorafenib AUC could be related to antitumor efficacy.
Patients and methods
From January 2008 to December 2009, consecutive patients with metastatic melanoma who progressed under previous therapeutic regimen containing one or more of the following: dacarbazine, fotemustine, interleukin-2, cisplatin, interferon or vaccine therapy, were offered sorafenib treatment in two academic cancer centres located in Paris, France (Cochin and Saint Louis Teaching Hospitals). At this time, vemurafenib was not available for patients with BRAF-mutated melanoma. BRAF mutation status was not assessed in our patients.
The schedule included an intra-patient dose escalation. A total of 30 patients with histological confirmed metastatic melanoma started sorafenib. All patients provided written informed consent, and the study was approved by the Local Ethics Committee.
Treatment plan
Patients were treated with sorafenib at a starting dose of 400 mg bid. In the absence of acute-limiting toxicity, intra-patient dose escalation of 200 mg bid every 2 weeks was planned. No maximum dose was specified. Sorafenib daily doses were only adjusted based on adverse events and not on plasma sorafenib exposure as the values of sorafenib AUC were not transmitted to clinicians.
Assessments
The primary endpoint was safety. Safety was assessed every 2 weeks during the whole-treatment period. In addition to summaries of adverse events classified and graded according to the National Cancer Institute Common Toxicity Criteria for Adverse Events version 3.0, term and category, safety analyses included evaluation of clinically significant laboratory test results and vital signs. A DLT was defined as any toxicity leading to dose reduction or to discontinuation of treatment. Tumour response was assessed by CT scan using one-dimensional measurements made at baseline, every 8 weeks thereafter and at the end of the treatment period if applicable. Treatment activity was evaluated using the revised RECIST guidelines (Therasse et al, 2000).
Plasma exposure to sorafenib
Sorafenib plasma concentrations were assessed in one sample drawn every 2 weeks (at the end of each period of dose escalation) by high-performance liquid chromatography (Blanchet et al, 2009). The accuracy, within-assay precision and inter-assay precision of this method were 96.9–104.0%, 3.4–6.2% and 7.6–9.9%, respectively. A specific bayesian estimator developed in our institution allowed estimating sorafenib AUC with a limited sampling strategy (Hornecker et al, 2011).
Statistical analyses
Overall survival (OS) was defined as the time from the treatment initiation to death (all causes). Survivors were censored at last follow-up. Progression-free survival (PFS) was defined as the time from the treatment initiation to the first recorded evidence of progression. Survivors without progression were censored at the date of last follow-up or death.
To retrospectively investigate the relation between clinical outcomes and drug exposure, different parameters were used: AUC measured 1 month after treatment initiation, mean and maximal AUC (AUCmax) over the whole-treatment period. As AUCs were not normally distributed AUCs between groups were compared using a Wilcoxon rank-sum test. The correlation between daily dose of sorafenib and AUC was computed with Spearman’s test. Response rate and toxicities were compared using Fisher’s exact test. Survival curves were estimated using Kaplan–Meier method and compared using log-rank test. Univariate Cox proportional hazard models for PFS and OS were built to compute the hazard ratios (HRs) with their 95% confidence intervals (95% CIs) of potential baseline predictors. Potential baseline predictors tested for OS were as follows: sex, WHO PS (⩾2), age (>59 years), AJCC stage, brain metastases, LDH baseline level (>ULN), time as metastatic disease (>15 months), number of previous treatment regimen (>2) and primary histological type. Variable tested for PFS included: sex, WHO PS (⩾2), age (>59 years), AJCC stage, brain metastases, BMI (>25 kg m−2), primary histological type, time as metastatic disease (>15 months), number of previous treatment regimen (>2), LDH baseline level (>ULN), AUCmax (⩾100 mg l−1 h−1), early grade⩾2 adverse events (at 2 months) including diarrhoea, hand–foot skin syndrome (HFSR), skin rash and hypertension considered separately or jointly. Then, multivariate analyses were conducted on all potential factors with P-value <0.2 in univariate analysis using a stepwise Cox model with enter variable with P-value <0.05 and remove if P-value >0.1. The median served as the cutoff point when continuous variables (mean and max AUCs) were separated into two groups.
Missing data were not estimated or carried forward in any statistical analyses. All analyses were performed using the JMP 8.0.2 (SAS Institute Inc., SAS Campus Drive, Cary, NC, USA). P-values were two tailed and considered significant when ⩽0.05.
Results
Patients characteristics
A total of 30 patients with histologically confirmed metastatic melanoma were treated with sorafenib. Baseline patients’ characteristics are summarised in Table 1. The median daily dose was 800 mg bid (range 400–2600), and 19 patients (63%) underwent dose escalation (range 600–2600 mg bid). The median duration of treatment was 2.9 months (range 0.4–16.3).
Response and survival
Two patients discontinued treatment owing to severe toxicity before the first evaluation. Therefore, 28 patients were evaluable for response. One complete response and five partial responses were observed, including three confirmed responses. The overall response rate was 21% (95% CI: 6.2–36.6). The objective responses were assessed early, with a median time from treatment initiation of 2.3 months (range: 1.3–3.4 months). In all, 3 of 10 patients (30%) with cerebral metastasis had cerebral partial responses. Median duration of confirmed response was 6.1 months. In total, 11 patients (39%) had stable disease with a median duration of 4.4 months, for an overall disease control rate (PR+SD) of 61% (95% CI: 42.6–78.8).
After a median follow-up of 10 months (range: 3–20), median PFS was 3.6 months (95% CI: 2.5–5.6 months; 18% censored) and median OS was 11 months (95% CI: 5–15 months; 21% censored). The 1-year survival rate was 33% (95% CI: 19–52%). Median survival in patients with brain metastases was 5.6 months (95% CI: 2.5–9,6, 0% censored). In univariate analysis, significant (P<0.05) prognostic factors were WHO PS ⩾2 (HR=3.72 (95% CI: 1.23–10.52)) and brain metastases (HR=2.68 (95% CI: 1.08–6.64)). The number of previous treatment regimens (>2) had P-value=0.18 and was added to the multivariate analysis. Only WHO PS ⩾2 had an independent prognosis value (HR=3.72 (95% CI: 1.32–10.58)) in multivariate analysis.
Safety
A total of 18 severe adverse events (grade ⩾3) occurred in 11 patients at the starting dose of 400 mg bid: 8 hand and foot skin reaction (HFSR), 5 skin rash, 2 stomatitis, 2 hypertension and 1 fatigue. Sorafenib was discontinued in the four patients who experienced both grade 3 rash and HFSR, and then reintroduced at 200 mg bid. Despite this daily dose adjustment, the severity of toxicity was unchanged; therefore the treatment was definitively discontinued. The four patients with isolated grade 3 HFSR were able to continue sorafenib for up to 5 months with a 50% dose decrease.
During the dose escalation, only two patients discontinued sorafenib because of toxicity: a symptomatic grade 3 pancreatitis in the first case, and a grade 4 diarrhoea in the second case. Dose escalation was associated with an increased rate of grade ⩾3 diarrhoea (26% vs 3%, P=0.03) and anorexia (26% vs 3%, P=0.03). None of the other severe adverse events, especially hypertension and HFSR, occurred more frequently during dose escalation (Table 2).
The early toxicities (HFSR, rash, diarrhoea and hypertension) that occurred during the first cycle (2 months) were associated with a better PFS (18 vs 12 weeks, P=0.024; HR=0.38 (95% CI: 0.15–0.98)). In univariate analyses, none of the specific early grade ⩾2 toxicity was associated with PFS gain (Table 3). Considering the whole-treatment period toxicities, patients experiencing either grade ⩾2 hypertension or HSFR had improved PFS (19 vs 9 weeks, P<0.0001; HR=0.13 (95% CI: 0.04–0.39)) but not patients experiencing either grade ⩾2 skin rash or diarrhoea (17 vs 13 weeks, P=0.3; HR=0.54 (95% CI: 0.17–2.02)).
Pharmacokinetics
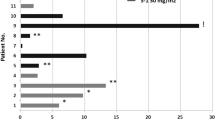
During the whole-study period, 216 sorafenib plasma concentrations were assessed (Supplementary Table 1). The median sorafenib AUC was 63 mg l−1 h−1 (range: 16–206). The median intra-patient variability was 31% (range: 7–71%) and inter-patient variability was 45% at 400 mg bid. Sorafenib exposure did increase with dose (Spearman’s test ρ=0.4, P<0.0001). Inter patient PK analysis showed that the median AUC was higher at all doses ranging from 600 to 1200 mg relative to 400 mg bid (Figure 1). Relative to 600 mg bid, the median AUC did not increase at higher doses. Intra patient PK analysis showed that dose escalation (range: 600–2600 mg bid) in 19 patients allowed achieving a greater sorafenib exposure in 13 (68%) of them (Figure 2). Two and four patients stable and decreasing exposure, respectively.

Effect of dose escalation on inter patient sorafenib AUC (mg l−1 h−1). A total 119 AUCs from 29 patients are represented. Wilcoxon’s P-value: *<0.05, **<0.005, NS >0.05.

Effect of dose escalation on intra patient sorafenib AUC (mg l−1 h−1). Median AUCs from 19 patients are represented. In red: increased exposure; in orange: stable exposure; in green: decreased exposure.
The long-term drug exposure monitoring showed that AUC rapidly reached its maximum after treatment initiation. Maximal AUC occurred during the first 2 months in 18/27 patients (67%) and the median time to reach the AUCmax was 36 days (range 8–161 days). Sorafenib exposure tended to decrease over time in case of prolonged treatment. In 11 patients receiving sorafenib for > 4 months, AUC had decreased in the last part of treatment (after 90 days; 77 vs 61 mg l−1 h−1, P=0.002).
One month after treatment initiation, sorafenib median AUC was greater in patients with grade⩾2 hypertension compared with those with normal blood pressure (82 vs 54 mg l−1 h−1, respectively, P=0.02). Each measurement of sorafenib was compared with the simultaneous safety report (n=194 pairs). The median AUC was greater in case of grade ⩾2 hypertension (84 vs 58 mg l−1 h−1, P<0.0001), and grade ⩾2 HFSR (76 vs 61 mg L.h, P=0.0008). Besides, AUC was not correlated with other adverse events such as diarrhoea, anorexia, allergic and non-allergic skin rash. The rate of severe adverse events (grade ⩾3) was not increased with AUCs ⩾100 mg l−1 h−1 (Table 3).
Concerning the relation between plasma sorafenib exposure and efficacy, it was first noticed that five of six responses occurred at 400 mg bid but these patients had high exposure at this dose (with AUC of 102, 101, 84 and 75 mg l−1 h−1 in four patients, and AUC not avalaible for the remaining patient). Then, the median AUCmax (100 mg l−1 h−1, range 51–206 mg l−1 h−1) was used to classify patients into high or low exposure groups. Patients with high exposure had a higher probability of tumour control on target lesions (86% vs 50%, P=0.04, Figure 3), RECIST partial response or stable disease (80% vs 33%, P=0.02) and PFS (21 vs 10 weeks, P=0.005; HR=0.28 (95% CI: 0.11–0.72); Figure 4; Table 3). The Youden index of the receiver operating characteristic (ROC) curve of the disease control relative to the AUCmax was 100 mg l−1 h−1 (data not shown). Maximal exposure had a positive impact on PFS in univariate analysis (Table 3) and confirmed by the multivariate analysis as AUCmax ⩾100 mg l−1 h−1 (HR=0.28 (95% CI: 0.11–0.72) was the only significant variable associated with PFS (Table 3).

Investigator-assessed tumour regression (i.e., maximum change from baseline in target lesions diameter). (n=27) Patients with RECIST progressive disease are indicated by an asterix. Clear grey: AUCmax <100 mg l−1 h−1; dark grey: AUCmax⩾100 mg l−1 h−1.

PFS probability according to maximal exposure to sorafenib (AUCmax). Dot line: patients with AUCmax <100 mg l−1 h−1/; solid line: patients with AUCmax⩾100 mg l−1 h−1.
Neither the AUC at 1 month after treatment initiation nor the mean AUC of the whole-treatment period were associated with a higher disease control rate (69% vs 46% P=0.4 and 54% vs 64% P=0.7, respectively) or a longer PFS (HRs=0.94 (95% CI: 0.40–2.28) and 0.51 (95% CI: 0.19–1.24), respectively). Thus, the discrepancies between the three pharmacokinetic parameters (AUC at 1 month, mean and max AUC) were investigated. Indeed, 6 (21%) and 8 (28%) patients were misclassified by the AUC at 1 month compared with the mean AUC and the AUCmax, respectively. Moreover, despite a low mean AUC, three responding patients had a high AUCmax, which could explain the clinical effect. Conversely, four patients with a mean AUC above the average but low AUCmax did not respond to the treatment.
Discussion
In this multi-institutional experience with sorafenib dose-escalation in patients with metastatic melanoma, the main results consisted in the positive correlation between AUCmax, objective response and PFS. Although modest in melanoma, sorafenib efficacy was directly correlated with exposure, as seen with sunitinib in RCC and GIST (Houk et al, 2010) or pazopanib in differentiated thyroid cancers (Bible et al, 2010). Consistently with results from the phase I trials (Awada et al, 2005; Clark et al, 2005; Moore et al, 2005; Furuse et al, 2008; Minami et al, 2008; Miller et al, 2009) AUC increased infra-proportionally to the dose. However, the dose-escalation schedule increased AUC in 68% (13/19) patients. In this series, dose adjustments could effectively correct drug under-exposure.
To go further, the changes in sorafenib clearance and bioavalability with doses >400 mg bid were described in a cohort of 71 patients treated with sorafenib in our institution, including the present series of melanoma patients (Hornecker et al, 2011). A one-compartment model with saturated absorption, first-order intestinal loss and elimination best described the pharmacokinetics of sorafenib. Absolute bioavailability significantly dropped with increasing daily doses of sorafenib. Area under the curve increased less than proportionally with increasing doses. Therefore, a split schedule three times a day might overcome absorption saturation, thereby leading to a higher exposure (Hornecker et al, 2011). Notably, tumour type did not seem to influence sorafenib pharmacokinetics. Only albumin was found to influence sorafenib clearance at standard doses (Tod et al, 2011). As well, in an independent cohort (Jain et al, 2011), no clinically important PK covariates were identified.
In this series, the highest AUC (AUCmax) was correlated with antitumor efficacy while the other PK parameters were biased by the dose-escalation schedule: the AUC at 1 month was too early and the mean AUC did not reflect periods of high exposure, shown to be correlated to antitumor efficacy in our study. The Youden index of the ROC curve of the disease control relative to the AUCmax was 100 mg l−1 h−1, suggesting that highest exposures are responsible for efficacy. These properties of antiangiogenic treatments have been previously described and represented by a bell-shaped dose–response curve (Reynolds, 2009). Strikingly, only 15% of samples assessed at 400 mg bid had an AUC over 90 mg l−1 h−1 vs 36% of samples at 600 mg bid and more (P=0.0003). With a target AUC of 90–100 mg l−1 h−1, theses results pinpoint that most patients are underexposed to sorafenib at 400 mg bid, and that individualised dose adjustments would be required. In line with these results, a recent study (Motzer et al, 2011) has shown the superiority of sunitinib 50 mg daily 4 weeks out of 6 over a continuous daily dosing of 37.5 mg, pinpointing the need to reach a threshold exposure.
Long-term pharmacokinetic follow-up allowed detecting that the AUC decreased over time, as previously described in hepatocellular carcinoma (Arrondeau et al, 2011). This unexpected result could explain the clinical efficacy of sorafenib dose escalation after failure at standard doses (Escudier et al, 2009) and argue for long-term pharmacokinetic follow-up. This decrease of AUC over time could result from increased expression of drug efflux pumps, as seen with imatinib (Burger et al, 2005). We therefore suggest validating in a prospective trial the AUC as a surrogate marker to tailor sorafenib dose adjustments, thereby avoiding increasing sorafenib dose until intolerable toxicity. This approach could probably improve the therapeutic index of sorafenib in approved indications such as hepatocellular carcinoma and renal cancer.
The limitations of this study include the limited number of patients, the limited sampling strategy and the proportion of patients in whom sorafenib standard dose was not tolerated. Dose escalation was feasible and no unexpected severe adverse event was seen, even in highly pretreated patients with brain metastasis. Only two patients discontinued sorafenib during dose escalation. Several hypotheses on the pathogenesis of sorafenib-related adverse events could be raised. Indeed, toxicities could be classified in three categories according to their correlation with dose and exposure. Diarrhoea and anorexia were related to sorafenib dose but not to its AUC. Regarding diarrhoea, this result is in line with a previous hypothesis assuming that intestinal toxicity may be due to a local effect of poorly absorbed drug. Indeed, the low solubility of sorafenib in aqueous media hampers its complete dissolution in digestive tract at high doses. Thus, the fraction of sorafenib not absorbed could exert a direct toxic effect on enterocytes. Interestingly, patients with abnormal gastrointestinal functions are prone to develop diarrhoea under sorafenib (Lauritano et al, 2009), and patients with abnormal liver functions have a highest rate of diarrhoea without elevated exposure (Miller et al, 2009; Michels et al, 2010). As a consequence, diarrhoea per se may decrease sorafenib exposure, due to reduced intestinal absorption and interruption of entero-hepatic cycle.
Regarding prediction of toxicity, hypertension and HFSR were related to the AUC in the present series. To date, only one pharmacodynamic study identified a rare polymorphism of VEGFR-2 as a predictor of HFSR and hypertension (Jain et al, 2010). Regarding prediction of efficacy, biomarkers have failed to select patients who would respond to sorafenib. The results of four independent trials conclude BRAFV600E mutation is not a predictive biomarker of response to sorafenib (Eisen et al, 2006; Flaherty et al, 2008; Amaravadi et al, 2009; Ott et al, 2010). We propose optimised maximal AUC (>90–100 mg l−1 h−1) as an alternative predictor for the activity of sorafenib, as illustrated presently in melanoma patients. Dose individualisation with drug monitoring might prevent under exposure to standard dose of sorafenib and favour antitumor activity in other tumour types. Dedicated phase II studies guided by pharmacokinetics are mandatory to prospectively confirm these results.
Change history
17 July 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alsina J, Gorsk DH, Germino FJ, Shih W, Lu SE, Zhang ZG, Yang JM, Hait WN, Goydos JS (2003) Detection of mutations in the mitogen-activated protein kinase pathway in human melanoma. Clin Cancer Res 9: 6419–6425
Amaravadi RK, Schuchter LM, McDermott DF, Kramer A, Giles L, Gramlich K, Carberry M, Troxel AB, Letrero R, Nathanson KL, Atkins MB, O'Dwyer PJ, Flaherty KT (2009) Phase II trial of temozolomide and sorafenib in advanced melanoma patients with or without brain metastases. Clin Cancer Res 15: 7711–7718
Amato RJ, Jac J, Harris P, Dalton S, Saxena S, Monzon F, Zhai J, Brady J, Willis JP (2008) A phase II trial of intra-patient dose-escalated sorafenib in patients (pts) with metastatic renal cell cancer (MRCC). J Clin Oncol 26 (Suppl): abstract 5122
Arrondeau J, Mir O, Boudou-Rouquette P, Coriat R, Ropert S, Dumas G, Rodrigues MJ, Rousseau B, Blanchet B, Goldwasser F (2011) Sorafenib exposure decreases over time in patients with hepatocellular carcinoma. Invest New Drugs doi:10.1007/s10637-10011-19764-10638
Awada A, Hendlisz A, Gil T, Bartholomeus S, Mano M, de Valeriola D, Strumberg D, Brendel E, Haase CG, Schwartz B, Piccart M (2005) Phase I safety and pharmacokinetics of BAY 43-9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br J Cancer 92: 1855–1861
Bible KC, Suman VJ, Molina JR, Smallridge RC, Maples WJ, Menefee ME, Rubin J, Sideras K, Morris JC, McIver B, Burton JK, Webster KP, Bieber C, Traynor AM, Flynn PJ, Goh BC, Tang H, Ivy SP, Erlichman C (2010) Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol 11: 962–972
Blanchet B, Billemont B, Cramard J, Benichou AS, Chhun S, Harcouet L, Ropert S, Dauphin A, Goldwasser F, Tod M (2009) Validation of an HPLC-UV method for sorafenib determination in human plasma and application to cancer patients in routine clinical practice. J Pharm Biomed Anal 49: 1109–1114
Burger H, van Tol H, Brok M, Wiemer EA, de Bruijn EA, Guetens G, de Boeck G, Sparreboom A, Verweij J, Nooter K (2005) Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Biol Ther 4: 747–752
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516
Clark JW, Eder JP, Ryan D, Lathia C, Lenz HJ (2005) Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin Cancer Res 11: 5472–5480
Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, Gibbens I, Hackett S, James M, Schuchter LM, Nathanson KL, Xia C, Simantov R, Schwartz B, Poulin-Costello M, O'Dwyer PJ, Ratain MJ (2006) Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer 95: 581–586
Escudier B, Szczylik C, Hutson TE, Demkow T, Staehler M, Rolland F, Negrier S, Laferriere N, Scheuring UJ, Cella D, Shah S, Bukowski RM (2009) Randomized phase II trial of first-line treatment with sorafenib versus interferon Alfa-2a in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 1280–1289
Flaherty KT, Schiller J, Schuchter LM, Liu G, Tuveson DA, Redlinger M, Lathia C, Xia C, Petrenciuc O, Hingorani SR, Jacobetz MA, Van Belle PA, Elder D, Brose MS, Weber BL, Albertini MR, O'Dwyer PJ (2008) A phase I trial of the oral, multikinase inhibitor sorafenib in combination with carboplatin and paclitaxel. Clin Cancer Res 14: 4836–4842
Furuse J, Ishii H, Nakachi K, Suzuki E, Shimizu S, Nakajima K (2008) Phase I study of sorafenib in Japanese patients with hepatocellular carcinoma. Cancer Sci 99: 159–165
Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, Corless CL, Li L, Li H, Sheng X, Cui C, Chi Z, Li S, Han M, Mao L, Lin X, Du N, Zhang X, Li J, Wang B, Qin S (2011) Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29: 2904–2909
Hornecker M, Blanchet B, Billemont B, Sassi H, Ropert S, Taieb F, Mir O, Abbas H, Harcouet L, Coriat R, Dauphin A, Goldwasser F, Tod M (2011) Saturable absorption of sorafenib in patients with solid tumors: a population model. Invest New Drugs doi:10.1007/s10637-10011-19760-z
Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ (2010) Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol 66: 357–371
Jain L, Sissung TM, Danesi R, Kohn EC, Dahut WL, Kummar S, Venzon D, Liewehr D, English BC, Baum CE, Yarchoan R, Giaccone G, Venitz J, Price DK, Figg WD (2010) Hypertension and hand-foot skin reactions related to VEGFR2 genotype and improved clinical outcome following bevacizumab and sorafenib. J Exp Clin Cancer Res 29: 95
Jain L, Woo S, Gardner ER, Dahut WL, Kohn EC, Kummar S, Mould DR, Giaccone G, Yarchoan R, Venitz J, Figg WD (2011) Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br J Clin Pharmacol 72: 294–305
Lauritano EC, Novi M, Rinninella E, Tortora A, Barbaro F, Piscaglia AC, Santoro M, Zocco MA, Gasbarrini A (2009) The pathogenic mechanims of sorafenib-related diarrhea: preliminary results. Dig Liv Dis 41S: A14
Michels J, Mir O, Blanchet B, Ropert S, Coriat R, Billemont B, Goldwasser F (2010) High incidence of severe sorafenib-induced diarrhea in patients with hyperbilirubinemia and/ or hypoalbuminemia. Ann Oncol 21 (Supplement 8): 1272P
Miller AA, Murry DJ, Owzar K, Hollis DR, Kennedy EB, Abou-Alfa G, Desai A, Hwang J, Villalona-Calero MA, Dees EC, Lewis LD, Fakih MG, Edelman MJ, Millard F, Frank RC, Hohl RJ, Ratain MJ (2009) Phase I and pharmacokinetic study of sorafenib in patients with hepatic or renal dysfunction: CALGB 60301. J Clin Oncol 27: 1800–1805
Min CJ, Liebes LF, Escalon J, Hamilton A, Yee H, Buckley MT, Wright JJ, Osman I, Polsky D, Pavlick AC (2008) Phase II trial of sorafenib (S [BAY 43-9006]) in metastatic melanoma (MM) including detection of BRAF with mutant specific-PCR (MS-PCR) and altered proliferation pathways-final outcome analysis. J Clin Oncol 26s, (Suppl): abstract 9072
Minami H, Kawada K, Ebi H, Kitagawa K, Kim YI, Araki K, Mukai H, Tahara M, Nakajima H, Nakajima K (2008) Phase I and pharmacokinetic study of sorafenib, an oral multikinase inhibitor, in Japanese patients with advanced refractory solid tumors. Cancer Sci 99: 1492–1498
Moore M, Hirte HW, Siu L, Oza A, Hotte SJ, Petrenciuc O, Cihon F, Lathia C, Schwartz B (2005) Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43-9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann Oncol 16: 1688–1694
Motzer RJ, Hutson TE, Olsen MR, Hudes GR, Burke JM, Edenfield WJ, Wilding G, Martell B, Hariharan S, Figlin RA (2011) Randomized phase II multicenter study of the efficacy and safety of sunitinib on the 4/2 versus continuous dosing schedule as first-line therapy of metastatic renal cell carcinoma: Renal EFFECT Trial. J Clin Oncol 29: LBA308
Ott PA, Hamilton A, Min C, Safarzadeh-Amiri S, Goldberg L, Yoon J, Yee H, Buckley M, Christos PJ, Wright JJ, Polsky D, Osman I, Liebes L, Pavlick AC (2010) A phase II trial of sorafenib in metastatic melanoma with tissue correlates. PLoS One 5: e15588
Reynolds AR (2009) Potential relevance of bell-shaped and u-shaped dose-responses for the therapeutic targeting of angiogenesis in cancer. Dose Response 8: 253–284
Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP (2005) Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res 65: 2412–2421
Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B (2007) Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12: 426–437
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216
Tod M, Mir O, Bancelin N, Coriat R, Thomas-Schoemann A, Taieb F, Boudou-Rouquette P, Ropert S, Michels J, Abbas H, Durand JP, Dauphin A, Vidal M, Goldwasser F, Blanchet B (2011) Functional and clinical evidence of the influence of sorafenib binding to albumin on sorafenib disposition in adult cancer patients. Pharm Res 28: 3199–3207
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O'Brien J, Bastian BC (2010) Mutations in GNA11 in uveal melanoma. N Engl J Med 363: 2191–2199
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7: 3129–3140
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumour progression and angiogenesis. Cancer Res 64: 7099–7109
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
FG has worked as paid consultant for Bayer Healthcare and Pfizer. OM has worked as paid consultant for Roche. The other authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Pécuchet, N., Lebbe, C., Mir, O. et al. Sorafenib in advanced melanoma: a critical role for pharmacokinetics?. Br J Cancer 107, 455–461 (2012). https://doi.org/10.1038/bjc.2012.287
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.287
Keywords
This article is cited by
-
Sorafenib administered using a high-dose, pulsatile regimen in patients with advanced solid malignancies: a phase I exposure escalation study
Cancer Chemotherapy and Pharmacology (2020)
-
Clinical development of targeted and immune based anti-cancer therapies
Journal of Experimental & Clinical Cancer Research (2019)
-
Alternative scheduling of pulsatile, high dose sunitinib efficiently suppresses tumor growth
Journal of Experimental & Clinical Cancer Research (2016)
-
How ‘Optimal’ are Optimal Sampling Times for Tyrosine Kinase Inhibitors in Cancer? Practical Considerations
Clinical Pharmacokinetics (2016)
-
Exposure–Toxicity Relationship of Sorafenib in Japanese Patients with Renal Cell Carcinoma and Hepatocellular Carcinoma
Clinical Pharmacokinetics (2014)
