
Abstract
Background:
Lapatinib is a dual inhibitor of epidermal growth factor receptor (EGFR) and human EGFR-2 (HER-2) tyrosine kinases. This study investigated the pharmacodynamic and clinical effects of lapatinib in patients with locally advanced squamous cell carcinoma of the head and neck (SCCHN).
Methods:
In total, 107 therapy-naive patients with locally advanced SCCHN were randomised (2 : 1) to receive lapatinib or placebo for 2–6 weeks before chemoradiation therapy (CRT). Endpoints included apoptosis and proliferation rates, clinical response, and toxicity.
Results:
Versus placebo, lapatinib monotherapy did not significantly increase apoptosis detected by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick-end labelling or caspase-3 assays. A statistically significant decrease in proliferation using Ki67 assay was observed (P=0.030). In a subset of 40 patients that received ⩾4 weeks of lapatinib or placebo, objective response rate (ORR) was 17% (n=4/24) vs 0% (n=0/16). In the lapatinib single-agent responders, all had EGFR overexpression, 50% had EGFR amplification, and 50% had HER2 expression by immunohistochemistry (including one patient with HER2 amplification). However, these patients showed variable modulation of apoptosis, proliferation, and phosphorylated EGFR on drug treatment. Following CRT, there was a statistically non-significant difference in ORR between lapatinib (70%) and placebo (53%). There was no clear correlation between changes in apoptosis or proliferation and response to chemoradiation. Mucosal inflammation, asthenia, odynophagia, and dysphagia were the most commonly reported adverse events with lapatinib.
Conclusion:
Short-term lapatinib monotherapy did not demonstrate apoptotic changes, but provided evidence of clinical activity in locally advanced SCCHN, and warrants further investigation in this disease.
Similar content being viewed by others
Main
The epidermal growth factor receptor (EGFR) is involved in normal cell growth and differentiation. EGFR is particularly important in the pathogenesis of squamous cell carcinoma of the head and neck (SCCHN) (Kalyankrishna and Grandis, 2006), with reported overexpression in approximately 90% of tumours (Grandis and Tweardy, 1993). EGFR promotes growth and survival through several oncogenic signalling pathways, and its overexpression in SCCHN correlates with poor prognosis, short disease-free survival, and increased locoregional recurrence (Ang et al, 2002; Eriksen et al, 2004; Hitt et al, 2005; Chung et al, 2006), and makes it an attractive therapeutic target for therapy (Bonner et al, 2002; Johns et al, 2003; Harari et al, 2007).
Lapatinib is a reversible dual inhibitor of both EGFR and human EGFR-2 (HER2) tyrosine kinases, which in turn inhibits activation of downstream signalling pathways such as Erk1/2 and Akt in cell lines and xenografts (Xia et al, 2002; Rusnak et al, 2007). Lapatinib elicits cytostatic or cytotoxic anti-tumour effects, depending on the cell type (Chu et al, 2005; Coley et al, 2006; Rusnak et al, 2007; Konecny et al, 2008), and has demonstrated clinical activity in several solid tumours (Spector et al, 2005; Burstein et al, 2008; Cameron et al, 2008; Ravaud et al, 2008).
Lapatinib has shown a single-agent activity in in vitro and in vivo xenograft studies in human head and neck cancer cell lines (GSK, data on file). In addition, lapatinib has been safely combined with chemoradiation in phase I study, with a recommended dose of 1500 mg day−1 for future trials (Harrington et al, 2009). A randomised-phase II study of chemoradiation plus lapatinib, followed by maintenance lapatinib versus chemoradiation plus placebo, followed by placebo, has shown a statistically non-significant 17%-point superiority in favour of lapatinib-treated patients for complete and overall response at 6 months post-chemoradiation (Harrington et al, 2010). Lapatinib is currently in phase III trial with chemoradiation in patients with high-risk features after surgical treatment of stage III/IV head and neck cancer.
Although the molecular effects of many targeted-anticancer agents are often characterised in vitro, correlation of such effects with clinical outcome has only started to be adopted in proof-of-concept trials (Goulart et al, 2007; Banerji et al, 2008). Such an approach often uses apoptosis or proliferation endpoints (Sarker and Workman, 2007; Doroshow and Parchment, 2008). Spector et al (2005) have recently shown that the 3-week treatment with lapatinib in patients with advanced malignancies resulted in increased tumour cell apoptosis, occurring in patients with evidence of tumour regression. The objectives of this study were to explore the biological effects of lapatinib on apoptosis and proliferation in pre- and post-treatment tumour tissues in patients with locally advanced SCCHN.
Materials and methods
This was a multinational, randomised, single-blinded, placebo-controlled study, conducted at 10 centres in six countries. The study was approved by independent ethics committees and regulatory agencies, and was carried out in accordance with the Declaration of Helsinki and good clinical practice. All patients gave written informed consent before enrolment.
Adults of at least 18 years of age with newly diagnosed stage III/IVA/IVB SCCHN undergoing chemoradiation therapy (CRT) were eligible. Other criteria included Eastern Cooperative Oncology Group performance status of 0, 1, or 2; adequate renal, hepatic, and bone marrow function; and normal left ventricular ejection fraction assessed by echocardiogram or multigated acquisition scan. Exclusion criteria included evidence of distant metastasis (stage IVC), earlier systemic chemotherapy, radiotherapy, or required concomitant use of cytochrome P450 3A4 inducers or inhibitors.
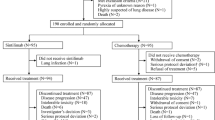
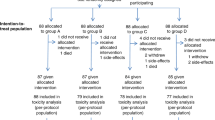
Figure 1A shows the study design. Patients were randomised 2 : 1 to receive lapatinib (1500 mg d−1) or placebo, and stratified by tumour site and performance status. Treatment with lapatinib/placebo continued for 2–6 weeks until the start of CRT. The ‘window of opportunity’ represented the period of time required for radiotherapy preparation. The initiation of CRT was not delayed in patients receiving either lapatinib or placebo, as the monotherapy phase lasted no longer than local standards allowed. The mean time between commencing lapatinib/placebo and initiating CRT was 25.8 days for both arms. Chemotherapy schedule and CRT was mandated as concomitant cisplatin and radiation, and followed the local standard. Conventional radiotherapy was given to a total of 66–70 Gy given over 6–7 weeks.

Study design and patient disposition. (A) Study design and allocated therapies are shown. (B) CONSORT diagram showing patient accountability. Abbreviations: CT=computed tomography; CTx=chemotherapy; ECOG=Eastern Cooperative Oncology Group; F/U=follow-up; ITT=intent-to-treat; LVEF=left ventricular ejection fraction; mITT=modified ITT; NPC=nasopharyngeal carcinoma; PS=performance status; RT=radiotherapy; SCCHN=squamous cell carcinoma of the head and neck.
Medications that inhibit or induce cytochrome P450 3A4 were prohibited. Lapatinib could be withheld for up to 1 week for any grade 3 or 4 toxicity, and permanently discontinued if grade 3 or 4 interstitial pneumonitis or cardiac dysfunction occurred.
Study assessments
Baseline assessments included demography, medical history, physical examination, performance status, panendoscopy, echocardiogram, multigated acquisition scan, haematology, and clinical chemistry. Clinical examination and laboratory tests were repeated during treatment and at follow-up. Adverse events and serious adverse events were collected throughout the study and were graded using the National Cancer Institute's Common Terminology Criteria for Adverse Events, v 3.0 (Cancer Therapy Evaluation Program, 2006).
Objective response rate (ORR) was assessed by performing radiologic examination at baseline, 8 and 12 weeks post-CRT. Additional scans were performed before the commencement of CRT for patients receiving at least 4 weeks of lapatinib/placebo. Efficacy was defined according to Response Evaluation Criteria in Solid Tumours criteria, version 1.0. (Therasse et al, 2000) All scans and clinical data were reviewed centrally by an independent review board (BioClinica, Newtown, PA, USA), and all readers were blinded to treatment. Follow-up beyond 3 months post-CRT was not part of the protocol.
A positron emission tomography (PET) substudy was conducted at participating centres. Thirty-five subjects who participated in the trial agreed to the substudy with fluorodeoxyglucose (FDG)–PET imaging at baseline and the follow-up time point. The sites that were qualified to conduct FDG–PET scans adhered to guidelines regarding the scans. The first scan of the head and neck was performed at screening, before biopsy, to obviate effects of the biopsy procedure on glucose uptake. The second scan was conducted at the end of the week-2 lapatinib treatment (again, before biopsy). The procedure further included: (1) administration of approximately 300–370 MBq FDG; (2) 60 minutes ±10 minutes of rest; (3) an attenuation scan for transmission correction; (4) whole-body emission scan; and (5) whole-body postcontrast computed tomography scan.
Analysis included acquisition of both quantitative measurements of standard uptake values (SUVs), as well as a qualitative assessment. The target and non-target lesions were analysed, based on SUV levels by a PET nuclear medicine expert. At baseline and post-monotherapy time points, approximately 300–370 MBq FDG was administered. Acquisition of SUV occurred after 60 minutes (±10 minutes) of rest.
Tissue acquisition
Fresh tumour biopsies were obtained at time of study enrolment (day 0) and after 2 weeks of study participation (day 14). Biopsies were immediately fixed in 10% neutral-buffered formalin containing phosphatase inhibitors before paraffin-embedded sections were prepared. Hematoxylin and eosin staining was used to confirm the presence of tumour.
Biological evaluations
Apoptosis was measured using immunohistochemistry (IHC) in paired tumour samples by both terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labelling (TUNEL) and active caspase-3 assays. The expression patterns of Ki-67, EGFR, HER2, phosphorylated-EGFR (pEGFR), p16, and p53 were also studied by IHC (details of antibody tests can be found in Supplementary Table 1). HER2 and EGFR gene amplification were assessed using fluorescence in situ hybridisation.
Statistical analysis
The primary endpoint was apoptotic index (AI), which was calculated at baseline and after treatment (percent stained nuclei/total nuclei). Secondary endpoints included proliferation rate given by proliferation index (PI) (percent proliferating cells/total number of cells), ORR, adverse events, and correlative biomarker analyses.
Using a two-sided significance level of 5 and 80% power, 90 patients were required to detect a 24% difference in apoptosis between the two groups. To allow for dropouts during tissue acquisition, a total of 107 patients were randomised.
The change from baseline in AI was analysed using an analysis of covariance model, adjusted for the baseline strata. All other endpoints were summarised as appropriate.
All clinical and biological analyses presented were based on the intent-to-treat (ITT) population, which comprised all patients randomised to study treatment. The evaluable population for radiologic assessment was defined as all patients who completed CRT and had baseline and follow-up scans. This population was used for a more accurate estimation of clinical benefit.
Results
Patient characteristics
Between March 2006 and July 2007, a total of 107 patients were randomised to receive lapatinib (n=71) or placebo (n=36). The study reached the last patient's last visit in December 2007. Figure 1B shows the flow of patients through the trial and highlights the specific patient populations that will be discussed throughout the manuscript: the ITT population comprised 107 patients randomised to either lapatinib or placebo; the modified ITT population included 84 patients in whom pre- and post-treatment biopsies were obtained for analysis of biological endpoints; the evaluable population was made up of 88 patients who had CT/MRI scans at 12 weeks post-chemoradiation, and who were evaluable for treatment response; the monotherapy efficacy population comprised a subgroup of 40 patients who underwent CT/MRI scanning after receiving at least 4 weeks of lapatinib/placebo before chemoradiation. A total of 84 and 88 patients were considered evaluable for apoptosis/proliferation and post-CRT clinical activity, respectively. Table 1 shows the baseline demography. The two treatment groups were generally well balanced. The incidence of p16-positive tumours (IHC 2+/3+) was 43% (35% for the lapatinib group and 60% for the placebo group). EGFR overexpression (IHC 2+/3+) was seen in 93 and 83% of lapatinib and placebo patients, respectively; EGFR gene amplification was seen in 28 and 39% of patients. EGFR amplification was highest in hypopharyngeal tumours (n=8/13 (62%)), and lowest in oral cavity (16%) and laryngeal tumours (17%). HER2 overexpression and gene amplification accounted for only 7% and 4% in total, respectively.
Eighty-two percent of lapatinib and 92% of placebo patients were at least 80% compliant. The planned radiotherapy dose was given to 88 and 81% of lapatinib and placebo patients, respectively. For the lapatinib and placebo groups, the median radiation doses delivered were 70.0 and 68.0 Gy, respectively. Similarly, 93 and 92% of patients in the lapatinib and placebo groups, respectively, completed at least two cycles of planned chemotherapy. The median cisplatin doses for lapatinib and placebo groups were 248.6 and 242.6 mg m−2, respectively. In regard to compliance with study drugs, the mean cumulative dose for lapatinib was 35 217.4 mg with a median value of 30 000.0 mg. The corresponding mean and median values for placebo were 34 916.7 and 31 500.0 mg, respectively.
Biological effects of lapatinib
The activation of EGFR (pEGFR, IHC 2+/3+) was observed in 28 (41%) and 15 (42%) patients in the lapatinib and placebo arms (Table 1), of which 24 and 15 patients, respectively, had post-treatment data available. Lapatinib reduced the level of pEGFR in 63% (n=15/24) of cases, versus 33% (n=5/15) in the placebo group (P=0.11).
Apoptosis induction
Apoptotic cells detected by TUNEL were frequent with lapatinib and placebo, both pre- and posttreatment (Figure 2A–D). Both groups showed similar rates of pretreatment apoptosis, with an AI mean of 3.8 (s.d.=3.38) and 3.2% (s.d.=3.14) for lapatinib and placebo, respectively. The post-treatment mean AI was 8.0% (s.d.=6.66) with lapatinib and 9.4% (s.d.=12.22) with placebo. The change in AI from pretreatment was not statistically significant for lapatinib versus placebo at the two-sided 5% significance level (difference=−1.7, s.e.=1.93, P=0.394, 95% confidence interval: −5.50%, 2.19%; see Supplementary Figure 1). AI was compared with T-stage, stage of disease, and tumour site; the results are shown in Supplementary Figures 2–4. In the caspase-3 assay, the change from pretreatment values with lapatinib was not statistically significant. The values for mean change in caspase-3 from baseline to post-treatment were 3.0 (s.d.=3.4) and 1.6 (s.d.=3.8) for the lapatinib- and placebo-treated patients, respectively. Data for AI from caspase-3 and TUNEL assays were concordant (data not shown).

AI by TUNEL assay. Representative IHC TUNEL staining pretreatment (A) and posttreatment (B) with lapatinib, and pretreatment (C) and posttreatment (D) with placebo.
Effect of lapatinib on proliferation
The pretreatment PI mean was 62.7 (s.d.=17.54) and 66.1% (s.d.=19.47) with lapatinib and placebo, respectively. Treatment with lapatinib decreased the PI, compared with placebo, with a mean of 56.7 (s.d.=17.49) and 64.4% (s.d.=16.54), respectively. The relative change from pretreatment was statistically significant at the two-sided 5% significance level (difference: −5.4%, s.e.=2.47, P=0.030, 95% confidence interval: −10.36% to −0.53%; Figure 3).

Effect of lapatinib on proliferation determined by Ki-67; box whisker plot of pretreatment and posttreatment proliferative index with lapatinib and placebo. The median values are presented to the left of each box. The mean values were: lapatinib pretreatment −62.7%, posttreatment −56.7%; placebo pretreatment −66.1%, posttreatment −64.4%.
Clinical outcome
Forty patients were assessed for response after the monotherapy phase and before CRT. The independently assessed ORR was 17 (n=4/24) versus 0% (n=0/16) in the ITT and evaluable populations, respectively, following approximately 4 weeks of lapatinib/placebo treatment. No progressive disease was observed with lapatinib, compared with 25% with placebo (see Table 2 and Figure 4A). From the lapatinib responders, all patients had EGFR overexpression; 50% had EGFR amplification; 50% HER2 expression by IHC, in one of these cases, HER2 gene was amplified; and variable modulation of apoptosis, proliferation, and pEGFR (details in Supplementary Table 2). Two of the responders were positive for p16.

(A) Waterfall plot of independently assessed ORR following lapatinib/placebo phase; (B) Bar chart of ORR following CRT versus tumour site; (C) Waterfall plot of the change from baseline in independently read SUVmax measurements. Abbreviations: R=responders (complete and partial response); NR=nonresponders (stable and progressive disease); HP=hypopharynx; L=larynx; OC=oral cavity; OP=oropharynx. *Percent within levels of tumour site.
The independently assessed ORR following CRT in the evaluable population was 86 and 63% in the lapatinib and placebo arms, respectively (see Table 2). Moreover, in the ITT, the response rate was 70 and 53%. A higher number of patients presented with progressive disease in the placebo arm (25%) than in the lapatinib arm (6%). Oropharyngeal and oral cavity tumours were characterised by highest response rate (95% and 91%, respectively) in the lapatinib arm, compared with larynx (70%), hypopharyngeal tumours (67%), or placebo arm (67%, 43%, 60%, and 67%, respectively; see Figure 4B).
In the PET substudy (n=35), maximum SUV (SUVmax) was reduced from baseline values in 75% of patients treated with lapatinib compared with 36% treated with placebo (see Figure 4C). The change in median SUVmax was −12.9 for lapatinib and +6.1 for placebo. The mean SUV change from baseline was also assessed, with a median −10.9 for lapatinib and +3.2 for placebo.
Safety
In the monotherapy phase, only patients receiving lapatinib reported rash (23%) and diarrhoea (22%), which were mainly grades 1 and 2 (see Supplementary Figure 5). Throughout the complete duration of the study, the lapatinib/CRT group showed a higher incidence of diarrhoea (26%) compared with 6% in the placebo/CRT group, and a slightly higher incidence of mucosal inflammation (70%) compared with 67% in the placebo/CRT group (see Supplementary Table 3). Grade 3 and 4 mucositis (a grouping of associated adverse events) were also higher in the lapatinib/CRT arm (46% and 4%, respectively), compared with placebo (41% grade 3; no grade 4 events).
The reported serious adverse events during or after CRT were higher in the placebo/CRT group (36%) than in the lapatinib/CRT group (19%), with mucosal inflammation, the most common serious adverse event (6% and 4%, respectively; see Supplementary Table 4).
Seven patients experienced cardiac-related events: 2 (3%) in the lapatinib arm and 5 (14%) in the placebo arm. The event was fatal for two patients (n=1 in each arm).
Twelve deaths were reported (10 and 14% of patients in the lapatinib and placebo arms, respectively). The primary causes were disease under study (6% lapatinib and 8% placebo), ventricular fibrillation (n=1, placebo), septicaemia (3%, placebo), and intestinal perforation, cardiac arrest, and sudden death (3% each, lapatinib). None of these fatal events was attributed to the study medication.
Discussion
This study set out to investigate whether the biological effects of lapatinib on cell survival and growth pathways predict clinical outcome and to identify any subgroup of patients that may benefit from lapatinib treatment. It was previously shown that activated EGFR modulates the proapoptotic and antiapoptotic pathways (Modjtahedi et al, 1998; Grandis et al, 2000; Ginsberg, 2007; Goel et al, 2007). Therefore, the primary objective assessed the effect of lapatinib on apoptosis. Apoptosis induction was not statistically different compared with placebo, and therefore, the primary endpoint was not met. Indeed, the AI by TUNEL staining increased, following both lapatinib and placebo treatment. Although the possibility that these data may have been affected by artifact introduced during post-treatment biopsy handling should be considered; we believe that the randomised nature of the study should have provided insurance against this risk. The lack of a clear apoptotic signal may be somewhat surprising, as lapatinib is a potent inhibitor of pEGFR and pHER2 in cell-free systems (Rusnak et al, 2001), and induces apoptosis in in vitro and in vivo models (Xia et al, 2002; Zhou et al, 2006) as well as clinical studies (Spector et al, 2005). However, similar values have previously been reported for spontaneous apoptosis and apoptosis induction (Hotz et al, 1999; Grabenbauer et al, 2003). Unfortunately, results regarding the prognostic and predictive significance of apoptosis are rather conflicting in SCCHN (Bartelink et al, 1999; Tsuchiya et al, 2001; Grabenbauer et al, 2003), which could be attributed, in part, to the methodologic complexities associated with these assays and the inherently asynchronous apoptotic process within a given tissue (Potten, 1996). TUNEL detects late apoptotic events, and despite the fact that positive signals are also found in necrotic cells and in some cells in which DNA fragmentation is later reparable (Collins et al, 1997), the co-analysis of the nuclear morphology provides a relatively accurate indicator of apoptosis (Sheridan et al, 1999; Willingham, 1999). Caspase-3 activation is an early event in both the intrinsic and extrinsic apoptotic pathways (Kaufmann and Vaux, 2003); nevertheless, the assay may not detect apoptosis independent of mitochondrial pathways (Gown and Willingham, 2002). The lack of apoptotic changes despite the observation of tumour shrinkage in some patients suggests that caspases may not be involved in lapatinib-induced cell death or that lapatinib does not induce apoptosis, and its mechanism of action is through some other pathway in locally advanced SCCHN.
Other biological effects with lapatinib treatment are intriguing and warrant further investigation. For example, the inhibition of pEGFR activity within 2 weeks of lapatinib treatment and the significant decrease in the proliferative activity may, in part, have contributed to the higher response rates to subsequent CRT documented in lapatinib-treated patients. This observation also supports a possible role for lapatinib in combination with radiotherapy or CRT as a means of preventing accelerated repopulation (Harrington et al, 2007). However, these data must be interpreted in light of the fact that 33% of placebo-treated patients also showed evidence of reduction of the pEGFR level. Future studies should address the issue of the reproducibility of these assays on serial samples.
Despite ambiguous biological results, this study conducted in a treatment-naive population with locally advanced disease showed an ORR of 17% after the monotherapy phase, including one complete response. Furthermore, there was a larger proportion of lapatinib-treated patients with a reduction in SUVmax in the PET substudy. Given the short lapatinib treatment, these findings are promising and indicate lapatinib activity in locally advanced SCCHN. These findings oppose a recent study by Abidoye et al, (2006) that reported minimal clinical activity in patients treated with lapatinib, who had recurrent or metastatic SCCHN.
Interestingly, all four monotherapy responders had tumours in which EGFR was overexpressed, two of which also showed HER2 expression and one that showed HER2 gene amplification. None of the other biological characteristics were predictive of response to lapatinib. However, the number of responding patients is too small to make any meaningful conclusions. More importantly, the post-treatment biopsy was obtained at earlier time points (day 14±3 days) than the radiologic scans (week 4 approximately), which may provide an explanation of why apoptotic changes were not also observed.
The ORR following CRT was higher in the lapatinib group compared with placebo in the ITT and evaluable populations. The fact that there was an excess of HPV-positive patients in favour of the placebo-treated group means that the absolute difference may have been greater if the study arms had been perfectly balanced. Although this study was not powered to test for differences in ORR between the study arms, the data suggest that lapatinib is worthy of further evaluation in patients with locally advanced head and neck cancer. The short duration of lapatinib before commencing CRT may have provided insufficient suppression of EGFR signalling to induce apoptosis, but may prime tumour cells for subsequent CRT-induced cell death through an unknown mechanism. This effect is unlikely to have been due to drug-induced G1 cell cycle arrest. Given the responses observed following monotherapy, the reduced tumour volume before commencing CRT in the lapatinib group may also provide an explanation.
Although EGFR overexpression seems to be predictive of response to lapatinib or lapatinib/CRT, EGFR gene amplification was not, which is in contrast to various reports with other EGFR inhibitors in SCCHN (Cohen et al, 2005; Erjala et al, 2006; Agulnik et al, 2007; Thomas et al, 2007). Although no clear correlation was demonstrated between HER2 overexpression and response to lapatinib or lapatinib/CRT, a functional role through heterodimerisation with EGFR cannot be excluded. Of interest is that two of the four monotherapy responders showed both EGFR and HER2 coexpression. The patient with 3+ overexpression for both EGFR and HER2 also demonstrated gene amplification for both receptors. Similar to a previous study (Harrington et al, 2009), the treatment was well tolerated and did not lead to significant modifications of CRT. The majority of patients received the planned radiotherapy and chemotherapy, which was generally similar to that seen in other studies (Cooper et al, 2004). Furthermore, an independent data-monitoring committee, which evaluates the safety in several ongoing phase II and III trials of lapatinib in SCCHN, has raised no safety concerns in this regard.
The present study provides a useful design that allowed inter- and intrapatient evaluation of the pharmacodynamic effects and putative predictors of response to targeted agents. It showed the feasibility of obtaining paired biopsies in locally advanced SCCHN. However, some limitations of study design should be addressed. First, there is no consensus on the appropriate timing for the second biopsy; therefore, the timing for this study was empirically chosen. Hence, the observed effects may not reflect the actual molecular events that lead to apoptosis or growth arrest. Second, the small number of patients means the results should be interpreted with caution. Third, there is a fairly short follow-up period, which did not allow for assessment of survival.
In summary, a short treatment period with lapatinib suggests that it may 1) not affect apoptosis; 2) lead to inhibition of pEGFR; 3) decrease proliferation; 4) induce tumour regression and enhance response to CRT; and 5) cause oropharyngeal and oral cavity tumours to respond favourably to treatment. It would be of value to confirm these hypotheses with large cohorts of patients. Work is ongoing to investigate other EGFR family members and their effect on downstream signalling pathways.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abidoye OO, Cohen EE, Wong SJ, Kozloff MF, Nattam SR, Stenson KM, Blair EA, Day S, Dancey JE, Vokes EE (2006) A phase II study of lapatinib (GW572016) in recurrent/metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN). J Clin Oncol 24 (suppl): 5568
Agulnik M, da Cunha Santos G, Hedley D, Nicklee T, Dos Reis PP, Ho J, Pond GR, Chen H, Chen S, Shyr Y, Winquist E, Soulieres D, Chen EX, Squire JA, Marrano P, Kamel-Reid S, Dancey J, Siu LL, Tsao MS (2007) Predictive and pharmacodynamic biomarker studies in tumor and skin tissue samples of patients with recurrent or metastatic squamous cell carcinoma of the head and neck treated with erlotinib. J Clin Oncol 25 (16): 2184–2190
Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, Fu KK, Milas L (2002) Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 62 (24): 7350–7356
Banerji U, de Bono J, Judson I, Kaye S, Workman P (2008) Biomarkers in early clinical trials: the committed and the skeptics. Clin Cancer Res 14 (8): 2512–2514
Bartelink H, Begg A, Martin JC, van Dijk M, van ‘t Veer L, van der Vaart P, Verheij M (1999) Towards prediction and modulation of treatment response. Radiother Oncol 50 (1): 1–11
Bonner JA, De Los Santos J, Waksal HW, Needle MN, Trummel HQ, Raisch KP (2002) Epidermal growth factor receptor as a therapeutic target in head and neck cancer. Semin Radiat Oncol 12 (3 Supplement 2): 11–20
Burstein HJ, Storniolo AM, Franco S, Forster J, Stein S, Rubin S, Salazar VM, Blackwell KL (2008) A phase II study of lapatinib monotherapy in chemotherapy-refractory HER2-positive and HER2-negative advanced or metastatic breast cancer. Ann Oncol 19 (6): 1068–1074
Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu CG, Chan S, Jagiello-Gruszfeld A, Kaufman B, Crown J, Chan A, Campone M, Viens P, Davidson N, Gorbounova V, Raats JI, Skarlos D, Newstat B, Roychowdhury D, Paoletti P, Oliva C, Rubin S, Stein S, Geyer CE (2008) A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 112 (3): 533–543
Cancer Therapy Evaluation Program. (2006) Common Terminology Criteria for Adverse Events, v3.0. National Cancer Institute, http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Published 9 August 2006. Accessed 4 January 2010.
Chu I, Blackwell K, Chen S, Slingerland J (2005) The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res 65 (1): 18–25
Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, Jarrett C, Carter J, Murphy BA, Netterville J, Burkey BB, Sinard R, Cmelak A, Levy S, Yarbrough WG, Slebos RJ, Hirsch FR (2006) Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol 24 (25): 4170–4176
Cohen EE, Lingen MW, Martin LE, Harris PL, Brannigan BW, Haserlat SM, Okimoto RA, Sgroi DC, Dahiya S, Muir B, Clark JR, Rocco JW, Vokes EE, Haber DA, Bell DW (2005) Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res 11 (22): 8105–8108
Coley HM, Shotton CF, Ajose-Adeogun A, Modjtahedi H, Thomas H (2006) Receptor tyrosine kinase (RTK) inhibition is effective in chemosensitising EGFR-expressing drug resistant human ovarian cancer cell lines when used in combination with cytotoxic agents. Biochem Pharmacol 72 (8): 941–948
Collins JA, Schandi CA, Young KK, Vesely J, Willingham MC (1997) Major DNA fragmentation is a late event in apoptosis. J Histochem Cytochem 45 (7): 923–934
Cooper JS, Pajak TF, Forastiere AA, Jacobs J, Campbell BH, Saxman SB, Kish JA, Kim HE, Cmelak AJ, Rotman A, Machtay M, Ensley JF, Chao KS, Schultz CJ, Lee N, Fu KK, Radiation Therapy Oncology Group 9501/Intergroup (2004) Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. New Engl J Med 350 (19): 1937–1944
Doroshow JH, Parchment RE (2008) Oncologic phase 0 trials incorporating clinical pharmacodynamics: from concept to patient. Clin Cancer Res 14 (12): 3658–3663
Eriksen JG, Steiniche T, Askaa J, Alsner J, Overgaard J (2004) The prognostic value of epidermal growth factor receptor is related to tumor differentiation and the overall treatment time of radiotherapy in squamous cell carcinomas of the head and neck. Int J Radiat Oncol Biol Phys 58 (2): 561–566
Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, Kulmala J, Pulkkinen J, Grenman R, Elenius K (2006) Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin Cancer Res 12 (13): 4103–4111
Ginsberg D (2007) EGFR signaling inhibits E2F1-induced apoptosis in vivo: implications for cancer therapy. Sci STKE 2007 (371): pe4
Goel S, Hidalgo M, Perez-Soler R (2007) EGFR inhibitor-mediated apoptosis in solid tumors. J Exp Ther Oncol 6 (4): 305–320
Goulart BH, Clark JW, Pien HH, Roberts TG, Finkelstein SN, Chabner BA (2007) Trends in the use and role of biomarkers in phase I oncology trials. Clin Cancer Res 13 (22 Part 1): 6719–6726
Gown AM, Willingham MC (2002) Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem 50 (4): 449–454
Grabenbauer G, Suckorada O, Niedobitek G, Rödel F, Iro H, Sauer R, Rödel C, Schultze-Mosgau S, Distel L (2003) Imbalance between proliferation and apoptosis may be responsible for treatment failure after postoperative radiotherapy in squamous cell carcinoma of the oropharynx. Oral Oncol 39 (5): 459–469
Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, Johnson DE, Huang L, He Y, Kim JD (2000) Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA 97 (8): 4227–4232
Grandis JR, Tweardy DJ (1993) Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res 53 (15): 3579–3584
Harari PM, Allen GW, Bonner JA (2007) Biology of interactions: antiepidermal growth factor receptor agents. J Clin Oncol 25 (26): 4057–4065
Harrington K, Jankowska P, Hingorani M (2007) Molecular biology for the radiation oncologist: the 5Rs of radiobiology meet the hallmarks of cancer. Clin Oncol 19 (8): 561–571
Harrington KJ, Berrier A, Robinson M, Remenar E, Housset M, Hurtado de Mendoza F, Compton N, Lau M, Legenne P, Kumar R (2010) Phase II study of oral lapatinib, a dual-tyrosine kinase inhibitor, combined with chemoradiotherapy (CRT) in patients (pts) with locally advanced, unresected squamous cell carcinoma of the head and neck (SCCHN). J Clin Oncol 28 (Suppl): 15s (abstract 5505)
Harrington KJ, El-Hariry IA, Holford CS, Lusinchi A, Nutting CM, Rosine D, Tanay M, Deutsch E, Matthews J, D’Ambrosio C, Turner SJ, Pandeshwara JS, Bourhis J (2009) Phase I study of lapatinib in combination with chemoradiation in patients with locally advanced squamous cell carcinoma of the head and neck. J Clin Oncol 27 (7): 1100–1107
Hitt R, Ciruelos E, Amador ML, Benito A, Sanchez JJ, Ballestin C, Cortes-Funes H (2005) Prognostic value of the epidermal growth factor receptor (EGRF) and p53 in advanced head and neck squamous cell carcinoma patients treated with induction chemotherapy. Eur J Cancer 41 (3): 453–460
Hotz MA, Bosq J, Zbaeren P, Reed J, Schwab G, Krajewski S, Brousset P, Borner MM (1999) Spontaneous apoptosis and the expression of p53 and Bcl-2 family proteins in locally advanced head and neck cancer. Arch Otolaryngol Head Neck Surg 125 (4): 417–422
Johns TG, Luwor RB, Murone C, Walker F, Weinstock J, Vitali AA, Perera RM, Jungbluth AA, Stockert E, Old LJ, Nice EC, Burgess AW, Scott AM (2003) Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci USA 100 (26): 15871–15876
Kalyankrishna S, Grandis JR (2006) Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol 24 (17): 2666–2672
Kaufmann SH, Vaux DL (2003) Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene 22 (47): 7414–7430
Konecny GE, Venkatesan N, Yang G, Dering J, Ginther C, Finn R, Rahmeh M, Fejzo MS, Toft D, Jiang SW, Slamon DJ, Podratz KC (2008) Activity of lapatinib a novel HER2 and EGFR dual kinase inhibitor in human endometrial cancer cells. Br J Cancer 98 (6): 1076–1084
Modjtahedi H, Affleck K, Stubberfield C, Dean C (1998) EGFR blockade by tyrosine kinase inhibitor or monoclonal antibody inhibits growth, directs terminal differentiation and induces apoptosis in the human squamous cell carcinoma HN5. Int J Oncol 13 (2): 335–342
Potten CS (1996) What is an apoptotic index measuring? A commentary. Br J Cancer 74 (11): 1743–1748
Ravaud A, Hawkins R, Gardner JP, von der Maase H, Zantl N, Harper P, Rolland F, Audhuy B, Machiels JP, Pétavy F, Gore M, Schöffski P, El-Hariry I (2008) Lapatinib versus hormone therapy in patients with advanced renal cell carcinoma: a randomized phase III clinical trial. J Clin Oncol 26 (14): 2285–2291
Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws Jr TF, Hudson-Curtis BL, Gilmer TM (2007) Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif 40 (4): 580–594
Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM (2001) The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther 1 (2): 85–94
Sarker D, Workman P (2007) Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res 96: 213–268
Sheridan MT, Cooper RA, West CM (1999) A high ratio of apoptosis to proliferation correlates with improved survival after radiotherapy for cervical adenocarcinoma. Int J Radiat Oncol Biol Phys 44 (3): 507–512
Spector NL, Xia W, Burris III H, Hurwitz H, Dees EC, Dowlati A, O’Neil B, Overmoyer B, Marcom PK, Blackwell KL, Smith DA, Koch KM, Stead A, Mangum S, Ellis MJ, Liu L, Man AK, Bremer TM, Harris J, Bacus S (2005) Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 23 (11): 2502–2512
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216
Thomas F, Rochaix P, Benlyazid A, Sarini J, Rives M, Lefebvre JL, Allal BC, Courbon F, Chatelut E, Delord JP (2007) Pilot study of neoadjuvant treatment with erlotinib in nonmetastatic head and neck squamous cell carcinoma. Clin Cancer Res 13 (23): 7086–7092
Tsuchiya K, Shirato H, Nishioka T, Yamazaki A, Hashimoto S, Kagei K, Oomori K, Yasuda M, Shindo M, Miyasaka K (2001) Pretreatment apoptotic scores do not predict response to radiation therapy in oropharyngeal squamous cell carcinoma. Oral Oncol 37 (2): 159–163
Willingham MC (1999) Cytochemical methods for the detection of apoptosis. J Histochem Cytochem 47 (9): 1101–1110
Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL (2002) Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 21 (41): 6255–6263
Zhou Y, Li S, Hu YP, Wang J, Hauser J, Conway AN, Vinci MA, Humphrey L, Zborowska E, Willson JK, Brattain MG (2006) Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res 66 (1): 404–411
Acknowledgements
This study was supported by GlaxoSmithKline (NCT00371566). All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support in the form copyediting, fact checking, collating author comments, and creation of tables and figures was provided by Michael Austin of Publication CONNEXION (Newtown, PA) and was funded by GlaxoSmithKline.
We thank the patients who participated and their families; also Phillip Murphy, Brandon Whitcher, Saba Khan-Wasti, John Farrell, and Natalie Franklin of GlaxoSmithKline and Aspasia Theodosiou, Sophie Gardiner, and Shirley Young, formerly of GlaxoSmithKline, for their support during the conduct of the study; Sarah Bacus from Therapeutic Medical Diagnostics for her expert advice on molecular markers; Hisham Mehanna and Sean James of InHanse for providing p16 analysis; and Andrea Perrone at Bioclinica for the centralised imaging analysis and interpretation.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part at the 33rd European Society for Medical Oncology Congress; 12–16 September 2008; Stockholm, Sweden. Abstract published in Ann Oncol 2008; 19 : viii217 (abstract 688O).
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
del Campo, J., Hitt, R., Sebastian, P. et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer 105, 618–627 (2011). https://doi.org/10.1038/bjc.2011.237
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.237
Keywords
This article is cited by
-
Targeted therapy for head and neck cancer: signaling pathways and clinical studies
Signal Transduction and Targeted Therapy (2023)
-
Clinical data mining reveals analgesic effects of lapatinib in cancer patients
Scientific Reports (2021)
-
Cardiac toxicities of lapatinib in patients with breast cancer and other HER2-positive cancers: a meta-analysis
Breast Cancer Research and Treatment (2017)
-
EGFR-targeted therapies in the post-genomic era
Cancer and Metastasis Reviews (2017)
-
Trends in Surgical Research in Head and Neck Cancer
Current Treatment Options in Oncology (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}