
Abstract
Background:
Prostate-specific antigen (PSA) screening has low specificity. Assessment of methylation status in body fluids may complement PSA screening if the test has high specificity.
Method:
The purpose of this study was to conduct a meta-analysis of the sensitivity and specificity for prostate cancer detection of glutathione-s-transferase–π (GSTP1) methylation in body fluids (plasma, serum, whole blood, urine, ejaculate, and prostatic secretions). We conducted a comprehensive literature search on Medline (Pubmed). We included studies if they met all four of the following criteria: (1) measurement of DNA methylation in body fluids; (2) a case-control or case-only design; (3) publication in an English journal; and (4) adult subjects. Reviewers conducted data extraction independently using a standardised protocol. Twenty-two studies were finally included in this paper. Primer sequences and methylation method in each study were summarised and evaluated using meta-analyses. This paper represents a unique cross-disciplinary approach to molecular epidemiology.
Results:
The pooled specificity of GSTP1 promoter methylation measured in plasma, serum, and urine samples from negative-biopsy controls was 0.89 (95% CI, 0.80–0.95). Stratified analyses consistently showed a high specificity across different sample types and methylation methods (include both primer sequences and location). The pooled sensitivity was 0.52 (95% CI, 0.40–0.64).
Conclusions:
The pooled specificity of GSTP1 promoter methylation measures in plasma, serum, and urine was excellent and much higher than the specificity of PSA. The sensitivity of GSTP1 was modest, no higher than that of PSA. These results suggest that measurement of GSTP1 promoter methylation in plasma, serum, or urine samples may complement PSA screening for prostate cancer diagnosis.
Similar content being viewed by others

Main
Prostate cancer is the most common cancer in men and the second leading cause of cancer-related death in both the United States and Western Europe. With a sensitivity of 80%, prostate-specific antigen (PSA) screening significantly increases the early diagnosis of prostate cancer (Catalona, 1994). However, the specificity of PSA screening is only 20% and may lead to many unnecessary biopsies and overtreatment. A highly specific circulating biomarker (using plasma, serum, or urine samples) that complements the traditional PSA test is therefore in great demand. A specific and non-invasive test would allow patients to avoid the physical pain and discomfort associated with biopsies, and avoid the adverse effects and unnecessary medical spending resulting from overtreatment. A blood draw is already essential for PSA screening and urine samples are easy to obtain; thus, in conjunction with measuring PSA levels, an additional measurement of plasma, serum, or urinary biomarkers does not place any extra burden on patients.
Gene promoter CpG island hypermethylation is one of the earliest somatic genome alterations during the development of several types of cancers. Studies have shown that glutathione-s-transferase—π (GSTP1) promoter hypermethylation is the most common somatic genome alteration during prostate cancer development (Lee et al, 1994; Goessl, 2000; Kang et al, 2004). If GSTP1 promoter hypermethylation can be detected in body fluids and if it accurately predicts prostate cancer, then this measurement has the potential to complement PSA screening. Research has shown that the prostate, as well as circulating phagocytic cells that have ingested prostate cancer cells, can release DNA into blood circulation (Nakayama et al, 2004). DNA can also appear in urine, ejaculates, and prostatic secretions after cells are shed into prostatic ducts. Therefore, detection of methylation status in body fluids (plasma, serum, whole blood, urine, ejaculates, and prostatic secretion) may complement PSA screening if the test has high specificity. Furthermore, although some studies have explored whether GSTP1 promoter hypermethylation in body fluids is associated with the patient's pathological stage, Gleason score, or PSA level, no meta-analysis has been carried out to summarise these results.
The purpose of this study was to conduct a meta-analysis on the sensitivity and specificity of GSTP1 methylation in body fluids on prostate cancer detection. We assessed the usefulness of several types of body fluids, including whole blood, plasma, serum, buffy coat, urine, ejaculates, and prostatic secretions. We also determined whether GSTP1 methylation was correlated with pathological stage, Gleason score, and/or PSA level among the cases.
Materials and methods
Study selection
We conducted a comprehensive literature search on Medline (Pubmed) of articles published between 1966 and January 30, 2010. We used the keywords ‘methylation’ and ‘prostate cancer’ in conjunction with any of the following terms: ‘whole blood’, ‘plasma’, ‘serum’, ‘urine’, ‘ejaculate’, and ‘prostate secrete’. Additional studies were found via the reference lists in the identified articles.
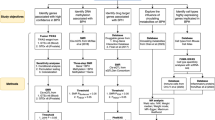
We included the studies that met all four of the following inclusion criteria: (1) measurement of DNA methylation in one of the following body fluids—whole blood, plasma, serum, buffy coat, urine, ejaculates, or prostate secretions; (2) a case-control or case-only study; (3) published in an English language journal; and (4) conducted in adults. We excluded studies that did not test GSTP1 methylation in body fluids. We also excluded studies in which men recently underwent a transurethral resection of the prostate or brachytherapy, or took medications (e.g., Finasteride), because these treatments or medications have the potential to reduce PSA levels. The selection process for studies included in our review is shown in Figure 1. Our search strategy and inclusion/exclusion criteria resulted in a total of 22 articles that were included in the systemic review (Suh et al, 2000; Cairns et al, 2001; Goessl et al, 2001a, 2001b; Jeronimo et al, 2002; Gonzalgo et al, 2003, 2004; Crocitto et al, 2004; Hoque et al, 2005; Papadopoulou et al, 2006; Rogers et al, 2006; Chuang et al, 2007; Reibenwein et al, 2007; Roupret et al, 2007, 2008; Altimari et al, 2008; Bastian et al, 2008; Bryzgunova et al, 2008; Ellinger et al, 2008; Woodson et al, 2008; Sunami et al, 2009). The age range of subjects in the 22 studies was 40–74 years. Details of each study such as mean age of case and control status are included in Supplementary Table 1.

Flow diagram for the selection of studies.
As evaluating the specificity of the methylation test was the major focus of this study, we rigorously classified our controls to remove potential heterogeneity, thereby minimising selection bias. It is well accepted that patients who are referred for biopsies typically have elevated PSA levels, an abnormal digital rectal exam, or related symptoms, and thus are completely different from randomly selected healthy controls. Healthy controls are most likely to introduce bias, resulting in a high specificity. Therefore, among the studies included, we classified controls into two categories: (1) patients who had negative biopsies (10 studies) (Goessl et al, 2001a, 2001b; Jeronimo et al, 2002; Gonzalgo et al, 2003; Crocitto et al, 2004; Bastian et al, 2008; Ellinger et al, 2008; Rossouw et al, 2008; Woodson et al, 2008; Payne et al, 2009) but had other diseases, including benign prostatic hyperplasia, other urologic symptoms (e.g., hematuria), or other types of cancers (e.g., lung cancer or colon cancer); and (2) healthy controls (9 studies) (Papadopoulou et al, 2006; Rogers et al, 2006; Reibenwein et al, 2007; Roupret et al, 2007; Altimari et al, 2008; Bryzgunova et al, 2008; Ellinger et al, 2008; Payne et al, 2009; Sunami et al, 2009). As prostatic intraepithelial neoplasia (PIN) was treated as a case in this study (see below), we excluded the data using PIN as a control in two studies (Goessl et al, 2001b; Woodson et al, 2008). We excluded Hoque et al (2005) from the first category because some of their controls did not have negative biopsies. Although we calculated the specificity separately for the two types of controls, our conclusions were not based on the results generated from healthy controls.
Biopsy-confirmed prostate cancer was treated as a case, except for one study in which biopsy-confirmed PIN was treated as a case. The timing of sample collection from cases varied widely. Some samples were collected prior to biopsies, some after biopsies, some after radical prostatectomy or surgery, and some after hormone therapy. As removing the prostate may influence the likelihood of cancer cells being released into circulation and because hormone treatments may change gene methylation status, we also stratified our analyses by treatment status when samples were collected from cases.
For methylation measurements, we classified them as follows: non-quantitative methylation-specific PCR (N-MSP), quantitative MSP (Q-MSP), methylation-sensitive restriction endonuclease-qPCR (MSRE-qPCR), and bisulfite genomic sequencing. Some methylation assays used a real-time PCR–SYBR Green approach to detect methylated and unmethylated DNA; however, the percentage of methylation is not truly quantified unless methylation and unmethylation control standards are included. Therefore, we considered those assays to be N-MSP. MSRE-qPCR uses a different approach from Q-MSP. It targets the non-bisulfite converted DNA sequence by methylation-sensitive/insensitive endonuclease without using primers specific for methylated and unmethylated sequences; therefore, it is not considered to be Q-MSP in the current manuscript.
As shown in Table 1, 12 studies (Cairns et al, 2001; Goessl et al, 2001a, 2001b; Jeronimo et al, 2002; Gonzalgo et al, 2003, 2004; Crocitto et al, 2004; Rogers et al, 2006; Reibenwein et al, 2007; Altimari et al, 2008; Roupret et al, 2008; Sunami et al, 2009) used N-MSP and the same set of methylated/unmethylated primers (∼74 to 170nt) to target the same CpG dinucleotides. Among those using Q-MSP (Chuang et al, 2007; Bastian et al, 2008; Bryzgunova et al, 2008; Ellinger et al, 2008), two targeted the region ‘29–168nt’, one targeted ‘179–305nt’, and one ‘7–122nt’. Bastian (2008), Bryzgunova (2008), Chuang (2007), and Ellinger (2008) used MSRE-qPCR or bisulfite sequencing to investigate the methylation status of CpG dinucleotides at the GSTP1 promoter. Although the sequence of primers in these studies (Chuang et al, 2007; Bastian et al, 2008; Bryzgunova et al, 2008; Ellinger et al, 2008) did not fall in the ‘+74 to 170 nt’ or ‘+7 to 305 nt’ regions, they were all located at the 5′ promoter region of GSTP1 (−80 to 400 nt from its transcriptional start site).
For our stratified analyses, we classified methylation methods based on their primer sequence, location, and PCR method. We categorised studies using N-MSP as one group and studies using Q-MSP, MSRE-qPCR, or bisulfite sequencing (plus one study (Papadopoulou et al, 2006) using gene scan) as another group, because there are not enough sample sizes to further stratify the second group by individual methylation methods.
Among the 22 studies, 11 studies used plasma or serum (Goessl et al, 2001a; Jeronimo et al, 2002; Papadopoulou et al, 2006; Chuang et al, 2007; Reibenwein et al, 2007; Altimari et al, 2008; Bastian et al, 2008; Bryzgunova et al, 2008; Ellinger et al, 2008; Sunami et al, 2009), and 11 studies used urine (Cairns et al, 2001; Goessl et al, 2001a, 2001b; Jeronimo et al, 2002; Gonzalgo et al, 2003, 2004; Hoque et al, 2005; Rogers et al, 2006; Roupret et al, 2007; Bryzgunova et al, 2008; Woodson et al, 2008; Payne et al, 2009), 1 study used whole blood (Roupret et al, 2008), 2 studies used ejaculates (Suh et al, 2000; Goessl et al, 2001a), and 2 studies used prostate secretions (Crocitto et al, 2004; Gonzalgo et al, 2004). Some studies collected more than one type of biospecimen.
Data extraction
Using a standardised data extraction form, two independent investigators (TW and PM) extracted and tabulated all data. Discrepancies were resolved by discussions with other co-authors. We included the author's last name, year of publication, sample size, mean subject age, cancer clinical classification, type of PCR method, and other relevant characteristics of the study population. We extracted the number of positive and negative results among cases and controls. Specifically, the primer location, sequence, and PCR method in each study are summarised in Table 1.
Statistical analysis
Pooled specificity and sensitivity
Sensitivity and specificity estimates from each study were analysed using random-effects models. To assess whether variation in the threshold definition of a positive result produced an association between sensitivity and specificity values across studies, we needed to establish whether there was an association between these parameters. The summary receiving operating characteristic (S-ROC) curve describes the extent of this relationship (Midgette et al, 1993). The S-ROC was obtained by estimating the linear regression of the log-odds ratio from each study on the sum of the logits of the true-positive and false-positive rates. When the regression between these quantities is null, independent analyses of pooled sensitivity and specificity using standard methods for binary data are appropriate. In these cases, data were analysed on the log-odds scale (e.g., for specificities, the effect size used was log(Spec/(1-Spec)), with approximate variance (1/R) + (1/(N-R)), where N and R are the number of negative (control) cases and the number of false positives, respectively, in the study. A continuity correction of 0.5 was added to each cell to allow calculations in the presence of zero cell counts.

To test for heterogeneity, the Cochrane Q test of heterogeneity (based on deviations of observed log-odds from the common log-odds under a fixed-effect model) was also performed for each analysis.
All analyses were performed using the MIXED procedure in SAS version 9.2. In particular, we observed significant heterogeneity among study outcomes (variation beyond chance expectation), which can complicate the interpretation of findings. However, this is rarely a valid reason for abandoning a meta-analysis altogether. Heterogeneity indicates that a single estimate for the parameter(s) of interest does not hold over all conditions that have been studied and that exploration of which conditions are associated with such variation is warranted. A meta-regression can be used to verify whether any suspected factors may contribute to this heterogeneity. We conducted a meta-regression to examine whether the specificity or sensitivity may be predicted by any of the following factors: age, methylation method, and sample type.
Pooled odds ratio (OR)among cases: Methylation associated with pathological stage, Gleason score, and PSA levels
We found that the sensitivity of GSTP1 promoter methylation varied considerably among the studies. We applied the random-effect model to directly analyse the pooled odds ratios of methylation associated with pathological stage, Gleason score, and PSA levels. We adjusted for age, sample type, and methylation method; if none of them appeared to be significant in the model, these variables were removed from the model. Our analyses were limited to samples collected prior to treatment.
Results
Individual specificity and pooled specificity of GSTP1 promoter methylation in all studies and in studies using different types of samples
Negative-biopsy controls (16 studies) are shown in Table 2. Most of the GSTP1 specificities were high and above 0.5, although the GSTP1 specificity in one study was below 0.5. The pooled specificity (Table 3) was 0.89 (95% CI, 0.80–0.95), suggesting that the GSTP1 methylation test has a much higher specificity than the PSA test. The P-value for heterogeneity was <0.001, indicating significant heterogeneity. Moreover, we performed stratified analyses on the studies according to sample type (plasma/serum or urine) and methylation method (N-MSP or other methods), as shown in Table 3. The specificity was similar between plasma/serum and urine samples regardless of methylation method. Four studies used biospecimens other than plasma/serum or urine. As the sample sizes from these studies were too small to further stratify by methylation method or location of primer, we analysed the pooled specificity of these four studies. The pooled specificity was 0.85 (95% CI, 0.48–0.97)(data not shown). The P-value for heterogeneity in each stratum was significant (Table 3).
Meta-regression analyses indicated a nonsignificant inverse association between age and specificity (β estimate=−0.22; P=0.2). No significant associations were observed between specificity and methylation method or between specificity and sample type.
Finally, the pooled specificity among the studies using healthy controls was high (0.92; 95% CI, 0.81–97), regardless of methylation method and sample type. The P-value for heterogeneity was 0.07, suggesting that there may still be variation not attributable to these covariates.
Individual and pooled sensitivity of GSTP1 promoter methylation
Unlike the relatively high specificity of GSTP1 found among most studies, the sensitivity of GSTP1 varied widely, from 0.05 to 1 (Supplementary Table 2, online only). The overall pooled sensitivity was 0.52 (95% CI, 0.40–0.64) (Table 4). No particular trend was observed among these studies in regard to methylation method or type of specimen (Table 4). The estimated pooled sensitivity for other types of specimens (ejaculate, prostate secretion, and others) was 0.66 (95% CI, 0.35–0.85). As only six studies used other types of specimens, we did not further stratify them by methylation method. The heterogeneity test P-values for the overall pooled sensitivity and the individual pooled sensitivity in each stratum were significant.
Upon further exploration, we found that sensitivity was higher in untreated samples (0.63; 95% CI, 0.50–0.75) than in treated samples (0.40; 95% CI, 0.25–0.78), regardless of the specimen type and the methylation method (Table 4). Further stratification by specimen type and methylation method among the treated and untreated groups revealed a higher sensitivity among the untreated (Table 5).
Meta-regression did not reveal any significant associations between sensitivity and age, between sensitivity and methylation method, or between sensitivity and sample type, but the meta-regression did show that sensitivity was significantly lower in the treated samples than in the untreated samples (beta=−1.22, P=0.02).
GSTP1 promoter methylation in relation to pathological stage, Gleason score, and PSA levels among prostate cancer cases
To further evaluate the odds ratios, we converted the pathological stage from an ordinal variable to a binary variable, that is, stages 3–4 vs 1–2. Likewise, we categorised Gleason scores as higher than 7 or lower than 7 and grouped PSA levels into higher or lower than 4 ngml−1. We limited our analyses to untreated samples and adjusted for methylation method and age; however, if any variable was not significant, it was removed from the model.
Six studies (Goessl et al, 2001b; Hoque et al, 2005; Ellinger et al, 2008; Roupret et al, 2008; Woodson et al, 2008; Payne et al, 2009) and additional unpublished data (provided Drs Roupret and James Catto) had sufficient information for analysing the association between gene methylation and pathological stage. We found that GSTP1 promoter methylation increased with prostate cancer pathological stage (stages 3–4 vs 1–2), with an odds ratio of 1.66 (95% CI, 0.86–3.19). The P-value for heterogeneity was 0.2 (Figure 2). We adjusted for age, sample type, and methylation method in the model; however, as none of these variables were significant, they were removed from the final model. When we excluded the study that used whole blood samples (Roupret et al, 2008), the odds ratio slightly increased (OR=1.80; 95% CI, 0.88–3.68; P for heterogeneity=0.2).

GSTP1 methylation in body fluids collected before treatment and risk of advanced stage of prostate cancer (comparing pathological stages 3–4 to stages 1–2). P-value for heterogeneity=0.2.
For similar reasons, we did not adjust for age, methylation method, or sample type for the following analyses. We found that GSTP1 promoter methylation was not associated with Gleason score (pooled odds ratio=1.05, 95% CI, 0.56–1.96; P-value for heterogeneity=0.4; 5 studies) (Goessl et al, 2001b; Hoque et al, 2005; Ellinger et al, 2008; Roupret et al, 2008; Woodson et al, 2008)(additional unpublished data provided by Drs Roupret and James Catto). Furthermore, GSTP1 promoter methylation was not associated with high-PSA levels (odds ratio=0.93, 95% CI, 0.77–1.02; P-value for heterogeneity=0.1; 5 studies)(Gonzalgo et al, 2004; Hoque et al, 2005; Rogers et al, 2006; Roupret et al, 2007; Woodson et al, 2008).
Discussion
The pooled specificity of GSTP1 was excellent (0. 89, 95% CI, 0.80–0.95) and much higher than the specificity of PSA. The specificity in each subgroup (stratified by sample type and methylation method) remained above 0.86. The sensitivity of GSTP1 was 0.63 (95% CI, 0.50–0.75) for samples collected before treatment and 0.40 (95% CI, 0.25–0.78) for samples collected after treatment; these sensitivities were not higher than the sensitivity of PSA screening. These results suggest that plasma, serum, or urine samples may complement PSA screening for prostate cancer diagnosis, although the positive link between GSTP1 methylation and pathological stage needs to be evaluated in more studies.
Collecting plasma/serum or urine samples is a non-invasive procedure, whereas invasive biopsy procedures may cause pain, anxiety, and increased medical costs. Urine samples were voided urine except the urine samples collected in the following studies: four were collected after a massage (Goessl et al, 2001b; Rogers et al, 2006; Woodson et al, 2008; Payne et al, 2009) and one after a biopsy (Rogers et al, 2006). High specificity remained even after we excluded the studies with urine collection after a massage.
This study highlights several important issues. First, we identified and systemically evaluated the methylation test at the GSTP1 promoter as an important potential test to complement PSA screening. As a complement rather than a replacement for PSA is needed, a high specificity is more important than a high sensitivity. To combine the strengths of both tests, they should be used sequentially, not simultaneously. The PSA test will be initially used to screen out potential patients, and the GSTP1 methylation test will then be given to those patients who have elevated PSA levels. Only those who have elevated PSA levels, followed by positive results on the GSTP1 methylation test, will undergo further biopsies. With its high specificity, the methylation test will exclude patients unlikely to have PCa but have elevated PSA levels. Using the two tests sequentially will reduce the number of unnecessary biopsies considerably, compared with using the PSA test alone. Serial testing has been used clinically for embolism and diarrhea (Fekety, 1997; Wells et al, 2001).
Second, unlike previous studies and reviews, we rigorously evaluated the specificity of GSTP1 by excluding healthy controls. In epidemiological research, we use controls that represent the population from which the cases were derived. As described above, randomly selected healthy controls usually do not have elevated levels of PSA or abnormal urological symptoms; therefore, they cannot represent patients who have high levels of PSA and undergo biopsy tests. Third, no previous studies have systemically evaluated the diagnostic value of measuring GSTP1 promoter methylation in different types of body fluids for prostate cancer diagnosis. This study indicates that the use of plasma/serum or urine samples for prostate cancer diagnosis is an important, non-invasive procedure that can complement PSA screening and minimise unnecessary biopsies.
Future assays that measure DNA methylation at gene promoters need to be standardised, simplified, and evaluated with external quality assurance programmes. Quantitative methods, such as pyrosequencing (<200 bp) and MassArrays (<600 bp), which truly quantify all the DNA methylation in the CpG islands and measure levels of all CpG dinucleotides, are also high-throughput technologies. Therefore, pyrosequencing and MassArrays are considered to be more efficient for validating the DNA methylation of gene promoters.
Of note, Figure 2 indicates that the DNA methylation test in whole blood samples is less sensitive to prostate cancer stages than the same test done in plasma, serum, or urine (Roupret et al, 2008). The methylated DNA in plasma, serum, or urine most likely derives from cancer cells whereas the methylated DNA detected in whole blood can be released from white blood cells as well. Therefore, the DNA methylation test in plasma, serum, or urine may be more accurate than the same test applied to whole blood in reflecting the severity of cancer stage. As most prostate cancer cases are detected at an early stage, the current PSA test does not predict specific prostate cancer stages. If GSTP1 methylation in plasma, serum, or urine samples is associated with pathological stage, this test will be even more appealing in addition to its high-specificity feature. More studies are warranted to confirm this finding.
The present study has several limitations. First, the validation assay of gene promoter methylation used in each study was different; some used N-MSP, others used Q-MSP, MSRE-Qpcr, or bisulfite sequencing, adding additional heterogeneity. Gonzalgo et al, 2004 (Gonzalgo et al, 2004) commented that primers selected at different regions on the same CpG island may have different sensitivities and specificities. Fortunately, the studies included in this paper all chose primers targeting the CpG dinucleotides in the 5′ promoter region starting from −80 to 400 nt from its transcriptional start site; hence findings in these studies are still believed to be appropriate for our analyses, as DNA methylation mostly occurs at the 5′ promoter region. Nevertheless, the specificity of GSTP1 across different methylation methods was consistently higher than the specificity of PSA, even though a wide range of sensitivities for GSTP1 was noted. This indicates the robustness of specificity of the GSTP1 methylation assay. Furthermore, our study did not find an association between sensitivity or specificity and methylation method.
Second, only 15 studies that did not use healthy controls could be used for specificity calculations. However, some of these studies used more than one specimen and gave us additional statistical power. Nevertheless, the evidence is compelling, in that the majority of individual GSTP1 specificities calculated here were higher than 0.8, and the overall pooled specificity was 0.89.
Third, the sample collection time varied widely among the studies. As mentioned above, because of our concern over the influence of treatment, we limited our analysis to untreated cases when analysing the associations between gene methylation and pathological stage and other factors, thereby decreasing our statistical power.
In summary, we summarised primer sequences, nucleotide position, and PCR methods in each study and evaluated them using a meta-analysis, which is a unique approach compared with a traditional review without any statistical analyses. Our study represents a new trend in epidemiology: a cross-disciplinary approach between molecular biology and epidemiology. Measuring DNA methylation at gene promoters has the potential to provide a new generation of biomarkers for prostate cancer diagnosis. Future studies should focus on the following tasks: (1) standardising the primers and the PCR protocols for each target gene; (2) using plasma, serum, or urine samples; (3) using patients with negative biopsies as controls rather than randomly selected healthy controls; and (4) collecting samples from cases before biopsies or at least before treatment to improve sensitivity. These tasks will reduce the heterogeneity among studies, enabling us to conduct an accurate meta-analysis to find a complement for the PSA test. Finally, more studies are needed to examine the association between gene methylation status and the stage and prognosis of prostate cancer. This will help avoid unnecessary treatment of some localised prostate cancers, as prostate cancer therapies are associated with significant adverse effects that impact patients’ health and quality of life.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Altimari A, Grigioni AD, Benedettini E, Gabusi E, Schiavina R, Martinelli A, Morselli-Labate AM, Martorana G, Grigioni WF, Fiorentino M (2008) Diagnostic role of circulating free plasma DNA detection in patients with localized prostate cancer. Am J Clin Pathol 129: 756–762
Bastian PJ, Palapattu GS, Yegnasubramanian S, Rogers CG, Lin X, Mangold LA, Trock B, Eisenberger MA, Partin AW, Nelson WG (2008) CpG island hypermethylation profile in the serum of men with clinically localized and hormone refractory metastatic prostate cancer. J Urol 179: 529–534; discussion 534–535
Bryzgunova OE, Morozkin ES, Yarmoschuk SV, Vlassov VV, Laktionov PP (2008) Methylation-specific sequencing of GSTP1 gene promoter in circulating/extracellular DNA from blood and urine of healthy donors and prostate cancer patients. Ann N Y Acad Sci 1137: 222–225
Cairns P, Esteller M, Herman JG, Schoenberg M, Jeronimo C, Sanchez-Cespedes M, Chow NH, Grasso M, Wu L, Westra WB, Sidransky D (2001) Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin Cancer Res 7: 2727–2730
Catalona WJ (1994) Management of cancer of the prostate. N Engl J Med 331: 996–1004
Chuang CK, Chu DC, Tzou RD, Liou SI, Chia JH, Sun CF (2007) Hypermethylation of the CpG islands in the promoter region flanking GSTP1 gene is a potential plasma DNA biomarker for detecting prostate carcinoma. Cancer Detect Prev 31: 59–63
Crocitto LE, Korns D, Kretzner L, Shevchuk T, Blair SL, Wilson TG, Ramin SA, Kawachi MH, Smith SS (2004) Prostate cancer molecular markers GSTP1 and hTERT in expressed prostatic secretions as predictors of biopsy results. Urology 64: 821–825
Ellinger J, Haan K, Heukamp LC, Kahl P, Buttner R, Muller SC, von Ruecker A, Bastian PJ (2008) CpG island hypermethylation in cell-free serum DNA identifies patients with localized prostate cancer. Prostate 68: 42–49
Fekety R (1997) Guidelines for the diagnosis and management of Clostridium difficile-associated diarrhea and colitis. American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol 92: 739–750
Goessl C (2000) Laser-fluorescence microsatellite analysis and new results in microsatellite analysis of plasma/serum DNA of cancer patients. Ann NY Acad Sci 906: 63–66
Goessl C, Muller M, Heicappell R, Krause H, Miller K (2001a) DNA-based detection of prostate cancer in blood, urine, and ejaculates. Ann NY Acad Sci 945: 51–58
Goessl C, Muller M, Heicappell R, Krause H, Straub B, Schrader M, Miller K (2001b) DNA-based detection of prostate cancer in urine after prostatic massage. Urology 58: 335–338
Gonzalgo ML, Nakayama M, Lee SM, De Marzo AM, Nelson WG (2004) Detection of GSTP1 methylation in prostatic secretions using combinatorial MSP analysis. Urology 63: 414–418
Gonzalgo ML, Pavlovich CP, Lee SM, Nelson WG (2003) Prostate cancer detection by GSTP1 methylation analysis of postbiopsy urine specimens. Clin Cancer Res 9: 2673–2677
Hoque MO, Topaloglu O, Begum S, Henrique R, Rosenbaum E, Van Criekinge W, Westra WH, Sidransky D (2005) Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol 23: 6569–6575
Jeronimo C, Usadel H, Henrique R, Silva C, Oliveira J, Lopes C, Sidransky D (2002) Quantitative GSTP1 hypermethylation in bodily fluids of patients with prostate cancer. Urology 60: 1131–1135
Kang GH, Lee S, Lee HJ, Hwang KS (2004) Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol 202: 233–240
Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG (1994) Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A 91: 11733–11737
Midgette AS, Stukel TA, Littenberg B (1993) A meta-analytic method for summarizing diagnostic test performances: receiver-operating-characteristic-summary point estimates. Med Decis Making 13: 253–257
Nakayama M, Gonzalgo ML, Yegnasubramanian S, Lin X, De Marzo AM, Nelson WG (2004) GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer. J Cell Biochem 91: 540–552
Papadopoulou E, Davilas E, Sotiriou V, Georgakopoulos E, Georgakopoulou S, Koliopanos A, Aggelakis F, Dardoufas K, Agnanti NJ, Karydas I, Nasioulas G (2006) Cell-free DNA and RNA in plasma as a new molecular marker for prostate and breast cancer. Ann NY Acad Sci 1075: 235–243
Payne SR, Serth J, Schostak M, Kamradt J, Strauss A, Thelen P, Model F, Day JK, Liebenberg V, Morotti A, Yamamura S, Lograsso J, Sledziewski A, Semjonow A (2009) DNA methylation biomarkers of prostate cancer: confirmation of candidates and evidence urine is the most sensitive body fluid for non-invasive detection. Prostate 69: 1257–1269
Reibenwein J, Pils D, Horak P, Tomicek B, Goldner G, Worel N, Elandt K, Krainer M (2007) Promoter hypermethylation of GSTP1, AR, and 14-3-3sigma in serum of prostate cancer patients and its clinical relevance. Prostate 67: 427–432
Rogers CG, Gonzalgo ML, Yan G, Bastian PJ, Chan DY, Nelson WG, Pavlovich CP (2006) High concordance of gene methylation in post-digital rectal examination and post-biopsy urine samples for prostate cancer detection. J Urol 176: 2280–2284
Rossouw JE, Cushman M, Greenland P, Lloyd-Jones DM, Bray P, Kooperberg C, Pettinger M, Robinson J, Hendrix S, Hsia J (2008) Inflammatory, lipid, thrombotic, and genetic markers of coronary heart disease risk in the women's health initiative trials of hormone therapy. Arch Intern Med 168: 2245–2253
Roupret M, Hupertan V, Catto JW, Yates DR, Rehman I, Proctor LM, Phillips J, Meuth M, Cussenot O, Hamdy FC (2008) Promoter hypermethylation in circulating blood cells identifies prostate cancer progression. Int J Cancer 122: 952–956
Roupret M, Hupertan V, Yates DR, Catto JW, Rehman I, Meuth M, Ricci S, Lacave R, Cancel-Tassin G, de la Taille A, Rozet F, Cathelineau X, Vallancien G, Hamdy FC, Cussenot O (2007) Molecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massage. Clin Cancer Res 13: 1720–1725
Suh CI, Shanafelt T, May DJ, Shroyer KR, Bobak JB, Crawford ED, Miller GJ, Markham N, Glode LM (2000) Comparison of telomerase activity and GSTP1 promoter methylation in ejaculate as potential screening tests for prostate cancer. Mol Cell Probes 14: 211–217
Sunami E, Shinozaki M, Higano CS, Wollman R, Dorff TB, Tucker SJ, Martinez SR, Mizuno R, Singer FR, Hoon DS (2009) Multimarker circulating DNA assay for assessing blood of prostate cancer patients. Clin Chem 55: 559–567
Wells PS, Anderson DR, Rodger M, Stiell I, Dreyer JF, Barnes D, Forgie M, Kovacs G, Ward J, Kovacs MJ (2001) Excluding pulmonary embolism at the bedside without diagnostic imaging: management of patients with suspected pulmonary embolism presenting to the emergency department by using a simple clinical model and d-dimer. Ann Intern Med 135: 98–107
Woodson K, O’Reilly KJ, Hanson JC, Nelson D, Walk EL, Tangrea JA (2008) The usefulness of the detection of GSTP1 methylation in urine as a biomarker in the diagnosis of prostate cancer. J Urol 179: 508–511; discussion 511–512
Acknowledgements
This project was partially supported by start-up funds provided to Dr Wu by the University of Cincinnati, by an Institutional Clinical and Translational Science Award, NIH/NCRR Grant Number 1UL1RR026314-01, by Dr Wu's KO7CA138714-01 award and ES006096. Research support for Dr Shuk-Mei Ho was provided by NIEHS grants P30-ES006096, 1RC2-ES018758, RC2-ES018789, ES018758, CA112532, and R01ES015584. Dr Wan-yee Tang is supported by K99ES016817.
We acknowledge the following investigators who kindly provided additional raw data for our analyses: Dr Olga Bryzgunova from the Siberian Division of the Russian Academy of Science; Dr Morgan Roupret from University Paris VI, Paris, France; and Dr James Catto, Institute of Cancer studies, University of Sheffield, United Kingdom.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wu, T., Giovannucci, E., Welge, J. et al. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: a meta-analysis. Br J Cancer 105, 65–73 (2011). https://doi.org/10.1038/bjc.2011.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.143
Keywords
This article is cited by
-
Early detection of the major male cancer types in blood-based liquid biopsies using a DNA methylation panel
Clinical Epigenetics (2019)
-
A urine-based DNA methylation assay, ProCUrE, to identify clinically significant prostate cancer
Clinical Epigenetics (2018)
-
A panel of DNA methylation markers for the detection of prostate cancer from FV and DRE urine DNA
Clinical Epigenetics (2018)
-
DNA methylation profiles in cancer diagnosis and therapeutics
Clinical and Experimental Medicine (2018)
-
Heterogeneous patterns of DNA methylation-based field effects in histologically normal prostate tissue from cancer patients
Scientific Reports (2017)
