
Abstract
Background:
In a phase I dose-escalation study, regorafenib demonstrated tolerability and antitumour activity in solid tumour patients. The study was expanded to focus on patients with metastatic colorectal cancer (CRC).
Methods:
Patients received oral regorafenib 60–220 mg daily (160 mg daily in the extension cohort) in cycles of 21 days on, 7 days off treatment. Assessments included toxicity, response, pharmacokinetics and pharmacodynamics.
Results:
Thirty-eight patients with heavily pretreated CRC (median 4 prior lines of therapy, range 0–7) were enrolled in the dose-escalation and extension phases; 26 patients received regorafenib 160 mg daily. Median treatment duration was 53 days (range 7–280 days). The most common treatment-related toxicities included hand–foot skin reaction, fatigue, voice change and rash. Twenty-seven patients were evaluable for response: 1 achieved partial response and 19 had stable disease. Median progression-free survival was 107 days (95% CI, 66–161). At steady state, regorafenib and its active metabolites had similar systemic exposure. Pharmacodynamic assessment indicated decreased tumour perfusion in most patients.
Conclusion:
Regorafenib showed tolerability and antitumour activity in patients with metastatic CRC. This expanded-cohort phase I study provided the foundation for further clinical trials of regorafenib in this patient population.
Similar content being viewed by others


Main
Globally, colorectal cancer (CRC) is the fourth most common cancer in men and the third most common cancer in women (Parkin et al, 2005). Despite effective screening, 20–25% of patients with CRC present with advanced, unresectable disease and are treated primarily with palliative systemic therapy (Hamilton and Grem, 1998).
5-Fluorouracil (5-FU)-based chemotherapy has long been the mainstay of systemic therapy for advanced or metastatic CRC (Cercek and Saltz, 2010). For first-line treatment, current guidelines recommend folinic acid+5-FU+oxaliplatin (FOLFOX) or folinic acid+5-FU+irinotecan (FOLFIRI) in combination with a targeted monoclonal antibody (bevacizumab, cetuximab or panitumumab; National Comprehensive Cancer Network, 2011). The use of the anti-epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab is not recommended for patients whose tumours harbour KRAS mutations. There is no standard therapy after failure of all these therapies, and new effective treatment options are urgently needed.
Regorafenib (BAY 73-4506; Bayer Pharma AG, Berlin, Germany) is an orally active, potent multi-kinase inhibitor that targets a broad range of angiogenic, stromal and oncogenic kinases, including vascular endothelial growth factor (VEGF) receptors 1, 2 and 3, tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2 (TIE-2), platelet-derived growth factor receptor-β, c-kit, ret, raf-1 and BRAF (both wild-type and the V600E mutant; Wilhelm et al, 2011). In preclinical studies, regorafenib demonstrated antitumour activity in a broad spectrum of xenograft models, including CRC (Wilhelm et al, 2011).
A first-in-human, open-label, non-randomised, phase I dose-escalation study in patients with advanced solid tumours demonstrated that single-agent regorafenib was well tolerated up to the recommended phase II dose of 160 mg once daily, given in cycles of 21 days on, 7 days off. Preliminary activity was observed in patients with CRC (Mross et al, 2012). This study was expanded to further evaluate regorafenib (160 mg once daily) in patients with advanced or metastatic CRC.
Patients and Methods
The study took place at three specialist cancer centres in Germany between July 2005 and June 2009. The protocol was approved by the independent ethics committee at each study centre. Before enrolment, all patients had to provide written, informed consent to participate. The study met all local legal and regulatory requirements and followed the Declaration of Helsinki.
Patients
In the dose-escalation phase of the study, patients with advanced, histologically or cytologically confirmed solid tumours (including CRC), non-Hodgkin's lymphoma or multiple myeloma were eligible. An additional extension cohort was planned, to focus on patients with cancer types that were considered to be promising targets for regorafenib, based on experience gained in the dose-escalation phase. In the event, CRC was identified as the tumour type of most interest for further analysis; thus the extension phase included patients with advanced, histologically confirmed CRC.
Patients had to be aged at least 18 years and have an Eastern Cooperative Oncology Group performance status of 0–2, life expectancy of at least 12 weeks, and adequate bone marrow, liver and renal function. Key exclusion criteria included a history of cardiac disease, congestive heart failure (New York Heart Association classes II–IV), uncontrolled hypertension, human immunodeficiency virus infection, active hepatitis B or C virus infection or other active clinically serious infections (grade >2 according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTC-AE) v3.0).
The participants had to have malignancies that were refractory to standard treatment or for which no standard therapy was available; in addition, patients could be included if a standard treatment was available but the patient had refused it. During the study, patients did not receive any other anticancer therapy.
Dosing and administration
In the initial dose-escalation phase, patients received once daily oral doses of regorafenib ranging from 60 to 220 mg in repeating 28-day cycles of 21 days on treatment, 7 days off treatment. In the extension cohort, patients received regorafenib 160 mg orally once daily (21 days on, 7 days off treatment).
Treatment continued until tumour progression, occurrence of unacceptable toxicity or withdrawal of consent. Interruptions in dosing or dose reductions were permitted if a patient experienced an adverse event that was considered to be related to the study medication and was at least grade 3 in severity, according to NCI CTC-AE v3.0. Individual patients could not have their dose increased.
Assessments
A pre-study examination was performed within 14 days before the first treatment dose. A complete end-of-treatment assessment was performed within 14 days of ending regorafenib treatment. Other assessments were undertaken as described below.
Safety evaluations included physical examinations, electrocardiogram, standard laboratory tests and measurement of vital signs. Adverse events were assessed according to NCI CTC-AE v3.0. Patients were reviewed for evidence of cumulative toxicity from repeated cycles of treatment. A follow-up visit was made 30±4 days after the last administration of regorafenib to collect information on ongoing toxicities or significant adverse events. Any adverse events that occurred within 30 days of the last dose of study medication were followed until resolution.
Tumour response and progression were evaluated using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 (Eisenhauer et al, 2009). All lesions were measured using X-ray, magnetic resonance imaging (MRI) or computed tomography at baseline, and were assessed and reported at the end of every second treatment cycle beginning at baseline, unless progression was noted.
Progression-free survival (PFS) was measured as the number of days from start of treatment to progression, death before progression (patient not censored) or last tumour evaluation at which the patient was known to have not progressed (patient censored). The Kaplan–Meier product-limit estimate of PFS was derived for all patients and for those with KRAS-mutated or wild-type tumours.
KRAS mutations were analysed using archival tumour or plasma samples. Using the QIAamp DNA Micro Kit (Qiagen, Crawley, UK), DNA was isolated from formalin-fixed, paraffin-embedded tissue sections of archival tumour biopsies collected at diagnosis, and mutational analysis was performed by Sequenom, Inc. (San Diego, CA, USA) using OncoCarta v1.0, which examines 238 mutational ‘hotspots’ in 19 different cancer-related genes. Plasma samples collected at baseline or during the study were analysed for mutations using the BEAMing technology (Inostics GmbH, Hamburg, Germany; Dressman et al, 2003).
Plasma concentrations of regorafenib and its pharmacologically active metabolites M2 (N-oxide metabolite; BAY 75–7495) and M5 (N-oxide/N-desmethyl metabolite; BAY 81–8752) were measured in blood samples collected on days 1 and 21 of cycle 1 (dose-escalation cohort) and day 21 of cycles 1 and 2 (extension cohort). Analyte concentrations were determined by high-performance liquid chromatography with tandem mass spectrometric detection, with a lower quantification limit of 2 μg l–1. Analyses were performed in accordance with the Food and Drug Administration guidelines on bioanalytical validation (Food and Drug Administration, 2001). Pharmacokinetic parameters were calculated using standard non-compartmental methods (WinNonlin Version 4.1; Pharsight Corporation, Mountain View, CA, USA) and summarised as geometric mean and geometric coefficient of variation (CV). Point estimators (least square means) and two-sided 90% confidence intervals for area under the concentration–time curve from 0 to 24 h (AUC0–24) and maximum concentration (Cmax) ratios (cycle 2:cycle 1 in extension cohort) were calculated using an analysis of variance model.
Tumour-perfusion properties were measured using dynamic contrast-enhanced (DCE) MRI at screening, on days 2 and 21 of cycle 1, on day 21 of cycles 2–4, on day 21 of every second cycle thereafter, and at the final visit. The area under the contrast agent concentration–time curve during the first 60 s after arrival of the contrast agent (iAUC60) was used as the DCE-MRI endpoint (Mross et al, 2009). The association between DCE-MRI endpoint and PFS was investigated by censored data linear rank statistics based on the Wilcoxon scores using the DCE-MRI endpoint as covariate.
Results
Results are presented for all 38 patients with metastatic CRC who were enrolled in this phase I study during the dose-escalation (n=15) and extension (n=23) phases. Patient demographic and baseline characteristics are shown in Table 1. Patients had previously received a median of 4 lines (range 0–7) of systemic therapy for metastatic disease, including oxaliplatin, irinotecan, bevacizumab and anti-EGFR antibodies; one patient with no prior treatment had refused to receive any systemic treatment before enrolling in this study. Twenty-eight patients had received regimens containing both oxaliplatin and irinotecan.
Table 2 shows the number of patients receiving regorafenib at each dose level and the duration of treatment. Reasons for discontinuation of regorafenib were progressive disease (n=19; 50%), consent withdrawal (n=5; 13%), treatment-related adverse events (n=11; 29%) and non-treatment-related adverse events (n=3; 8%).
Safety
Treatment-emergent, drug-related adverse events were reported in 32 (84%) of the 38 patients with CRC (Table 3). All treatment-related adverse events were grade 3 or lower, except one case of clinically asymptomatic grade 4 thrombocytopenia. No grade 5 adverse events were reported. Dose reduction or interruption of regorafenib was necessary in 25 patients (66%). The most common adverse event leading to dose reduction was hand–foot skin reaction. Overall, regorafenib was permanently discontinued in 11 patients (29%) due to a treatment-related adverse event. Among the 25 patients treated at the 160 mg dose level, 6 patients permanently discontinued regorafenib due to treatment-related adverse events (hand–foot skin reaction n=1; hypertension n=1; fatigue n=1; fatigue and other constitutional symptoms n=1; thrombocytopenia n=1; duodenal ulcer n=1).
Efficacy
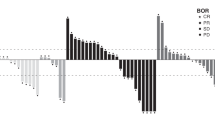
Eleven patients were not evaluable for tumour response for the following reasons: discontinuation of treatment due to adverse events before the first tumour assessment at the end of cycle 2 (n=8); consent withdrawal before the tumour assessment at the end of cycle 2 (n=1); diagnosis of brain metastasis 1 day after the start of treatment (n=1); and missing tumour assessment (n=1). As a result, 27 patients were evaluable for response. Best responses included a confirmed partial response in one patient (4%) and stable disease in 19 patients (70%), giving a disease control rate (partial response or stable disease lasting for at least 2 months) of 74%. Seven patients (26%) had progressive disease as best response. Patients achieving partial response or stable disease had received previous treatment with cetuximab or panitumumab (45%), bevacizumab (45%), oxaliplatin (85%) or irinotecan (80%). The maximum percentage change in tumour size from baseline for each patient is shown in Figure 1. Tumour shrinkage (of any percentage at any time during the study) was observed in 13 patients.

Maximum percentage change in target lesion sum from baseline (n=27).
Median PFS was 107 days (95% CI, 66–161). At data cutoff, 13 patients had PFS of >100 days (Figure 2).

Progression-free survival for individual patients, showing KRAS mutational status.
KRAS mutation analysis
KRAS mutations were found in 19 of 37 patients with tumour or plasma samples; 18 patients had wild-type KRAS and 1 had no available sample (see Figure 2). The Kaplan–Meier analysis shows no clear difference in PFS between KRAS wild-type and mutant group, even though median PFS was 84 days in the mutant group and 161 days in the wild-type group. The data from this exploratory mutational analysis performed in this relatively small number of subjects with a relatively high number of censored data indicate that the presence of a KRAS mutation does not preclude regorafenib activity.
Pharmacokinetic assessments
Plasma concentration–time profiles at steady state (day 21 of cycles 1 and 2) of regorafenib and its metabolites M2 and M5 are shown in Figure 3. The difference between Cmax and minimum concentration (Cmin) within the dosing interval of 24 h was small, with mean Cmax:Cmin ratios of ∼2–3 for both regorafenib and M2, and ∼1.5 for M5. Steady-state pharmacokinetic profiles demonstrated similar mean systemic exposure of regorafenib and its metabolites. Ratios for cycle 2:cycle 1 are shown in Table 4. AUC0–24 and Cmax at steady state showed pronounced variability between patients, with inter-individual CV of ∼60–90% for regorafenib and M2, and >100% for M5. The intra-individual CVs for Cmax and AUC0–24 (n=14) were 32–34% for regorafenib, 37–46% for M2 and 52–63% for M5 (see Table 4). Intra-individual variability was therefore markedly reduced compared with inter-individual variability. At the 160 mg dose, plasma exposure at steady state (AUC0–24, geometric mean) of the metabolites M2 (48 mg h l–1) and M5 (65–79 mg h l–1) was similar to, or slightly greater than, that of regorafenib (45–50 mg h l–1). The terminal half-life of M2 (25 h) was comparable to that of regorafenib (26–28 h), although the elimination of M5 was slower, with an estimated half-life of 51–64 h.

Concentration–time profile of regorafenib and its metabolites M2 (N-oxide metabolite) and M5 (N-oxide/N-desmethyl metabolite).
Pharmacodynamic assessments
Tumour perfusion in representative lesions was measured by DCE-MRI and the iAUC60 data are shown in Figure 4. The median ratio to baseline was 0.803 (range 0.367–1.799,n=26) on day 2 of cycle 1, 0.661 (range 0.129–1.143, n=22) on day 21 of cycle 1 and 0.507 (range 0.031–1.53, n=20) on day 21 of cycle 2. There was no apparent association between iAUC60 (natural logarithm of ratio to baseline) and PFS (P=0.74).

Dynamic contrast-enhanced magnetic resonance imaging: ratio to baseline of the area under the contrast agent concentration–time curve during the first 60 s after arrival of the contrast agent.
Discussion
The current study evaluated regorafenib in 38 patients with heavily pretreated, advanced or metastatic CRC. Regorafenib was well tolerated in this study. The most common treatment-related adverse events included hand–foot skin reactions, fatigue, voice changes, anorexia and diarrhoea. In general, adverse events were manageable in an outpatient setting with dose reduction or interruption. These findings are consistent with the safety profile observed in patients with other solid tumours (Mross et al, 2012) and with results from a phase II study of regorafenib in patients with untreated metastatic or unresectable renal cell carcinoma (Eisen et al, 2011).
In this study, regorafenib demonstrated clinical efficacy in patients with metastatic CRC. According to RECIST, a disease control rate of 74% was achieved (4% partial response, 70% stable disease). Such activity is encouraging, given that, with one exception (a patient who had refused chemotherapy), these patients had been heavily pretreated: the vast majority of patients had failed standard therapies containing 5-FU, oxaliplatin and irinotecan; approximately half of the patients had also received either anti-VEGF or anti-EGFR agents (including some patients who appear to have received anti-EGFR therapy despite having KRAS-mutant tumours—possibly at a time when the importance of KRAS status was not as universally understood as it is now). Best supportive care or participation in a clinical trial is usually recommended in this setting (Cercek and Saltz, 2010; National Comprehensive Cancer Network, 2011). Although only one confirmed partial response was observed in this study, most patients achieved disease stabilisation, which has been shown to be clinically meaningful (Akbulut et al, 2004; Bitossi et al, 2008; van Cutsem et al, 2008). One group estimated that in patients with chemorefractory metastatic CRC receiving panitumumab ∼80% of the treatment effect on PFS was due to disease stabilisation (van Cutsem et al, 2007). Therefore, it is possible that the high disease control rate observed with regorafenib may translate into a survival benefit. However, it must be borne in mind that this was primarily a dose-finding and safety study and the true survival impact needs to be verified in large randomised controlled trials.
Angiogenesis is a validated therapeutic target for CRC. Bevacizumab, an anti-VEGF-A antibody, in combination with standard chemotherapy improves PFS and overall survival compared with chemotherapy alone in metastatic CRC (Hurwitz et al, 2004); to date, bevacizumab is the only antiangiogenic agent approved for the treatment of CRC. Consistent with preclinical data, pharmacodynamic assessment by DCE-MRI in the present phase I study indicated reduced tumour perfusion after regorafenib treatment in a majority of patients with metastatic CRC, suggesting that regorafenib may have antiangiogenic effects. However, DCE-MRI has not been fully validated as a pharmacodynamic marker for antiangiogenic agents. In this study, no apparent correlation was observed between DCE-MRI change and PFS. Given the broad kinase inhibitory spectrum of regorafenib, inhibition of other, non-angiogenic kinases may have contributed to the reported clinical activity. Further investigations are required to fully elucidate the mechanism of action of regorafenib (angiogenic and non-angiogenic) in CRC.
Mutation in the KRAS gene is a well established biomarker in CRC that clearly predicts lack of activity with the anti-EGFR agents cetuximab and panitumumab (Cercek and Saltz, 2010; National Comprehensive Cancer Network, 2011). An exploratory mutational analysis was undertaken in the present study to investigate whether KRAS status might have an impact on response to regorafenib. Kaplan–Meier analysis showed no statistically significant difference in PFS between patients with KRAS wild-type and mutant tumours. However, the small number of patients (with a relatively high number of censored data) resulted in wide confidence intervals. Thus, no firm conclusions can be drawn about the impact of KRAS status on response to regorafenib therapy from the present study. Further investigations in larger patient populations are needed to provide more conclusive answers to this question.
Systemic exposures of regorafenib and its active metabolites M2 and M5 at steady state demonstrated markedly lower intra-individual than inter-individual variability. Compared with the parent drug, M2 and M5 had similar or slightly greater systemic exposure, and similar or slower elimination. In preclinical in vitro and xenograft studies, M2 and M5 have been shown to be pharmacologically active, with efficacies similar to the parent compound (Zopf et al, 2010). Therefore, the M2 and M5 metabolites of regorafenib are likely to contribute to clinical activity.
In conclusion, regorafenib 160 mg orally once daily in cycles of 21 days on, 7 days off treatment was tolerable, had a manageable adverse event profile and demonstrated clinical activity in heavily pretreated patients with metastatic CRC. As a result of the outcomes from this study, it was deemed appropriate to consider CRC as a focus for further investigation of regorafenib, including a randomised, double-blind, placebo-controlled, phase III study of best supportive care plus either regorafenib or placebo in patients with metastatic CRC, who have progressed after standard therapy (the CORRECT study; ClinicalTrials.gov identifier: NCT01103323).
Change history
23 January 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Akbulut H, Icli F, Yalcin B, Demirkazik A, Onur H, Buyukcelik A, Utkan G (2004) Activity of irinotecan, cisplatin and dacarbazine (CPD) combination in previously treated patients with advanced colorectal carcinoma. Exp Oncol 26: 149–152
Bitossi R, Sculli CM, Tampellini M, Alabiso I, Brizzi MP, Ferrero A, Ottone A, Bellini E, Gorzegno G, Berruti A, Dogliotti L (2008) Gemcitabine and protracted 5-fluorouracil infusion as third-line chemotherapy in refractory colorectal cancer patients. Anticancer Res 28: 3055–3060
Cercek A, Saltz L (2010) Evolving treatment of advanced colorectal cancer. Curr Oncol Rep 12: 153–159
Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B (2003) Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci USA 100: 8817–8822
Eisen T, Joensuu H, Nathan P, Harper P, Wojtukiewicz M, Nicholson S, Bahl B, Tomczak P, Wagner A, Quinn D (2011) Phase II trial of the oral multikinase inhibitor regorafenib (BAY 73-4506) as first-line therapy in patients with metastatic or unresectable renal cell cancer (RCC). Eur J Cancer 47 (Suppl): Abstract 7. 141
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247
Food and Drug Administration (2001) Guidance for Industry. Bioanalytical Method Validation. Food and Drug Administration: Silver Spring, MD, USA
Hamilton J, Grem J (1998) Lower gastrointestinal tract cancer. In Current Cancer Therapeutics Kirkwood JM, Lotze MT, Yasko JM (eds) 3rd edn, pp 156–160. Churchill Livingstone, Current Medicine Inc.: Philadelphia, PA, USA
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342
Mross K, Fasol U, Frost A, Benkelmann R, Kuhlmann J, Büchert M, Unger C, Blum H, Hennig J, Milenkova TP, Tessier J, Krebs AD, Ryan AJ, Fischer R (2009) DCE-MRI assessment of the effect of vandetanib on tumor vasculature in patients with advanced colorectal cancer and liver metastases: a randomized phase I study. J Angiogenes Res 1: 5
Mross K, Frost A, Steinbild S, Hedborn S, Büchert M, Fasol U, Unger C, Kraetzschmar J, Heinig R, Boix O, Christensen O (2012) A phase I dose escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res; e-pub ahead of print 20 April 2012
National Comprehensive Cancer Network (2011) NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network: Fort Washington, PA, USA
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108
Van Cutsem E, Siena S, Humblet Y, Canon JL, Maurel J, Bajetta E, Neyns B, Kotasek D, Santoro A, Scheithauer W, Spadafora S, Amado RG, Hogan N, Peeters M (2008) An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol 19: 92–98
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25: 1658–1664
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D (2011) Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129: 245–255
Zopf D, Heinig R, Thierauch K-H, Hirth-Dietrich C, Hafner F-T, Christensen O, Lin T, Wilhelm S, Radtke M (2010) Regorafenib (BAY 73-4506): preclinical pharmacology and clinical identification and quantification of its major metabolites (17–21 April) (2010). At the American Association for Cancer Research 101st Annual Meeting, Washington, DC, USA
Acknowledgements
We thank Dr Frank-Thorsten Hafner for the bioanalytical work on regorafenib and metabolites, as well as Katherine Wilson and Sara Black for writing assistance in the preparation and revision of the draft manuscript. Bayer Pharma AG (Berlin, Germany) sponsored this study and provided funding for medical writing assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Strumberg, D., Scheulen, M., Schultheis, B. et al. Regorafenib (BAY 73-4506) in advanced colorectal cancer: a phase I study. Br J Cancer 106, 1722–1727 (2012). https://doi.org/10.1038/bjc.2012.153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.153
Keywords
This article is cited by
-
Platform combining statistical modeling and patient-derived organoids to facilitate personalized treatment of colorectal carcinoma
Journal of Experimental & Clinical Cancer Research (2023)
-
Appropriate dose of regorafenib based on body weight of colorectal cancer patients: a retrospective cohort study
BMC Cancer (2023)
-
Effects of regorafenib on the mononuclear/phagocyte system and how these contribute to the inhibition of colorectal tumors in mice
European Journal of Medical Research (2023)
-
Phase II dose titration study of regorafenib in progressive unresectable metastatic colorectal cancer
Scientific Reports (2023)
-
Defining regorafenib as a senomorphic drug: therapeutic potential in the age-related lung disease emphysema
Experimental & Molecular Medicine (2023)
