
Abstract
Background:
Despite focused research in conventional therapies and considerable advances in the understanding of the molecular carcinogenesis of head and neck squamous cell carcinoma (HNSCC), the 5-year survival rate for patients with advanced disease remains ∼15–20%. The major causes of HNSCC-related deaths are cervical node and distant metastasis. E-cadherin has a key role in epithelial intercellular adhesion and its downregulation is a hallmark of epithelial–mesenchymal transition (EMT), which is associated with invasion, metastasis, and poor prognosis. Epithelial–mesenchymal transition is the major mechanism responsible for mediating invasiveness and metastasis of epithelial cancers. Recently, we reported the role of E-cadherin transcriptional repressors in the inflammation-induced promotion of EMT in HNSCC, which is mediated by COX-2. These findings suggest that therapies targeting the cyclooxygenase pathway may diminish the propensity for tumour metastasis in HNSCC by blocking the PGE2-mediated induction of E-cadherin transcriptional repressors.
Methods:
Herein, we evaluate the efficacy of the COX-2 inhibitor, apricoxib, in HNSCC cell lines. Apricoxib is effective in preventing tumour cell growth in three-dimensional, and anchorage-independent growth assays, as well as decreasing the capacity for tumour cell migration.
Results:
Herein, we evaluate the efficacy of the COX-2 inhibitor, apricoxib, in HNSCC cell lines. Apricoxib is effective in preventing tumour cell growth in three-dimensional, and anchorage-independent growth assays, as well as decreasing the capacity for tumour cell migration. Treatment of HNSCC cells with apricoxib also causes greater upregulation of E-cadherin and Muc1 expression and downregulation of vimentin, as compared with celecoxib treatment. This has significant implications for targeted chemoprevention and anti-cancer therapy because E-cadherin expression has been implicated as a marker of sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor and other therapies. We show for the first time the molecular mechanisms underlying the efficacy of apricoxib in HNSCC cells.
Conclusion:
In addition to reversing EMT via inhibition of COX-2, apricoxib upregulates 15-prostaglandin dehydrogenase and the prostaglandin transporter, thereby reducing the levels of active PGE2 by both suppressing its synthesis and increasing its catabolism. These findings have significant implications for metastasis and tumour progression in HNSCC.
Similar content being viewed by others


Main
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide with 600 000 cases diagnosed per year (Brennan et al, 1995). As a potential investigational approach for prevention and treatment of HNSCC, there has been extensive study of several types of molecular-targeted agents, including epidermal growth factor receptor (EGFR)-selective tyrosine kinase inhibitors (TKIs), and COX-2 inhibitors. These two types of agents act on different biological targets: tyrosine phosphorylated EGFR and COX-2, respectively. Both targets have been shown to contribute to HNSCC carcinogenesis (Breyer et al, 2001). In spite of extensive research in EGFR and COX-2 inhibitors, their combined use for HNSCC is still in its developmental stage. Despite advances in multimodality therapy, the overall 5-year survival rate is 40–50%, emphasising the importance of new therapeutic strategies (Buchanan et al, 2003). Clinical challenges to improving survival in HNSCC include frequency of late-stage diagnosis, locoregional and metastatic recurrence, and secondary primary tumours.
Studies have shown that the EGFR and COX-2 have an important role in the biology of HNSCC. Overexpression of COX-2 is associated with a poor prognosis in HNSCC, and COX-2 inhibitors have demonstrated synergy when combined with EGFR inhibitors in preclinical models (Chen et al, 2004; Chung et al, 2011). Inflammatory mediators can promote epithelial–mesenchymal transition (EST) and increase resistance to EGFR TKIs in HNSCC (Ferlay et al, 2001). These studies provide a strong rationale for combining a COX-2 inhibitor with an EGFR TKI.
Levels of COX-2 and its catalytic product PGE2 are increased in a variety of malignancies, including HNSCC (Buchanan et al, 2003; Dannenberg and Subbaramaiah, 2003; Cooper et al, 2004). PGE2 can stimulate cell proliferation, motility, and angiogenesis while inhibiting apoptosis and immune surveillance (Buchanan et al, 2003; Cooper et al, 2004). COX-2-derived PGE2 may also promote metastasis by stimulating EMT and cell invasion (Ferlay et al, 2001; Dohadwala et al, 2006). It has been reported that PGE2 is transported or passed through the cell membrane via prostaglandin-specific transporters, including the prostaglandin transporter (PGT, an influx transporter). Intratumoural PGE2 levels depend not only upon the rate of production, but also on the rate of degradation. Inactivation of PGE2 located in the developing tumour microenvironment has been suggested to occur by a two-step model (Haddad et al, 2009). The first step is mediated by the PGT, which engages carrier-mediated membrane transport of prostaglandins, including PGE2, PGF2α, and PGD2 (Haddad et al, 2009), from the extracellular milieu to the cytoplasm. This transporter belongs to the organic anion superfamily of transporting polypeptides that contain 12 transmembrane-spanning domains. The second step of PGE2 inactivation occurs in the cytoplasm, where 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catabolises and thus inactivates PGE2 (Haddad et al, 2009). Studies have shown that 15-PGDH expression is frequently reduced in several other epithelial cancers as well (Ichikawa et al, 1996; Holla et al, 2008), suggesting that abnormalities in catabolism of PGE2 may have an important role in the development of these cancers. Quidville et al have shown that in thyroid cancer the silencing of 15-PGDH by RNA interference enhances proliferation, and 15-PGDH is involved in the anti-proliferative effect of COX inhibitors (Investigator’s Brochure-Apricoxib, 2011).
Recent work has suggested a role for COX-2 inhibitors in HNSCC prevention and treatment. Celecoxib, in conjunction with erlotinib and reirradiation, was shown to be a feasible and clinically active regimen in a population of patients with recurrent HNSCC who had a poor prognosis (Jaeckel et al, 2001). However, the majority of data suggest a limited role for celecoxib in head and neck cancer therapy, either due to toxicity or lack of efficacy (Dannenberg and Subbaramaiah, 2003). Celecoxib was ineffective in controlling oral premalignant lesions in a recent randomised controlled trial (Kao et al, 2011). COX2 inhibition has a chemopreventive effect, but its application as a treatment of HNSCC in a clinical setting still requires further research to overcome its limited anti-cancer effects (Kim et al, 2010).
In comparison with celecoxib, in preclinical models apricoxib has shown greater efficacy in several epithelial carcinomas, with a lower side-effect profile (Dohadwala et al, 2006). Apricoxib is a selective COX-2 inhibitor with preclinical data showing analgesic, anti-inflammatory, and anti-tumour effects. Beyond pain and inflammation, apricoxib has shown anti-tumour effects in epithelial malignancies in animal cancer models. In comparison with celecoxib, apricoxib shows greater efficacy and, in some systems, the magnitude of the difference is greater than might be expected solely on respective in vitro (or ex vivo) IC50 values for enzyme inhibition (Dohadwala et al, 2006). The preclinical safety profile of apricoxib is consistent with a selective COX-2 inhibitor with minimal gastrointestinal toxicity at exposures demonstrating anti-inflammatory and antitumour activity. Recently, apricoxib plus erlotinib was tested in a Phase I study in non-small cell lung cancer and was found to be well tolerated with a 60% disease control rate (Lee et al, 2007).
On the basis of the increased efficacy of apricoxib in epithelial malignancies, we questioned whether apricoxib might alter PGE2levels by altering the eicosanoid pathway by other mechanisms in addition to COX-2 inhibition. In the present study, we show for the first time that the efficacy of apricoxib over celecoxib is likely mediated by an increase in PGT and 15-PGDH levels in addition to its anti-COX-2 effect. Our collective data suggest that the increased efficacy of apricoxib in treating epithelial cancers is mediated by the novel mechanism of coordinated up- and down-regulation of genes involved in PGE2 transport and metabolism. Our work supports a potential role for apricoxib in modulating early stages of carcinogenesis and treatment of progressive disease in HNSCC.
Materials and methods
Reagents and cell lines
HNSCC cells utilised in this study included: Tu686, Tu212 (generously provided by Dr D Shin Papadimitrakopoulou et al, 2008), and OSC, HOC, and TSU (generously provided by Dr M Nagayama Peebles et al, 2007). The HNSCC cell lines Tu686, Tu212, and HOC are used for all of experiments presented unless otherwise specified. All cell lines used are genotyped and checked for mycoplasma contamination on a regular schedule. For three-dimensional spheroid culture experiments, 24-well plates were coated with Engelbreth-Holm-Swarm extracellular matrix extract, resulting in reduction of growth factor (Trevigen, Gaithersburg, MD, USA). In all, 50 000 cells were then added to each well in 500 μl of growth medium. Twenty-four hours later later, the growth medium was replaced with fresh medium containing apricoxib (5 μ M) and 10% Engelbreth-Holm-Swarm. Every other day the medium was changed adding fresh apricoxib (5 μ M) and 10% Engelbreth-Holm-Swarm. Quantification in the spheroid model was performed by counting the number of compact spheroids/HPF.
Total RNA preparation, cDNA synthesis, and real-time PCR
To analyse the regulation of mRNA expression, total RNA from 1 × 106 control and treated HNSCC cells were extracted using Trizol reagent according to the manufacturer’s instructions (Invitrogen, Grand Island, NY, USA). The cDNA was prepared with a kit (Invitrogen) according to the manufacturer’s instructions. mRNA levels were quantified by real-time RT–PCR using the SYBR Green quantitative PCR kit from Bio-Rad in a MyiQ Cycler (Bio-Rad, Hercules, CA, USA) following the manufacturer’s protocol. Amplification was carried out in a total volume of 20 μl for 40 cycles of 15 s at 95 °C, 20 s at 60 °C, and 30 s at 72 °C. Samples were run in triplicate and their relative expression was determined by normalising expression of each target either to glyceraldehyde-3-phosphate dehydrogenase or β-actin. These were then compared with the normalised expression in a reference sample to calculate a fold change value. Primers were designed as previously described (Ferlay et al, 2001).
Western blot analysis
HNSCC cells were washed with PBS and whole-cell lysate was prepared with modified RIPA buffer. Proteins were resolved by SDS—PAGE and analysed by western blot using PVDF membranes (Millipore, Bedford, CA, USA). The membranes were probed with anti-E-cadherin, anti-vimentin, anti-15-PGDH, anti-PGT, or anti-Tubulin antibody (BD Biosciences Pharmingen/Transduction Laboratories, San Jose, CA, USA) at 1 : 5000 dilution; or with anti-p38 or anti-P-p38 antibody (Cell Signaling Technology, Danvers, MA, USA) at 1 : 1000 dilution in TBST containing 1.0% nonfat dry milk. The membranes were developed by the ECL chemiluminescence system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Quantitative analysis was performed by ImageJ software (NIH, Bethesda, MD, USA).
Invasion and migration assay
The invasion assays were performed in Cytoselect 24-well cell invasion chambers (Basement membrane, colorimetric format, Cell Biolabs Inc., San Diego, CA, USA) according to the manufacturer’s protocol. For migration assays, BD Falcon cell culture inserts ( # 353097) were used.
Soft agar assay
In brief, 1.2% noble agar was melted, then cooled to 50 °C in a water bath. Equal volumes of this 1.2% agar solution and 2 × DMEM/20% FBS medium (1 : 1) were then mixed. Immediately, 50 μl of the mixture was added to each well of a 96-well flat-bottom microplate to form a base agar layer. The plate was stored at room temperature to allow the agar to solidify for 1 h. HNSCC Cells were harvested and resuspended in culture medium. Equal volumes of 1.2% agar: 2 × DMEM/20% FBS medium: cell suspension (1 : 1 : 1) were then mixed in a tube and left at 40 °C. At this step, the indicated drugs were added. After 7 days, 12.5 μl alamarblue was added to each well and the cells were incubated at 37 °C overnight. Plates were then read using a Bio-Rad Benchmark plate reader.
Statistics
All experiments were repeated at least three times and measurements were performed in triplicate. When applicable, data are presented as the mean±s.d. Group comparisons were performed using the Student’s t-test or Fisher’s exact test as appropriate. The Spearman rank correlation was used to assess the level of association between the staining intensities of pairs of markers. A P-value of <0.05 was considered significant. The S-plus version 6 (Insightful, Corvallis, OR, USA) was utilised for all statistical analyses.
Results
Apricoxib upregulates 15-PGDH in HNSCC cells
The catabolism of PGE2 has been shown to play an important role in the development of some head and neck cancers (Investigator’s Brochure-Apricoxib, 2011). 15-hydroxyprostaglandin dehydrogenase is known to catabolise and thus inactivate PGE2 (Ichikawa et al, 1996; Haddad et al, 2009). We examined the effects of apricoxib treatment (5 μ M) on 15-PGDH expression in HNSCC cells. Apricoxib treatment resulted in a robust upregulation of 15-PGDH expression (Figure 1A).

Apricoxib (Apr) upregulates 15-PGDH in HNSCC cells. OSC, Tu212, and HOC HNSCC cells were treated with 5 μ M Apr for 18 h. Protein from whole cell lysates were analysed for 15-PGDH expression by western blot as described in Materials and Methods. (A) Apr upregulates 15-PGDH expression at the protein level in all HNSCC cell lines studied. (B) Apr enhances the influence of erlotinib on upregulating 15-PGDH.
To further define the efficacy of apricoxib in a clinical context, we examined the influence of apricoxib+erlotinib on 15-PGDH levels. We then compared this with the influence of celecoxib+erlotinib on 15-PGDH. We noted that apricoxib is more effective than celecoxib at enhancing the effects of erlotinib on 15-PGDH (Figure 1B).
Apricoxib is more effective than celecoxib at upregulating PGT in HNSCC and NSCLC cells
Inactivation of PGE2 located in the tumour microenvironment has been suggested to occur by a two-step process (Haddad et al, 2009). In addition to the inactivation of PGE2 by 15-PGDH, the PGT transports PGE2 from the extracellular milieu to the cytoplasm (Jaeckel et al, 2001). Tu686, Tu212, and A549 NSCLC cells were treated with apricoxib or celecoxib. We noted that apricoxib is more effective than celecoxib at increasing PGT levels in HNSCC and NSCLC cells (Figure 2A). In addition to assessing PGT expression using western blot analysis, we determined PGT mRNA expression by real-time RT–PCR in control and apricoxib-treated HNSCC cell lines (OSC and Tu212). When these lines were exposed to apricoxib, PGT mRNA expression levels were elevated, (Figure 2B). These data demonstrate that apricoxib is more efficacious than celecoxib in upregulating PGT.

Apricoxib (Apr) upregulates PGT in HNSCC cells. OSC, Tu686, and Tu212 HNSCC cells, and A549 NSCLC cells were treated with 5 μ M Apr or 5 μ M celecoxib (Cel) for 18 h. Protein from whole-cell lysates was analysed for PGT expression by western blot as described in Materials and Methods. (A) Apr is more effective than Cel at upregulating PGT in HNSCC and NSCLC cells. (B) OSC and Tu212 HNSCC cells were treated with the indicated concentrations of Apr for 18 h and levels of mRNA for PGT were evaluated by quantitative real-time PCR analysis as described in Materials and Methods. Upon addition of Apr, PGT mRNA expression levels were elevated. Data shown for mean of three experiments; bars, s.e. P<0.05.
Apricoxib is more effective than celecoxib at increasing E-cadherin levels and reversing EMT in HNSCC and NSCLC cells
Epithelial–mesenchymal transition is a process that has important roles in normal organ development and in cancer progression. Epithelial–mesenchymal transition is characterised by the combined loss of E-cadherin and gain of mesenchymal markers, such as vimentin, and increased invasion and migration (Ferlay et al, 2001; Quidville et al, 2004). Loss of E-cadherin is closely correlated with poor prognosis (Breyer et al, 2001; Ferlay et al, 2001; Dannenberg and Subbaramaiah, 2003; Quidville et al, 2004). We examined the effects of apricoxib treatment (5 μ M) on E-cadherin expression in Tu212 and OSC HNSCC cells. Apricoxib caused the upregulation of E-cadherin expression in HNSCC and NSCLC cell lines (Figure 3A). In addition to assessing E-cadherin expression using western blot analysis, we determined E-cadherin mRNA expression by real-time RT–PCR in control and apricoxib-treated HNSCC cell lines. When these lines were exposed to apricoxib, E-cadherin mRNA expression levels were elevated (Figure 3B).

Apricoxib (Apr) increases E-cadherin (E-cad) levels in HNSCC cells. OSC and Tu212 HNSCC cells were treated with 5 μ M Apr for 18 h. Protein from whole-cell lysates was analysed for E-cad expression by western blot as described in Materials and Methods. (A) Apr upregulates E-cad expression at the protein level in all HNSCC cell lines studied. (B) OSC and Tu212 HNSCC cells were treated with the indicated concentrations of Apr for 18 h and levels of mRNA for E-cad were evaluated by quantitative real-time PCR analysis as described in Materials and Methods. Upon addition of Apr, E-cad mRNA expression levels were elevated. Data shown for mean of three experiments; bars, s.e. P<0.05. (C) Apr is more effective than celecoxib (Cel)at increasing E-cad levels in HNSCC and NSCLC cells. OSC and Tu212 HNSCC cells, and A549 NSCLC cells were treated with 5 μ M Apr or 5 μ M Cel for 18 h. E-cad levels were higher in Apr-treated cells. (D) Apr is more effective than Cel at affecting other EMT markers (MUC1 and vimentin).
To further define the efficacy of apricoxib, we compared cells treated with apricoxib to those treated with celecoxib in terms of the effect on E-cadherin and other EMT markers. We noted that apricoxib is more effective than celecoxib at increasing E-cadherin levels in HNSCC (Tu686 and Tu212) and NSCLC (A549) cells (Figure 3C). We went on to look at other EMT markers (MUC1 and vimentin) and noted that apricoxib is also more effective than celecoxib at affecting the levels of other EMT markers (MUC1 and Vimentin, Figure 3D). These data support that apricoxib is more efficacious than celecoxib in reversing EMT, and thus reducing tumour invasion and metastasis.
Apricoxib enhances the effect of erlotinib on inhibiting invasion and migration, and anchorage-independent-growth
Erlotinib (Tarceva, Astellas Inc., Northbrook, IL, USA) is an EGFR-targeting TKI that has shown promise in clinical trials with HNSCC and is currently in advanced stages of clinical testing for treatment of this disease (Sackett et al, 2008; Reckamp et al, 2011). We examined the effect of apricoxib on HNSCC cells using anchorage-independent growth on soft agar. The addition of apricoxib to erlotinib further inhibited anchorage-independent growth in HNSCC (Figure 4B). The addition of apricoxib to erlotinib also exhibited significantly diminished cell migration and invasion compared with control cells in Transwell invasion assays (Figure 4C).

Apricoxib (Apr) prevents HNSCC cell growth and enhances the influence of erlotinib (Erl) on inhibiting invasion and migration. (A) Apr is effective in preventing HNSCC cell growth in spheroid culture. (B) Apr enhances the influence of Erl on the inhibition of anchorage-independent growth. (C) Apr enhances the influence of Erl on inhibiting invasion and migration. Abbreviation: Veh=vehicle.
Apricoxib is effective in preventing HNSCC cell growth in spheroid culture
In addition to driving a change in the expression of EMT markers, treatment with apricoxib conferred a less metastatic phenotype in a spheroid model (Figure 4A). Multicellular spheroids (MCS) have been used as an in vitro model system of micrometastases, useful in studying cell adhesion-dependent morphological changes (Sharafinski et al, 2010). In untreated Tu686 cells, there is decreased E-cadherin, resulting in a more fibroblastoid phenotype (control). When Tu686 cells are treated with apricoxib, the cells undergo mesenchymal–epithelial transition (MET) and revert to a more epithelial phenotype, with functional adhesion molecules (E-cadherin), and they are able to maintain a tight, compact phenotype (Figure 4A).
Discussion
The use of tobacco products is the primary cause of head and neck carcinoma (Brennan et al, 1995; Celis et al, 2008). Carcinogens in tobacco products can induce molecular changes throughout the entire upper aerodigestive tract. Head and neck cancer results from a multistep carcinogenesis process in which increasing degrees of mucosal changes and cellular atypia occur over large areas of the carcinogen-exposed upper aerodigestive tract epithelium. The major causes of head and neck cancer-related deaths are persistent or recurrent disease (Celis et al, 2008).
Numerous challenges and costs associated with clinical trials of therapeutic agents underscore the importance of preclinical studies to provide a biological rationale for use of a given agent as therapy. In vitro studies are important in identifying molecular alterations involved in carcinogenesis that are affected by a specific agent. In the current study, we investigated the efficacy of apricoxib as compared with celecoxib in HNSCC and NSCLC models. Additionally, we examined the synergistic effects of apricoxib and erlotinib. The data demonstrated three interrelated modes of action that potentially account for the greater efficacy of apricoxib as compared with celecoxib: COX-2 inhibition, 15-PGDH upregulation, and PGT upregulation. Thus, levels of pro-tumourigenic PGE2 are reduced at multiple levels: synthesis is decreased, active extracellular pools are depleted by PGT-mediated transport, and intracellular concentrations are reduced by 15-PDGH-mediated catabolism (Figure 5).

Model for collateral pathways of apricoxib activity. We propose that one explanation for the increased efficacy of apricoxib is that it has three interrelated modes of action: COX-inhibition, 15-PGDH upregulation, and PGT upregulation. Thus, levels of pro-tumourigenic PGE2 are reduced at multiple levels: synthesis is decreased; active extracellular pools are depleted by PGT-mediated transport; and intracellular concentrations are reduced by 15-PDGH-mediated catabolism.
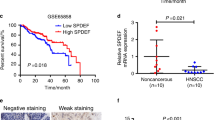
The current study presents the first evidence indicating that PGT and 15-PGDH are upregulated in head and neck and lung cancer cell lines in response to apricoxib treatment. Increased PGE2 in neoplasia results in part from reduced expression of genes involved in PGE2catabolism. Extracellular PGE2 can be transported into the cell, where it can be inactivated or act indirectly via specific nuclear receptors (Vermorken et al, 2007). Quidville had previously reported that the silencing of 15-PGDH by RNA interference enhances proliferation. Our current observation of increased expression of PGT and 15-PGDH in apricoxib-treated HNSCC cell lines could help account for decreased local PGE2 levels in the tumour microenvironment, thus minimising tumour invasion and metastasis.
We also show that apricoxib is effective in preventing tumour cell growth in three-dimensional, anchorage-independent growth assays, as well as decreasing the capacity for tumour cell migration. Treatment of HNSCC cells with apricoxib also causes greater upregulation of E-cadherin expression and downregulation of vimentin, as compared with celecoxib treatment. This has significant implications for targeted chemoprevention and anti-cancer therapy because E-cadherin expression has been implicated as a marker of sensitivity to EGFR TKI.
Epithelial–mesenchymal transition is a major mechanism for carcinoma progression and metastasis. Several studies of E-cadherin in oral and head and neck carcinomas have shown that its reduction or loss is generally related to features that correlate with tumour aggressiveness, such as poorly differentiated or anaplastic histology and widely invasive growth (Breyer et al, 2001; Ferlay et al, 2001; Dannenberg and Subbaramaiah, 2003; Quidville et al, 2004). Distant metastasis-free survival was significantly worse in tumours showing reduced E-cadherin expression (Ferlay et al, 2001; Quidville et al, 2004). Our data on spheroid cell morphology, cell migration and invasion, and the expression of lineage marker proteins clearly indicated that apricoxib is able to promote the transition of HNSCC cells from mesenchymal cells to epithelial cells (Figures 3 and 4). Additionally, we noted that the apricoxib-treated cells grew much more slowly in an anchorage-independent growth assay than the parental cells (Figure 4).
Our current observations demonstrate that apricoxib treatment antagonises EMT in HNSCC cells, indicating an important metastasis-suppressive role of apricoxib and a promising prospect for apricoxib as a valuable therapeutic and chemopreventive drug in HNSCC. The metastasis-suppressive effect may be a consequence of an apricoxib-induced reversal of the mesenchymal phenotype of HNSCC cells (Figures 3 and 4). On the basis of our data, we hypothesise that apricoxib stimulates the reversion of malignant cells towards a relatively normal phenotype. In addition, apricoxib drives MET, increasing the expression of E-cadherin and downregulating vimentin. As elevated levels of E-cadherin are associated with increased sensitivity to other chemotherapeutic agents, this has added benefit in patient therapy.
In summary, we report the Phase 2 COX-2 inhibitor apricoxib displayed superior activity to celecoxib in HNSCC in vitro, due to upregulation of PGE2catabolic pathways in concert with COX-2 inhibition. Apricoxib also reversed EMT in HNSCC lines, which may underlie its synergy with erlotinib (Vermorken et al, 2007; St John et al, 2009). Recently, apricoxib plus erlotinib was tested in a Phase I study (non-small cell lung cancer) and was found to be well tolerated with a 60% disease control rate (Reckamp). The current study justifies studying apricoxib as a strategy for chemoprevention and therapy of HNSCC. If apricoxib is found to have clinical activity in humans, it will represent an important therapeutic opportunity for patients who, either due to premalignant lesion, or a prior diagnosis of HNSCC, are at high risk for this often fatal malignancy for which there is currently no approved preventive therapy.
Change history
30 July 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Brennan JA, Boyle JO, Koch WM (1995) Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N Engl J Med 332: 712–717
Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001) Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41: 661–690
Buchanan FG, Wang D, Bargiacchi F, DuBois RN (2003) Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem 278: 35451–35457
Celis JE, Gromov P, Cabezón T, Moreira JM, Friis E, Jirström K, Llombart-Bosch A, Timmermans-Wielenga V, Rank F, Gromova I (2008) 15-prostaglandin dehydrogenase expression alone or in combination with ACSM1 defines a subgroup of the apocrine molecular subtype of breast carcinoma. Mol Cell Proteomics 7: 1795–1809
Chen Z, Zhang X, Li M, Wang Z, Wigand HS, Grandis JR, Shin DM (2004) Simultaneously targeting epidermal growth factor receptor tyrosine kinase and cyclooxygenase-2, an efficient approach to inhibition of squamous cell carcinoma of the head and neck. Clin Can Res 10: 5930–5939
Chung JH, Rho JK, Xu X, Lee JS, Yoon HI, Lee CT, Choi YJ, Kim HR, Kim CH, Lee JC (2011) Clinical and molecular evidences of epithelial to mesenchymal trans ition in acquired resistance to EGFR-TKIs. Lung Cancer 73: 176–182
Cooper JS, Pajak TF, Forastiere AA (2004) Postoperative concurrent radio- therapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med 350: 1937–1944
Dannenberg AJ, Subbaramaiah K (2003) Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell 4: 431–436
Dohadwala M, Yang SC, Luo J (2006) Cyclooxygenase-2–dependent regulation of E-cadherin: prostaglandin E2 induces transcriptional repressors ZEB1 and Snail in non–small cell lung cancer. Cancer Res 66: 5338–5345
Ferlay J, Bray F, Pisani P, Parkin DM (2001) GLOBOCAN 2000: Cancer Incidence, Mortality and Prevalence Worldwide, Version 1.0. IARC Cancer Base 5. WHO Press: Switzerland
Haddad Y, Choi W, McConkey DJ (2009) Delta-crystallin enhancer binding factor 1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous cell carcinoma lines. Clin Cancer Res 15: 532–542
Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN (2008) Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res 1: 93–99
Ichikawa A, Sugimoto Y, Negishi M (1996) Molecular aspects of the structures and functions of the prostaglandin E receptors. J Lipid Mediat Cell Signal 14: 83–87
Investigator’s Brochure-Apricoxib (2011) Sponsor: Tragara Pharmaceutical, Inc. Version Number 3
Jaeckel EC, Raja S, Tan J, Das SK, Dey SK, Girod DA, Tsue TT, Sanford TR (2001) Correlation of expression of cyclooxygenase-2, vascular endothelial growth factor, and peroxisome proliferator-activated receptor delta with head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 127: 1253–1259
Kao J, Genden EM, Chen CT, Rivera M, Tong CC, Misiukiewicz K, Gupta V, Gurudutt V, Teng M, Packer SH (2011) Phase 1 trial of concurrent erlotinib, celecoxib, and reirradiation for recurrent head and neck cancer. Cancer 117: 3173–3181
Kim YY, Lee EJ, Kim YK, Kim SM, Park JY, Myoung H, Kim MJ (2010) Anti-cancer effects of celecoxib in head and neck carcinoma. Mol Cells 29: 185–194
Lee GY, Kenny PA, Lee EH, Bissell MJ (2007) Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 4 (4): 359–365
Papadimitrakopoulou VA, William WN, Dannenberg AJ, Lippman SM, Lee JJ, Ondrey FG, Peterson DE, Feng L, Atwell A, El-Naggar AK, Nathan CA, Helman JI, Du B, Yueh B, Boyle JO (2008) Pilot randomized phase II study of celecoxib in oral premalignant lesions. Clin Cancer Res 14 (7): 2095–2101
Peebles KA, Lee JM, Mao JT, Hazra S, Reckamp KL, Krysan K, Dohadwala M, Heinrich EL, Walser TC, Cui X, Baratelli FE, Garon E, Sharma S, Dubinett SM (2007) Inflammation and lung carcinogenesis: applying findings in prevention and treatment. Expert Rev Anticancer Ther 7: 1405–1421
Quidville V, Segond N, Pidoux E, Cohen R, Jullienne A, Lausson S (2004) Tumor growth inhibition by indomethacin in a mouse model of human medullary thyroid cancer: implication of cyclooxygenases and 15-hydroxyprostaglandin dehydrogenase. Endocrinology 145: 2561–2571
Reckamp K, Gitlitz B, Chen LC, Patel R, Milne G, Syto M, Jezior D, Zaknoen S (2011) Biomarker-based phase I dose-escalation, pharmacokinetic, and pharmacodynamic study of oral apricoxib in combination with erlotinib in advanced nonsmall cell lung cancer. Cancer 117: 809–818
Sackett MK, Bairati I, Meyer F, Jobin E, Lussier S, Fortin A, Gélinas M, Nabid A, Brochet F, Têtu B (2008) Prognostic significance of cyclooxygenase-2 overexpression in glottic cancer. Clin Cancer Res 14 (1): 67–73
Sharafinski ME, Ferris RL, Ferrone S, Grandis JR (2010) Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck. Head Neck 32: 1412–1421
St John MA, Dohadwala M, Luo J, Wang G, Lee G, Shih H, Heinrich E, Krysan K, Walser T, Hazra S, Zhu L, Lai C, Abemayor E, Fishbein M, Elashoff DA, Sharma S, Dubinett SM (2009) Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin Cancer Res 15: 6018–6027
Vermorken JB, Trigo J, Hitt R (2007) Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol 25 (16): 2171–2177
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
St John, M., Wang, G., Luo, J. et al. Apricoxib upregulates 15-PGDH and PGT in tobacco-related epithelial malignancies. Br J Cancer 107, 707–712 (2012). https://doi.org/10.1038/bjc.2012.203
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.203
Keywords
This article is cited by
-
Glycogen synthase kinase-3β mediated regulation of matrix metalloproteinase-9 and its involvement in oral squamous cell carcinoma progression and invasion
Cellular Oncology (2018)
-
New Insights on COX-2 in Chronic Inflammation Driving Breast Cancer Growth and Metastasis
Journal of Mammary Gland Biology and Neoplasia (2015)
