
Abstract
Despite substantial gains in our understanding of the genomics of acute myelogenous leukemia (AML), patient survival remains unsatisfactory especially among the older age group. T cell-based therapy of lymphoblastic leukemia is rapidly advancing; however, its application in AML is still lagging behind. Bispecific antibodies can redirect polyclonal effector cells to engage chosen targets on leukemia blasts. When the effector cells are natural-killer cells, both antibody-dependent and antibody-independent mechanisms could be exploited. When the effectors are T cells, direct tumor cytotoxicity can be engaged followed by a potential vaccination effect. In this review, we summarize the AML-associated tumor targets and the bispecific antibodies that have been studied. The potentials and limitations of each of these systems will be discussed.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML), characterized by the infiltration of bone marrow, blood, and other tissues by malignant cells of the myeloid lineage is the most common acute hematologic malignancy of adults. In patients diagnosed before 60 years of age, AML is curable in 35–40% of cases, whereas only 5–15% of those presenting later in life can be cured.1 The treatment of AML that has remained essentially unchanged over the last three decades consists of intensive induction therapy followed by hematopoietic stem cell transplantation (HSCT). Many novel therapeutic agents, both small molecules targeting signaling pathways and immunologics are actively being investigated as salvage therapies or as alternatives to the standard of care. One class of immunotherapeutic agents is that of bispecific antibodies.
Bispecific antibodies combine the binding specificities and biologic functions of two antibodies into one molecule, one for a tumor-associated surface antigen, and the other for a surface antigen on the effector cells, such as T cells or natural-killer (NK) cells. Through the dual specificities, tumor cells are brought into close proximity to the effectors. Most importantly, if binding to the second specificity is agonistic, the cytotoxic functions of effectors can be activated at close proximity to the leukemic cells. Various combinations of whole antibodies and their fragments have yielded more than 60 different formats of such AML bispecific antibodies (examples in Figure 1).2 The immunologic properties and clinical potentials of each of these AML-associated targets are summarized in Table 1. Besides, a list of clinical trials investigating bispecific antibodies in myeloid leukemia is mentioned in Table 2. Characteristics of the bispecific antibodies (molecular weight, affinity, EC50 and parental clone) are summarized in Table 3.

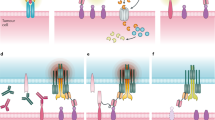
Different bispecific antibody formats. Heavy chain sequences are depicted in dark blue, dark red and dark gray, whereas corresponding light chains are in similar but lighter colors. Linkers are shown by continuous lines and disulfide bonds, when shown, are illustrated in dotted lines. Antigen epitopes are shown by triangles and semicircles. scFv, single-chain Fv fragments; scBsTaFv, single-chain bispecific tandem fragment variable; BiTE, Bispecific T-cell Engager; bsscFv, bispecific single-chain Fv; BiKE, bispecific killer-cell engager; DART, Dual-Affinity Re-Targeting; TandAb, Tandem Diabodies; sctb, Single-chain Fv Triplebody; BIf, Bispecific scFv Immunofusion; DVD-Ig, Dual-Variable-Domain Immunoglobulin; VH, variable heavy chain; VL, variable light chain; CL, constant light chain; CH1-3, constant heavy chains 1 to 3.
In this review, we summarize those AML targets for which bispecific antibodies have been developed, in descending order of clinical relevance: CD33, CD123, Wilms’ tumor protein (WT1), CD13, CD15, CD30, CD45, CD47, C-type lectin-like molecule 1 (CLL1), Fms-like tyrosine kinase 3 (FLT-3) and angiogenic growth factors.
CD33
CD33 or Siglec-3, a 67-kDa glycoprotein, is a member of the sialic acid-binding immunoglobulin-like lectin (Siglec) family, which in turn belongs to the immunoglobulin superfamily. The extracellular domain of CD33 comprises one amino-terminal sialic acid-binding V-type and one C2-type immunoglobulin-like domain connected by a helical transmembrane sequence to a cytoplasmic tail containing two immunoreceptor tyrosine-based inhibitor motifs (ITIM). Phosphorylation of the tyrosine residues by the Src family kinases is involved in the recruitment and activation of the Src homology-2 (SH2) domain-containing tyrosine phosphatases SHP1 and SHP2.3
CD33 is a receptor that appears during commitment of the hematopoietic stem cell to the myelomonocytic lineage. It is expressed on myeloid progenitors, monocytes, myeloid dendritic cells and less so, on macrophages and granulocytes.3 Although it is restricted mainly to the myeloid lineage, low levels of CD33 expression has been reported on some lymphoid cells, including the earliest precursors of human fetal thymocytes and human CD34(+) postnatal thymocytes.4 In addition, activated NK cells and T cells can also express CD33.5 CD33 is expressed on the majority of AML cells and the level of CD33 seems to correlate with the disease prognosis.6
The first bispecific T-cell engager (BiTE) developed against CD33 is AMG 330 (Amgen Inc., Thousand Oaks, CA, USA), which binds to a linear epitope containing the amino acid sequence IPYYDKN in the CD33 V-type domain.7, 8, 9 This BiTE activates and expands T cells from autologous clinical samples of patients with AML and mediates in vitro lysis of primary AML and normal myeloid cells in a dose dependent manner at concentrations as low as 1 ng/ml (EC50=0.35–2.7 pm).8, 10 However, it is noteworthy that activated T and NK cells can also express CD33.5 CD33-independent activation with an anti-CD19 BiTE led to CD33 neo-expression on a minor subset (<6%) of T cells, associated with their fratricide but with minimal effect on AML-directed cytotoxic T-cell function.9 Importantly, soluble CD33 found in the blood of some patients with AML does not affect the AMG 330-mediated T-cell activation or cytotoxicity.9 Antibody-mediated endocytosis of antigens could reduce the availability of cell surface targets for antibody therapy. In contrast to bivalent anti-CD33 IgG antibodies, AMG 330 is not endocytosed and does not modulate surface expression of CD33. In addition, the function of this BiTE is neither affected by common CD33 single nucleotide polymorphisms nor by the adenosine triphosphate-binding cassette transporter activity. In vivo experiments showed that AMG 330, in the presence of activated human T cells, could suppress the growth of a subcutaneous AML cell line xenograft in humanized mice improving survival.8, 9
To better understand the immunobiology of AMG 330 BiTE, human leukemic cell lines were engineered to express the inhibitory (PD-L1 and PD-L2) or activating (CD80 and CD86) ligands to interact with their respective receptors (that is, CD28 and PD-1), It was found that the expression of PD-L1 and PD-L2 inhibitory molecules on target cells reduced the in vitro cytotoxicity of AMG 330 BiTE in the presence of healthy donor T cells at low effector:target (E:T) ratios. This inhibition correlated with the level of ligand expression on target cells.11 On the other hand, CD80 and CD86 activating molecules expressed on tumor cells increased the cytolytic activity of AMG 330. Treatment with anti-CD28 activating antibodies enhanced the cytotoxicity of AMG 330 against human-AML cell lines and primary AML samples from patients with refractory leukemia.11
In a subset of patients, T-cell activation and cytotoxicity were suboptimal,6, 8, 10 possibly because of inhibition by the PD-1/PD-L1 pathway. Although the majority of primary AML cells do not express PD-L1, it can be upregulated by the proinflammatory cytokines released during the activation of T-cells by AMG 330. The addition of anti-PD-1/PD-L1 antibodies restored T-cell expansion, their interferon-γ (IFNγ) secretion and the lysis of leukemic cells. Importantly, whereas low E:T ratios and low CD33 expression on targets impeded T-cell cytotoxicity, extending the in vitro incubation time substantially improved tumor killing.10, 12 In addition, the use of epigenetic modifiers such as panobinostat and azacitidine could increase the expression of CD33 to enhance AMG 330-induced cytotoxicity.10 These observations are clinically relevant since leukemic stem cells express lower amounts of CD33 when compared with the bulk of the leukemia population.6 These observations have prompted the use of prolonged administration schedule of AMG 330 to improve its anti-leukemic effect. However, as CD33 is present on normal hematopoietic stem cells (although lower than that on leukemic stem cells6), such an infusion schedule could prolong also cytopenia.13 A recent study showed that the effectiveness of AMG 330 was inferior against AML samples from patients with relapsed/refractory leukemia when compared with samples taken from patients at the time of first diagnosis. These data suggest the existence of possible resistance mechanisms among AML blasts.14
A second bispecific antibody format against CD3 and CD33 attaches the VH of an anti-CD3 to the VL of an anti-CD33 antibody, whereas connecting the VH of the anti-CD33 to the VL of the anti-CD3 antibody, with the two tandem single-chain Fv fragments (scFv) joined by a glycine-serine (G4S)5 linker. The resulting construct was initially complicated by the formation of fragmented nonfunctional proteins because of DNA recombination triggered by sequence homology. This problem was resolved by rearranging the variable regions in a way such that the variable heavy (VH) of CD33 and VH of CD3 were linked to their cognate variable light (VL) regions. In addition, the (G4S)5 linker was replaced by a new helical sequence (amino acid sequence: EKEALKKIIEDQQESLNK) in joining the CD33 and CD3 binding moieties. The newly generated CD33 × CD3 single-chain bispecific tandem fragment variable (scBsTaFv) construct was able to lyse CD33-expressing target cells in the presence of peripheral blood mononuclear cells (PBMC) obtained from healthy donors.15, 16 When a green fluorescent protein was incorporated into the bispecific antibody, the formation of immunological synapse between T cells and target cells was visualized.17 When the CD33-CD3 scBsTaFv bispecific antibody was further humanized, it retained its ability to arm T cells to kill AML blasts at picomolar concentrations (EC50=15 pm).18, 19 When tested by in vitro colony forming assays, the multilineage reconstitution potential of hematopoietic stem cells treated with the humanized antibody was not compromised.18 To further improve the potency of this bispecific antibody, 4-1BB ligand (4-1BBL) co-stimulatory molecule was incorporated into the construct in a modular manner. The novel modular system outperformed the original humanized CD33 × CD3 scBsTaFv in killing CD33low AML cells at low E:T ratios in vitro.19
One obstacle in the use of these small-size bispecific antibody fragments is their rapid clearance through kidney, necessitating their continuous infusion for up to several weeks.20 To tackle this problem, mesenchymal stromal cells (MSC) were engineered to continuously secrete the humanized CD33-CD3 scBsTaFv, and to express 4-1BBL for T-cell co-stimulation. T cells, AML cells and scBsTaFv-engineered MSC cells were co-cultured for 24 h before injection into immunodeficient mice. Results showed that antibody-engineered MSC cells could redirect T cells toward AML cells and extend the survival of leukemic mice.21 Further experiments will be necessary to assess whether this therapeutic modality can target neoplastic cells in established leukemia animal models, and to determine the optimal delivery method for the engineered MSCs, as well as to investigate if MSCs can home to tumor sites and activate T cells without inducing immunosuppressive effects.
Recently, a panel of bispecific tandem diabodies (TandAb) specific for CD3 and CD33 have been generated of which one (AMV-564) was chosen for further clinical development. The AML lysis mediated by this construct in the presence of T cells depended on antibody dose and E:T ratio. Furthermore, the TandAb-induced cytotoxicity was similar against AML samples from newly-diagnosed patients, or from those with refractory/relapsed leukemia. No correlation was found between the antibody-mediated cytotoxicity and CD33 expression level on target cells. This TandAb was also successful against AML subcutaneous xenografts in mice.22
Moving beyond CD3 on T cells, other activating ligands or antibodies designed for lymphocytes have been tested against human-AML. Since both NK and CD8(+)T cells express the NKG2D activating receptor, a dual specific scFv-ligand was designed where the NKG2D activating ligand, UL16 binding protein 2 (ULBP2), was fused to the anti-CD33 scFv via an 18 amino acid (EKEALKKIIEDQQESLNK) linker. The ULB-CD33 scFv construct redirected the cytotoxicity of pre-activated NK cells and PBMCs toward AML targets. Although administration of this scFv-ligand to freshly-isolated CD8(+)cells in the presence of AML blasts enhanced IFNγ secretion, no killing of leukemic cells was observed. Furthermore, although co-administration of ULB-CD33 scFv and CD33 × CD3 scBsTaFv augmented the IFNγ and tumor necrosis factor-α (TNFα) secretion by CD8(+)T cells, the anti-leukemic cytotoxicity was not superior when compared with CD33 × CD3 scBsTaFv alone. Co-stimulation through the NKG2D receptor could reinforce the CD3-mediated signaling on T cells, but it was not enough to increase the lysing of target cells.16
Although many efforts have focused on bispecific antibodies to retarget T cells in cancer immunotherapy, the engaging of the innate immune system (that is, NK cells through the CD16 Fc-receptor) has also shown promise. A chemical conjugate of anti-CD16 and anti-CD33 monoclonal antibodies was able to redirect cytotoxicity of resting or activated NK cells, as well as peripheral blood lymphocytes toward AML leukemic blasts, with maximum cytotoxicity requiring 10 ng/ml concentrations of antibody.23 Nevertheless, chemical conjugates generally yield a heterogeneous mixture of cross-linked products, of which only a subset has the desired target specificity. Using recombinant DNA technology, large scale and efficient production of bispecific antibodies has since taken over.24 A fully-humanized scFv of CD16 and CD33 were joined together using a 20-amino acid linker derived from the human muscle aldolase. This BiKE could activate NK cells to secret IFNγ and TNFα, and to lyse CD33(+) AML targets. Treatment of NK cells with an ADAM17 inhibitor and CD16 × CD33 BiKE prevented CD16 shedding from NK-cell surface, thereby enhancing NK-cell cytokine secretion and cytotoxicity.25 This BiKE was further tested using NK-cell samples from patients with myelodysplastic syndrome (MDS) where the NK-cell frequency and their CD16 expression were low. Upon treatment with the CD16 × CD33 BiKE, NK cells from these patients produced IFNγ and TNFα, and lysed a CD33(+) AML tumor cell line as well as CD33(+) myeloid derived suppressor cells. The latter cells possess NK and T-cell suppressing properties, and are significantly increased in patients with MDS.26
A further improvement of BiKE attempted to improve in vivo expansion of NK cells. Instead of using interleukin-2 (IL2), which induces regulatory T cells (Tregs) that suppress NK-cell function,27 interleukin-15 (IL15) can be used to enhance NK-cell homeostasis in a trispecific killer-cell engager (TriKE). Here, the human IL15 sequence, flanked by two linkers, was inserted between the scFvs of the CD16 × CD33 BiKE.28 The IL15 TriKE induced similar NK-cell mediated killing when compared with a mixture of IL15 and BiKE. In a murine AML xenograft model using human NK cells, TriKE was superior to BiKE.28
Instead of the IL15 sequence, a third scFv can be inserted to create a triple scFv antibody. Two anti-CD33 scFv fragments attached in tandem via a flexible 20-amino acid linker (Gly4Ser)4 to a single anti-CD16 scFv (sctb: single-chain Fv triplebody) was compared with the single anti-CD33 scFv construct (bsscFv: bispecific single-chain Fv). CD33 avidity of the sctb was 3.5 fold higher when compared with that of the bsscFv, even though their affinity was comparable. Both antibody constructs in the presence of human PBMC mediated lysis of CD33(+) AML cell lines with EC50 values in picomolar ranges (212–426 pm for bsscFv, 1.8–18 pm for sctb). Importantly, the sctb was 10–200-fold more potent than the bsscFv in killing human leukemic cell lines.29 The logic of this enhancement was extended to targeting more than one tumor antigen on the surface of cancer cells. For acute leukemia with mixed-lineage phenotypes, a sctb with dual target specificities (anti-CD33 scFv and anti-CD19 scFv) was attached to an anti-CD16 scFv. Compared with the CD33 × CD16 or CD19 × CD16 bsscFv, the CD33 × CD16 × CD19 sctb redirected cytotoxicity of MNCs against a double-positive cell line at 23 and 1.4 fold lower concentrations, respectively.30 In another series of experiments three sctb antibodies were generated. Two of the sctb antibodies targeted a single antigen on leukemia cells (either CD33 or CD123), whereas the third sctb targeted both leukemia antigens. Although both single and double antigen formats were able to mediate lysis of AML targets in the presence of PBMC, the latter was more potent, showing the advantage of simultaneous targeting of more than one tumor antigen.31
CD64 (FcγRI), which is expressed on monocytes, macrophages and neutrophils, has an important role in antibody-dependent cell mediated cytotoxicity (ADCC). A bispecific antibody was generated by chemical conjugation of CD33 and CD64 and was able to redirect cytokine-activated monocyte cytotoxicity toward CD33 leukemic cells. IFNγ, granulocyte-colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) increased the expression of CD64 on monocytes and enhanced their cytotoxic potential.32 Treatment of target cells with IFNγ also increased their killing by the bispecific antibody.33 Although the treatment of AML cells with monospecific anti-CD33 or anti-CD64 antibodies for 48 h inhibited AML cell growth, the inhibitory potency mediated by the bispecific molecule was higher. Furthermore, the bispecific antibody augmented the anti-tumor effects of monocytes in a murine tumor model.34
CD123
CD123 is the alpha subunit of the interleukin-3 (IL-3) receptor. This 75 kDa single-pass type-I membrane protein contains three extracellular, one transmembrane and one intracellular domain. It is expressed on monocytes, B cells, megakaryocytes, plasmacytoid dendritic cells, and hematopoietic stem and progenitor cells.35 CD123 expression in B lymphoid and myeloid progenitors is high, whereas erythroid progenitors and primitive hematopoietic cells express either low or undetectable levels.36 Overexpression of CD123 can promote cell proliferation and may induce leukemogenesis.37 CD123 was once thought to be an AML stem cell marker.38 AML cells with high CD123 expression had higher proliferation and were more resistant to apoptosis induced by growth factor deprivation. Higher expression level of CD123 among AML blasts is associated with lower complete remission rates and poorer overall survival.39
To redirect T cells toward CD123 leukemic cells, the scFv of an anti-CD123 antibody (CD123scFv) was fused to the N terminus of the truncated human IgG1-Fc (CH2-CH3) connected in turn to an anti-CD3 scFv (CD3 scFv) at its C terminus, a platform named bispecific scFv immunofusion (BIf). The C-terminal location of the CD3 scFv reduced its affinity for CD3(+) T cells by two orders of magnitude. The BIf was able to mediate lysis of CD33(+) target cells by T cells at low picomolar EC50 (EC50=8 pm).40 Another bispecific tandem scFv construct was built comprising the scFv fragments of anti-CD123 and anti-CD16 antibodies. The bispecific construct at picomolar concentrations (EC50=211–364 pm) triggered the lysis of leukemic cells in the presence of PBMC.41 As mentioned earlier, a CD16 scFv-based sctb against CD123 demonstrated potent anti-leukemic activity, but inferior to a similar but double-targeting sctb simultaneously targeting CD33 and CD123.31
A dual-affinity re-targeting (DART) molecule named MGD006 (MacroGenics, Rockville, MD, USA), was built using two independent chains, one consisting of the humanized VL of anti-CD3 attached in tandem to the VH of anti-CD123, and the other consisting of the VL of anti-CD123 attached to the VH of anti-CD3 antibody. The two polypeptides were attached via disulfide bonds between the C terminus of the VH fragments. This DART molecule was able to activate and expand T cells and suppress AML xenografts in mice reconstituted with human PBMC. Because of the small size of the molecule (59 kD), continuous infusion over seven days was necessary. In cynomolgus macaques, MGD006 produced transient IL-6 release and reversible decrease in the red blood cell mass.42 In humanized NSG mice, MGD006 was effective against xenografts derived from a leukemic cell line or from primary AML samples.43 A first in human study is ongoing in AML patients (ClinicalTrials.gov identifier NCT02152956).
WT1
WT1 is a zinc finger transcription factor that has important roles in cell survival and development. Several isoforms of WT1 exist, each with specific inhibitory or activating functions on a variety of important genes and cellular pathways. Mutations in the WT1 gene have been associated with human diseases. In AML, WT1 is considered an oncogene and its expression was found on the majority of AML samples.44 Importantly, higher WT1 gene expression is associated with lower complete remission rates and decreased survival.45 Most therapeutic antibodies recognize intact antigens expressed on the cell surface; however, the majority of leukemia-associated antigens are intracellular. When degraded, their peptides are presented on human leukocyte antigens (HLA) as peptide–MHC (major histocompatibility complex) complexes on the cell surface. Recently, a BiTE construct comprised of the scFv fragments of antibodies specific for CD3 and WT1 epitope RMF in the context of HLA-A*02:01 (WT1-BiTE) was made.46 This BiTE could bind to WT1(+) and HLA-A*02:01(+)AML targets and activate T cells to proliferate and kill the leukemic cells in vitro and in immunodeficient mice. No binding to CD34(+)cells was found. Furthermore, WT1-BiTE therapy induced long-term T-cell responses against tumor-associated epitopes other than WT1 because of epitope spreading, which could greatly enhance the therapeutic efficacy of this bispecific construct. However, cross reactivity of these T-cell receptor (TCR)-like antibodies with other peptides on other HLA antigens could be a major hurdle for clinical development.47
CD13
CD13, also known as aminopeptidase-N or gp150, is a myeloid membrane-bound zinc-dependent metalloprotease with an extracellular enzymatic moiety that cleaves N-terminal amino acid residues from oligopeptides.48 CD13 contains an extracellular domain joined via a helical transmembrane sequence to a small cytoplasmic domain. CD13 has several roles including tumor cell invasion, adhesion, differentiation, proliferation, apoptosis, motility, phagocytosis and angiogenesis.49 Monoclonal antibodies targeting CD13 result in apoptosis of primary AML cells and cell lines.50 In an attempt to target CD13 in AML, a bispecific antibody was generated by chemically conjugating the anti-CD3 and anti-CD13 Fab’ fragments. This bispecific antibody markedly enhanced the cytotoxicity of IL2- or IL7-stimulated PBMCs against CD13(+) AML cells. Although the bispecific construct inhibited granulocyte-macrophage and mixed cell colonies of normal bone marrow progenitors, the inhibition was less than that for AML blast colonies.51
CD15
CD15 (3 alpha-fucosyl-N-Acetyl lactosamine), also called Lewis X or stage-specific embryonic antigen 1 (SSEA-1), is a carbohydrate antigen expressed on the majority of AML cells and on some NK cells, T cells, monocytes, neutrophils and eosinophils.52 CD15 expression on AML blast was associated with favorable prognosis such as continuous complete remission and longer survival.53 In an attempt to design a bispecific antibody, the fragment of antigen-binding (Fab) of an anti-CD64 antibody was chemically conjugated with whole IgM monoclonal antibody against CD15. This bispecific construct was able to redirect in vitro cytotoxicity of IFNγ-activated monocytes toward CD15(+)AML cells in the presence of human serum as a source of complement. The antibody conjugate was assessed in a phase I clinical trial of four patients with CD15(+) cancer. Six doses of the drug over a period of two weeks were administered. In a patient with AML, a 30–60% transient reduction in peripheral blasts was observed when peak serum level of the antibody was as low as 50 ng/ml. No toxicity was reported.54
CD30
CD30, member 8 (TNFRSF8) of the tumor necrosis factor receptor superfamily, is a 120-kDa type-I transmembrane glycoprotein. It is expressed on resting human CD8(+) T cells, activated B and T cells and their leukemias and lymphomas. CD30 has a co-stimulatory, mitogenic and activation effects on T cells.55 CD30 is also expressed with variable intensity on 36%56 to 50%57 of patient AML and its expression is associated with FLT-3-ITD mutations and leucocytosis.57 Upon cleavage from the extracellular domain, CD30 is released as a soluble form that can be detected in the blood of patients with Hodgkin and non-Hodgkin lymphoma and it is associated with poorer prognosis.58, 59 A TandAb containing two binding sites for CD30 and CD16A has been generated. Unlike an anti-CD30 IgG, the TandAb did not bind to the non-activating CD16B receptor. Furthermore, binding of the TandAb to CD30(+) target cells was higher than that of the anti-CD30 IgG, resulting in improved killing of target cells. Although not tested against AML blasts, this TandAb exhibited cytotoxicity against CD30(+) non-Hodgkin lymphoma cells. In the absence of CD30(+) target cells, TandAb did not activate NK cells,60 an important consideration for the clinical application of such bispecific antibodies.
CD45
CD45 is a protein tyrosine phosphatase enzyme expressed in various isoforms. It contains an extracellular sequence connected via a helical transmembrane domain to a cytoplasmic tail. CD45 is expressed on all hematopoietic cells except mature red blood cells and platelets.61 A recombinant bispecific antibody was generated comprising a scFv against the radiometal complex with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, a metal chelating agent) and a CD45 binding moiety containing the light chain of an anti-CD45 antibody and the heavy chain of rituximab.62 In vivo targeting was performed in human-AML-bearing mice after injecting the antibody construct, followed by administering a clearing agent after 22 h and finally injecting the yttrium‐90-labeled DOTA. Biodistribution studies revealed favorable antibody retention in tumor sites, whereas the normal organs showed minimum uptake. The therapeutic potential of this bispecific agent was not reported.
CD47
CD47 is a 50-kDa glycoprotein belonging to the immunoglobulin superfamily. It contains an extracellular IgV domain next to a five-fold-passing transmembrane domain and a short cytoplasmic sequence.63 CD47 binding to signal regulatory protein-α expressed on dendritic cells and macrophages delivers a signal which inhibits phagocytosis.64 CD47 is a leukemia stem cell (LSC) marker and its overexpression in AML has been associated with poor survival.65 Recently, a bispecific antibody was generated to target CD47 and CD20 to treat lymphoma. Here, the C terminus of the VH and VL of an anti-CD20 antibody were joined by short peptide linkers to the N terminus of the respective chains in the anti-CD47 IgG to generate a bispecific dual-variable-domain immunoglobulin (DVD-Ig) format. Affinity of the DVD-Ig relative to the parental monoclonal antibody for CD47 was diminished 15–20-fold. In comparison to a 12-amino acid linker, DVD-Ig using the shorter 5-amino acid linker showed weaker binding to CD47. Fortunately this lower affinity allowed the 5-amino acid linker form to prefer the double-positive CD47(+)CD20(+)cells and avoid being trapped in a CD47 antigen sink. The bispecific short-linker construct improved the survival of lymphoma-bearing mice and recapitulated the therapeutic benefit of anti-CD47 and anti-CD20 antibody combination therapy.66
CLL1
CLL1, also known as C-type lectin domain family 12 member A (CLEC12A), is a pan-myeloid antigen containing an extracellular, a transmembrane and a cytoplasmic domain. CLL1 is expressed on myeloid cells and peripheral blood monocytes, dendritic cells and granulocytes. Thanks to its absence on normal hematopoietic stem cells and its expression on LSC, CLL1 was explored as a marker for minimal residual disease in AML.67 To test the effect of targeting CLL1 in AML, the Fab fragments of anti-CCL1 and anti-CD3 antibodies were site-specifically modified using the unnatural amino acid, p-acetylphenylalanine and then conjugated to generate a bispecific Fab (BiFab). Compared with a BiFab against CD33 and CD3 generated using the same methodology, the CLL1-CD3 antibody-mediated stronger in vitro cytotoxicity and superior in vivo anti-tumor activity in immunodeficient mice with established leukemia xenografts receiving expanded T cells.68 Another laboratory also successfully demonstrated the in vitro functionality of a CLL1-CD3 BiTE in the presence of T cells against AML cells.69
FLT-3
FLT-3, also known as CD135, is a receptor-type tyrosine kinase and has an important role in the development of hematopoietic progenitor cells and dendritic cells. FLT-3 contains an extracellular sequence with an Ig-like C2-type domain, a helical transmembrane part and a cytoplasmic domain with protein kinase activity. FLT-3 is expressed on committed lymphoid and myeloid progenitors and on monocytes.70, 71 Activating FLT-3 mutations are present in about 30% of patients with AML.72 Furthermore, the level of FLT-3 expression on AML cells is increased and is associated with higher percentage of bone marrow blasts and high leukocyte counts.70, 73 Two bispecific antibody formats have been designed to redirect T cells towards FLT-3(+)AML cells. One of the constructs, termed Fabsc, contained an anti-FLT-3 Fab fused to a scFv specific for CD3 via a CH2 linker modified to prevent dimerization and binding to Fc receptors. In the other construct, scFv fragments of the two antibodies were fused together using a BiTE format. Whereas both constructs were able to lyse FLT-3(+)target cells, the Fabsc format had higher affinity for FLT-3, lower aggregate formation and superior production yield.71
Angiogenic growth factors
Neoangiogenesis has a crucial role in pathogenesis of cancers including AML. The expression of two angiogenic agents, vascular endothelial growth factor-A (VEGF-A) and the angiopoietin-2 (Ang-2) on AML cells is associated with poorer patient outcome.74, 75 Circulating levels of Ang-2 and VEGF are also strong predictors of poor survival in AML patients.76, 77, 78 The expression of VEGF-A and its receptors on AML cells is increased, raising the possibility of an autocrine proliferation mechanism in AML.79 Bispecific antibodies have been generated to simultaneously target VEGF-A and Ang-2. In one approach called CrossMab technology, a heterodimeric IgG1-based bispecific antibody with monovalent binding moieties for VEGF-A and Ang-2 antigens was constructed. In this platform, monomeric heavy chains of anti VEGF-A and Ang-2 antibodies are preferentially paired via the Knobs-into-holes methodology to make a heterodimeric heavy chain, whereas the correct pairing of light chains is guaranteed by crossover of the heavy chain constant-1 (CH1) and light chain constant (CL) domains.80 Although not yet tested against AML targets, this CrossMab antibody showed in vivo functionality against various cell lines including breast, lung, prostate, gastric, pancreatic and colon cancer, inhibited hematogenous spread of tumor cells, and was able to suppress corneal angiogenesis in mice.80, 81 Using another approach, so-called bispecific CovX-bodies were chemically synthesized by first linking the chemically synthesized VEGF and Ang-2 binding peptides by means of an azetidinone linker and then fusing these heterodimeric peptides to a scaffold antibody in a site-specific manner. In vivo functionality studies showed that the CovX-body imparted anti-angiogenic effects and reduced the growth of colon, breast and skin cancer xenografts.82
Perspective and conclusions
T lymphocytes are highly efficient professional killers proven successful clinically in a variety of human cancer types. Although this recognized potential of T cells is just beginning to be exploited for immunotherapy of leukemia, NK cells offer unique opportunities in the AML setting, both in terms of its trafficking pattern and its functional competency. In contrast to solid cancers leukemia is generally a blood borne disease where NK and T cells have an easier access to the cancer cells. Since leukemic blasts are mainly present in the bone marrow and blood, effector cells have better access to cancer cells. Unlike T cells, NK cells do not penetrate all tissues.83 Hence, although T-cell-bispecific antibodies may be necessary for solid tumors, both NK and T-cell bispecifics could be used for blood born malignancies such as leukemia. The important role of NK cells and the killer-cell immunoglobulin-like receptor)–ligand interactions in preventing relapse following allogeneic transplantation for AML is well documented.84 As higher E:T ratio improves the function of bispecific antibodies,10, 13, 85 a robust endogenous effector population or the adoptive transfer of exogenous effector cells should increase the efficacy of these therapies. Application of allogeneic effector cells as an off-the-shelf immunotherapy has been extensively investigated after myeloablative preconditioning.86 With age, thymic function involutes and if T cells are further damaged by myeloablative conditioning,87 they need to be resuscitated with growth factors, such as interleukin-7, interleukin-15, keratinocyte growth factor or sex steroid hormone inhibition.88 Alternatively, the administration of allogeneic precursor T cells could improve T and NK-cell reconstitution without inducing graft-versus-host disease (GvHD) at least in mouse models;89 however, their potential for human application is still uncertain. Unlike T cells, NK-cell recovery does not require the thymus, and is relatively fast after dose-intensive or myeloablative chemotherapy.90 Unlike allogeneic T cells that induce GvHD, allogeneic NK cells do not generate and may even reduce GvHD.91 Hence, administrating such NK cells to enhance effector-target ratio should be clinically safe. Although CD3 is a proven activating receptor on T cells for bispecific antibodies, the clinical utility of agonistic receptors other than CD16 on NK cells is still investigational. NKG2D is one such activating receptor92 and MICA, MICB, ULBP1-6 are the other known activating ligands.93, 94, 95 Activating NK cells using anti-CD16 antibodies allows selective activation of FcRIII (CD16), unlike IgGs which stimulate all FcR receptors of the activating (CD16,CD32A, CD64) and inhibitory classes (for example, CD32B). In addition, activation of CD16 through specific anti-CD16 antibody bypasses the genetic polymorphism among patients with differential FcR affinity for IgG.60, 96, 97 With increasing sophistication in the genetic engineering of antibodies, multi-specific formats to include antibodies that will neutralize the inhibitory receptors may be possible, including inhibitory KIRs (2DL2/2DL3, 3DL1),98, 99 PD-1 (B7-H1),100 and B7-H3.101
Selecting the optimal leukemia-associated target antigen is as critical as choosing the proper class of effector cells or the particular antibody platform. Because of the importance of LSC in the recurrence of AML, bispecific antibody constructs need to engage antigens expressed on LSCs. Some of the potential LSC candidate antigens for generation of bispecific antibodies have been discussed by other investigators, including CD44,102 CD52,103 CD96,104 CD300f105 and TIM-3.106 A subset of leukemia called biphenotypic or mixed-lineage leukemia are composed of a heterogeneous mixtures of lymphoid and myeloid blasts. In addition, lineage switching could happen de novo in neonates, or over the natural course of the disease or following ralapse after treatment.107 Bispecific antibodies simultaneously targeting CD3 (on T cells) and a single marker shared by both lymphoid and myeloid stem cells (for eample, CD9(refs 108, 109)) or trispecific antibodies targeting two separate markers on the leukemia blast (CD33 for myeloid and CD19 for lymphoid) could prevent escape of biphenotypic LSC. Furthermore, the carbohydrate antigen Lewis Y (CD174) has been introduced as a new marker for AML blasts and has been targeted with CARs in a clinical trial.110 Development of bispecific antibodies against this antigen in AML merits attention. CD33 may have a theoretical advantage over other myeloid markers for building bispecific antibodies since it carried on myeloid suppressor cells, an important population in the tumor microenvironment that impacts host T-cell immunity.111
Antigen density on leukemic cells can affect antibody binding and effectiveness of bispecific antibodies. When antigens are expressed at low levels, like WT1-peptide-MHC complexes, higher-affinity antibodies may be necessary for optimal cytotoxicity.112 However, in the case of antigens with abundant expression on tumor cells, standard-affinity antibodies should be adequate for killing leukemic cells while sparing normal cells with low levels of the antigen. In this context, high affinity could be counterproductive given the sequestration of such antibodies by antigen sinks in normal tissues or by affinity barriers.113, 114
Several bispecific antibody platforms have been tested in AML. Each platform has its unique strengths and weaknesses. BiTEs, diabodies and DARTs have small molecular size (~50 kD) below the renal clearance threshold. Although suitable for imaging because of their fast clearance from the blood, continuous infusion is necessary for their therapeutic applications. Other formats, such as Fc-fusion proteins or IgG-scFv have molecular sizes above the renal threshold and hence longer serum half-lives. None of the platforms presented in this review are able to cross the blood brain barrier, which permits only certain molecules with small molecular weights (⩽600 Da) and water–lipid partition coefficients, unless the bispecific antibody is piggybacked onto transcytosis pathways such as the transferrin receptor.115 In addition to their size, the valency of these platforms could be important for their biologic effect. Higher avidity for their cognate antigen usually translates into better binding and cytotoxicity, although cross-linking antigens can also lead to internalization of some antigens (for eample, CD33). Bispecific antibodies that engage T cells stimulate the release of proinflammatory cytokines which act like a double-edged sword. Such cytokines can increase the anti-leukemia efficacy by direct cytotoxicity and by activation and recruitment of immune cells into the tumor site. However, they also increase the risk of adverse effects including the life-threatening cytokine release syndrome.116 Moreover, lessons learned from bone marrow transplantation would suggest that application of bispecific antibodies at the time of minimal residual disease should maximize the chance of cure besides reducing the risk of tumor lysis syndrome or the accompanying cytokine storm.
Generally, antibodies recognize cell surface antigens. Nonetheless, as the majority of leukemia-associated antigens are intracellular proteins whose peptide epitopes are presented outwardly in the context of HLA molecules, developing bispecific antibodies against these peptide–HLA complexes can unveil a treasure trove of potential novel tumor-specific targets. However, the field of TCR-like antibody is still in its infancy and many challenges remain. Cross reactivity of such antibodies with other peptides and with other HLA molecules could risk adverse side effects. Furthermore, as these antibodies only recognize antigens in the context of HLA molecules, they can only be applied in patients with specific HLA types.
Although not covered in this review, highly encouraging results of chimeric antigen receptor (CAR)-armed T cells in lymphoblastic leukemia have been widely published. Yet the cost and complexity of cell preparation protocols, as well neurotoxicity could hamper the universal application of CAR technology beyond specialized centers with well-trained cytotherapy staff. In comparison, bispecific antibodies could be more cost-effectively generated in pharmaceutical scales for conventional transport and distribution with less geographic restrictions. Despite these advantages, the future of bispecific antibodies for AML will depend highly on their clinical efficacy and toxicity as more patients are being treated with these modalities.
References
Dohner H, Weisdorf DJ, Bloomfield CD . Acute myeloid leukemia. N Engl J Med 2015; 373: 1136–1152.
Spiess C, Zhai QT, Carter PJ . Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67: 95–106.
Laszlo GS, Estey EH, Walter RB . The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev 2014; 28: 143–153.
Marquez C, Trigueros C, Franco JM, Ramiro AR, Carrasco YR, Lopez-Botet M et al. Identification of a common developmental pathway for thymic natural killer cells and dendritic cells. Blood 1998; 91: 2760–2771.
Hernandez-Caselles T, Martinez-Esparza M, Perez-Oliva AB, Quintanilla-Cecconi AM, Garcia-Alonso A, Alvarez-Lopez DMR et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. J Leuk Biol 2006; 79: 46–58.
Krupka C, Kufer P, Kischel R, Zugmaier G, Bogeholz J, Kohnke T et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2014; 123: 356–365.
Walter RB . Biting back: BiTE antibodies as a promising therapy for acute myeloid leukemia. Expert Rev Hematol 2014; 7: 317–319.
Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia 2013; 27: 1107–1115.
Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther 2014; 13: 1549–1557.
Laszlo GS, Gudgeon CJ, Harrington KH, Dell'Aringa J, Newhall KJ, Means GD et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014; 123: 554–561.
Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB . T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J 2015, 5.
Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia 2015; 30: 484–491.
Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia 2016; 30: 484–491.
Harrington KH, Gudgeon CJ, Laszlo GS, Newhall KJ, Sinclair AM, Frankel SR et al. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS One 2015; 10: e0135945.
Stamova S, Cartellieri M, Feldmann A, Arndt C, Koristka S, Bartsch H et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol Immunol 2011; 49: 474–482.
Stamova S, Cartellieri M, Feldmann A, Bippes CC, Bartsch H, Wehner R et al. Simultaneous engagement of the activatory receptors NKG2D and CD3 for retargeting of effector cells to CD33-positive malignant cells. Leukemia 2011; 25: 1053–1056.
Stamova S, Feldmann A, Cartellieri M, Arndt C, Koristka S, Apel F et al. Generation of single-chain bispecific green fluorescent protein fusion antibodies for imaging of antibody-induced T cell synapses. Anal Biochem 2012; 423: 261–268.
Arndt C, von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia 2013; 27: 964–967.
Arndt C, Feldmann A, von Bonin M, Cartellieri M, Ewen EM, Koristka S et al. Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: description of a novel modular targeting system. Leukemia 2014; 28: 59–69.
Handgretinger R, Zugmaier G, Henze G, Kreyenberg H, Lang P, von Stackelberg A . Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia 2011; 25: 181–184.
Aliperta R, Cartellieri M, Feldmann A, Arndt C, Koristka S, Michalk I et al. Bispecific antibody releasing-mesenchymal stromal cell machinery for retargeting T cells towards acute myeloid leukemia blasts. Blood Cancer J 2015; 5: e348.
Reusch U, Harrington K, Gudgeon C, Fucek I, Ellwanger K, Weichel M et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res 2016; 22: 5829–5838.
Silla LM, Chen J, Zhong RK, Whiteside TL, Ball ED . Potentiation of lysis of leukaemia cells by a bispecific antibody to CD33 and CD16 (Fc gamma RIII) expressed by human natural killer (NK) cells. Br J Haematol 1995; 89: 712–718.
Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J et al. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012; 4: 653–663.
Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res 2013; 19: 3844–3855.
Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33(+) targets. Blood 2014; 123: 3016–3026.
Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun 2015; 56: 66–80.
Vallera DA, Felices M, McElmurry RT, McCullar V, Zhou X, Schmohl J et al. IL-15 trispecific killer engagers (TriKEs) make natural killer cells specific to CD33+ targets while also inducing in vivo expansion, and enhanced function. Clin Cancer Res 2016; 22: 3440–3450.
Singer H, Kellner C, Lanig H, Aigner M, Stockmeyer B, Oduncu F et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J Immunother 2010; 33: 599–608.
Schubert I, Kellner C, Stein C, Kugler M, Schwenkert M, Saul D et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. mAbs 2011; 3: 21–30.
Kugler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol 2010; 150: 574–586.
Chen J, Zhou JH, Ball ED . Monocyte-mediated lysis of acute myeloid leukemia cells in the presence of the bispecific antibody 251 x 22 (anti-CD33 x anti-CD64). Clin Cancer Res 1995; 1: 1319–1325.
Balaian L, Ball ED . Direct effect of bispecific anti-CD33 × anti-CD64 antibody on proliferation and signaling in myeloid cells. Leuk Res 2001; 25: 1115–1125.
Balaian L, Ball ED . Inhibition of acute myeloid leukemia cell growth by mono-specific and bi-specific anti-CD33 × anti-CD64 antibodies. Leuk Res 2004; 28: 821–829.
Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y . CD123 and its potential clinical application in leukemias. Life Sci 2015; 122: 59–64.
Huang S, Chen Z, Yu JF, Young D, Bashey A, Ho AD et al. Correlation between IL-3 receptor expression and growth potential of human CD34(+) hematopoietic cells from different tissues. Stem Cells 1999; 17: 265–272.
Steelman LS, Algate PA, Blalock WL, Wang XY, Prevost KD, Hoyle PE et al. Oncogenic effects of overexpression of the interleukin-3 receptor on hematopoietic cells. Leukemia 1996; 10: 528–542.
Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000; 14: 1777–1784.
Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P et al. Elevated expression of IL-3 R alpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002; 100: 2980–2988.
Kuo SR, Wong L, Liu JS . Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng Des Sel. 2012; 25: 561–569.
Stein C, Kellner C, Kugler M, Reiff N, Mentz K, Schwenkert M et al. Novel conjugates of single-chain Fv antibody fragments specific for stem cell antigen CD123 mediate potent death of acute myeloid leukaemia cells. Br J Haematol 2010; 148: 879–889.
Chichili GR, Huang L, Li H, Burke S, He L, Tang Q et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci Transl Med 2015; 7: 289ra82.
Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016; 127: 122–131.
Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S et al. Presence of Wilms-Tumor gene (Wt1) transcripts and the Wt1 nuclear-protein in the majority of human acute leukemias. Leukemia 1995; 9: 1060–1067.
Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H et al. Wt1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute-leukemia. Blood 1994; 84: 3071–3079.
Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 2015; 33: 1079–1086.
Ataie N, Xiang J, Cheng N, Brea EJ, Lu W, Scheinberg DA et al. Structure of a TCR-mimic antibody with target predicts pharmacogenetics. J Mol Biol 2016; 428: 194–205.
Ashmun RA, Look AT . Metalloprotease activity of CD13/aminopeptidase N on the surface of human myeloid cells. Blood 1990; 75: 462–469.
Mina-Osorio P . The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med 2008; 14: 361–371.
Piedfer M, Dauzonne D, Tang RP, N'Guyen J, Billard C, Bauvois B . Aminopeptidase-N/CD13 is a potential proapoptotic target in human myeloid tumor cells. Faseb J 2011; 25: 2831–2842.
Kaneko T, Fusauchi Y, Kakui Y, Masuda M, Akahoshi M, Teramura M et al. A bispecific antibody enhances cytokine-induced killer-mediated cytolysis of autologous acute myeloid leukemia cells. Blood 1993; 81: 1333–1341.
Ball ED, Schwarz LM, Bloomfield CD . Expression of the CD15 antigen on normal and leukemic myeloid cells: effects of neuraminidase and variable detection with a panel of monoclonal antibodies. Mol Immunol 1991; 28: 951–958.
Schwarzinger I, Valent P, Koller U, Marosi C, Schneider B, Haas O et al. Prognostic-significance of surface marker expression on blasts of patients with de novo acute myeloblastic-leukemia. J Clin Oncol 1990; 8: 423–430.
Ball ED, Guyre PM, Mills L, Fisher J, Dinces NB, Fanger MW . Initial trial of bispecific antibody-mediated immunotherapy of CD15-bearing tumors: cytotoxicity of human tumor cells using a bispecific antibody comprised of anti-CD15 (MoAb PM81) and anti-CD64/Fc gamma RI (MoAb 32). J Hematother 1992; 1: 85–94.
Horie R, Watanabe T . CD30: expression and function in health and disease. Semin Immunol 1998; 10: 457–470.
Zheng W, Medeiros LJ, Hu Y, Powers L, Cortes JE, Ravandi-Kashani F et al. CD30 expression in high-risk acute myeloid leukemia and myelodysplastic syndromes. Clin Lymphoma Myeloma Leuk 2013; 13: 307–314.
Fathi AT, Preffer FI, Sadrzadeh H, Ballen KK, Amrein PC, Attar EC et al. CD30 expression in acute myeloid leukemia is associated with FLT3-internal tandem duplication mutation and leukocytosis. Leuk Lymphoma 2013; 54: 860–863.
Casasnovas RO, Mounier N, Brice P, Divine M, Morschhauser F, Gabarre J et al. Plasma cytokine and soluble receptor signature predicts outcome of patients with classical Hodgkin's lymphoma: a study from the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2007; 25: 1732–1740.
Purdue MP, Lan Q, Martinez-Maza O, Oken MM, Hocking W, Huang WY et al. A prospective study of serum soluble CD30 concentration and risk of non-Hodgkin lymphoma. Blood 2009; 114: 2730–2732.
Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. mAbs 2014; 6: 728–739.
Omary MB, Trowbridge IS, Battifora HA . Human homolog of murine T200 glycoprotein. J Exp Med 1980; 152: 842–852.
Kenoyer AL, Lin Y, Orozco JJ, Balkin E, Hamlin D, Hernandez AH et al. Pre-targeted radioimmunotherapy employing a recombinant bispecific antibody using a murine xenograft model of human leukemia. Blood 2014; 124: 3749.
Oldenborg PA . CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol 2013; 2013: 614619.
Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009; 138: 271–285.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009; 138: 286–299.
Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. mAbs 2015; 7: 946–956.
van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007; 110: 2659–2666.
Lu H, Zhou Q, Deshmukh V, Phull H, Ma J, Tardif V et al. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew Chem 2014; 53: 9841–9845.
Noordhuis P, Terwijn M, Rutten AP, Smit L, Ossenkoppele GJ, Schuurhuis GJ . Targeting of CLEC12A in acute myeloid leukemia by antibody-drug-conjugates and bispecific CLL-1 x CD3 BiTE antibody. Blood 2010; 116: 1191.
Hofmann M, Grosse-Hovest L, Nubling T, Pyz E, Bamberg ML, Aulwurm S et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012; 26: 1228–1237.
Durben M, Schmiedel D, Hofmann M, Vogt F, Nubling T, Pyz E et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol Therapy 2015; 23: 648–655.
Moreno I, Martin G, Bolufer P, Barragan E, Rueda E, Roman J et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica 2003; 88: 19–24.
Kuchenbauer F, Kern W, Schoch C, Kohlmann A, Hiddemann W, Haferlach T et al. Detailed analysis of FLT3 expression levels in acute myeloid leukemia. Haematologica 2005; 90: 1617–1625.
Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U et al. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol 2005; 23: 1109–1117.
Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, Keating M et al. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood 1999; 94: 3717–3721.
Schliemann C, Bieker R, Thoennissen N, Gerss J, Liersch R, Kessler T et al. Circulating angiopoietin-2 is a strong prognostic factor in acute myeloid leukemia. Leukemia 2007; 21: 1901–1906.
Aref S, El Menshawy N, Azmy E, El-Refaie M . Soluble angiopoietin-2/sTie2 receptor ratio is an independent prognostic marker in adult acute myeloid leukemia. Indian J Hematol Blood Transfus 2009; 25: 17–22.
Aref S, El Sherbiny M, Goda T, Fouda M, Al Askalany H, Abdalla D . Soluble VEGF/sFLt1 ratio is an independent predictor of AML patient out come. Hematology 2005; 10: 131–134.
Fiedler W, Graeven U, Ergun S, Verago S, Kilic N, Stockschlader M et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood 1997; 89: 1870–1875.
Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci USA 2011; 108: 11187–11192.
Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res 2013; 19: 6730–6740.
Doppalapudi VR, Huang J, Liu D, Jin P, Liu B, Li L et al. Chemical generation of bispecific antibodies. Proc Natl Acad Sci USA 2010; 107: 22611–22616.
Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol 2014; 192: 3805–3815.
Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med 2012; 367: 805–816.
Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M et al (eds).Construction and Characterization of Novel CD33/CD3 Tandem Diabodies (TandAbs®) for the Treatment of Acute Myeloid Leukemia (AML). ASCO Annual Meeting Proceedings, 2015.
Roddie C, Peggs KS . Donor lymphocyte infusion following allogeneic hematopoietic stem cell transplantation. Expert Opin Biol Therapy 2011; 11: 473–487.
Fry TJ, Mackall CL . Immune reconstitution following hematopoietic progenitor cell transplantation: challenges for the future. Bone Marrow Transplant 2005; 35: S53–S57.
Chaudhry MS, Velardi E, Dudakov JA, van den Brink MR . Thymus: the next (re)generation. Immunol Rev 2016; 271: 56–71.
Zakrzewski JL, Suh D, Markley JC, Smith OM, King C, Goldberg GL et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat Biotechnol 2008; 26: 453–461.
Petersen SL, Ryder LP, Bjork P, Madsen HO, Heilmann C, Jacobsen N et al. A comparison of T-, B- and NK-cell reconstitution following conventional or nonmyeloablative conditioning and transplantation with bone marrow or peripheral blood stem cells from human leucocyte antigen identical sibling donors. Bone Marrow Transplant 2003; 32: 65–72.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295: 2097–2100.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D . A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res 2013; 73: 1777–1786.
Kellner C, Gunther A, Humpe A, Repp R, Klausz K, Derer S et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. Oncoimmunology 2016; 5: e1058459.
Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001; 14: 123–133.
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285: 727–729.
Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113: 3716–3725.
Turini M, Chames P, Bruhns P, Baty D, Kerfelec B . A FcgammaRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget 2014; 5: 5304–5319.
Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM et al. cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol 2008; 180: 6392–6401.
Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014; 123: 678–686.
Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One 2015; 10: e0134715.
Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci USA 2004; 101: 12640–12645.
Jin LQ, Hope KJ, Zhai QL, Smadja-Joffe F, Dick JE . Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006; 12: 1167–1174.
Blatt K, Herrmann H, Hoermann G, Willmann M, Cerny-Reiterer S, Sadovnik I et al. Identification of Campath-1 (CD52) as Novel Drug Target in Neoplastic Stem Cells in 5q-Patients with MDS and AML. Clin Cancer Res 2014; 20: 3589–3602.
Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA 2007; 104: 11008–11013.
Korver W, Zhao X, Singh S, Pardoux C, Zhao J, Guzman ML et al. Monoclonal antibodies against IREM-1: potential for targeted therapy of AML. Leukemia 2009; 23: 1587–1597.
Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010; 7: 708–717.
Dorantes-Acosta E, Pelayo R . Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res 2012; 2012: 406796.
Menendez J, Jin L, Poeppl A, Sayegh D, O'Reilly K, Ceric N et al. Anti-CD9 antibody, AR40A746.2.3, inhibits tumor growth in pancreatic and breast cancer models and recognizes CD9 on CD34+CD38- leukemic cancer stem cells. Cancer Res 2008; 68: 3993.
Yamazaki H, Xu CW, Naito M, Nishida H, Okamoto T, Ghani FI et al. Regulation of cancer stem cell properties by CD9 in human B-acute lymphoblastic leukemia. Biochem Biophys Res Commun 2011; 409: 14–21.
Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 2013; 21: 2122–2129.
Zhang H, Li ZL, Ye SB, Ouyang LY, Chen YS, He J et al. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother 2015; 64: 1587–1599.
Velders MP, van Rhijn CM, Oskam E, Fleuren GJ, Warnaar SO, Litvinov SV . The impact of antigen density and antibody affinity on antibody-dependent cellular cytotoxicity: relevance for immunotherapy of carcinomas. Br J Cancer 1998; 78: 478–483.
Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res 2001; 61: 4750–4755.
Weinstein JN, van Osdol W . Early intervention in cancer using monoclonal antibodies and other biological ligands: micropharmacology and the "binding site barrier". Cancer Res 1992; 52: 2747s–2751s.
Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med 2014; 6: 261ra154.
Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013; 121: 5154–5157.
Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G et al. Expression of the target receptor CD33 in CD34(+)/CD38(−)/CD123(+) AML stem cells. Eur J Clin Invest 2007; 37: 73–82.
Laszlo GS, Estey EH, Walter RB . The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev 2014; 28: 143–153.
Pollard JA, Alonzo TA, Loken M, Gerbing RB, Ho PA, Bernstein ID et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012; 119: 3705–3711.
Sun H, Li Y, Zhang ZF, Ju Y, Li L, Zhang BC et al. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int J Hematol 2015; 102: 579–586.
Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005; 106: 4086–4092.
van der Velden VHJ, te Mervelde JG, Hoogeveen PG, Bernstein ID, Houtsmuller AB, Berger MS et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001; 97: 3197–3204.
Abdool A, Yeh CH, Kantarjian H, O'Brien S, Bruey J, Giles F et al. Circulating CD33 and its clinical value in acute leukemia. Exp Hematol 2010; 38: 462–471.
Larson RA, Sievers EL, Stadtmauer EA, Lowenberg B, Estey EH, Dombret H et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005; 104: 1442–1452.
Walter RB, Appelbaum FR, Estey EH, Bernstein ID . Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012; 119: 6198–6208.
Munoz L, Nomdedeu JF, Lopez O, Carnicer MJ, Bellido M, Aventin A et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001; 86: 1261–1269.
Sato N, Caux C, Kitamura T, Watanabe Y, Arai K, Banchereau J et al. Expression and factor-dependent modulation of the interleukin-3 receptor subunits on human hematopoietic-cells. Blood 1993; 82: 752–761.
Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014; 123: 2343–2354.
Hosen N, Shirakata T, Nishida S, Yanagihara M, Tsuboi A, Kawakami M et al. The Wilms' tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia 2007; 21: 1783–1791.
Bergmann L, Miething C, Maurer U, Brieger J, Karakas T, Weidmann E et al. High levels of Wilms' tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 1997; 90: 1217–1225.
Ramani P, Cowell JK . The expression pattern of Wilms' tumour gene (WT1) product in normal tissues and paediatric renal tumours. J Pathol 1996; 179: 162–168.
Thalhammer-Scherrer R, Mitterbauer G, Simonitsch I, Jaeger U, Lechner K, Schneider B et al. The immunophenotype of 325 adult acute leukemias: relationship to morphologic and molecular classification and proposal for a minimal screening program highly predictive for lineage discrimination. Am J Clin Pathol 2002; 117: 380–389.
Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL et al. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol 1994; 47: 43–47.
Griffin JD, Davis R, Nelson DA, Davey FR, Mayer RJ, Schiffer C et al. Use of surface marker analysis to predict outcome of adult acute myeloblastic-leukemia. Blood 1986; 68: 1232–1241.
Buttler D, Mai JK, Ashwell KW, Andressen C . Clonogenic CD15 immunoreactive radial glial cells from the developing human lateral ganglionic eminence. Curr Pharm Biotechnol 2013; 14: 29–35.
Chadburn A, Inghirami G, Knowles DM . The kinetics and temporal expression of T-cell activation-associated antigens CD15 (LeuM1), CD30 (Ki-1), EMA, and CD11c (LeuM5) by benign activated T cells. Hematol Pathol 1992; 6: 193–202.
Deutsch E, Cheung NKV . Non-coordinating buffers. I. Synthesis and characterization of water soluble derivatives of 2,6-di-tert-butyl-pyridine. J Org Chem 1973; 38: 1123.
The Human Protein Atlashttp://www.proteinatlas.org/ENSG00000120949-TNFRSF8/tissue. 2016. Available from: http://www.proteinatlas.org/ENSG00000120949-TNFRSF8/tissue.
Vanderjagt RHC, Badger CC, Appelbaum FR, Press OW, Matthews DC, Eary JF et al. Localization of radiolabeled antimyeloid antibodies in a human acute-leukemia xenograft tumor-model. Cancer Res 1992; 52: 89–94.
Caldwell CW, Patterson WP, Toalson BD, Yesus YW . Surface and cytoplasmic expression of Cd45 antigen isoforms in normal and malignant myeloid cell-differentiation. Am J Clin Pathol 1991; 95: 180–187.
Witte KE, Ahlers J, Schafer I, Andre M, Kerst G, Scheel-Walter HG et al. High proportion of leukemic stem cells at diagnosis is correlated with unfavorable prognosis in childhood acute myeloid leukemia. Pediatr Hemat Oncol 2011; 28: 91–99.
Bakker ABH, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res 2004; 64: 8443–8450.
Levis M, Small D . FLT3: ITDoes matter in leukemia. Leukemia 2003; 17: 1738–1752.
Trujillo A, McGee C, Cogle CR . Angiogenesis in acute myeloid leukemia and opportunities for novel therapies. Journal of oncology. 2012; 2012: 128608.
Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci USA 2013; 110: 5145–5150.
Bachmann M, Stamova S . Anti-CD33 antibodies and use thereof for immunotargeting in treating CD33-associated illnesses. Google Patents 2014.
Stamova S, Cartellieri M, Feldmann A, Arndt C, Koristka S, Bartsch H et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol Immunol 2011; 49: 474–482.
Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 × 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res 2013; 19: 3844–3855.
McCall AM, Adams GP, Amoroso AR, Nielsen UB, Zhang L, Horak E et al. Isolation and characterization of an anti-CD16 single-chain Fv fragment and construction of an anti-HER2/neu/anti-CD16 bispecific scFv that triggers CD16-dependent tumor cytolysis. Mol Immunol 1999; 36: 433–445.
Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res 2016; 22: 3440–3450.
Kuo SR, Wong L, Liu JS . Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng Design Select 2012; 25: 561–569.
Kuo SR, Alfano RW, Frankel AE, Liu JS . Antibody internalization after cell surface antigen binding is critical for immunotoxin development. Bioconjug Chem 2009; 20: 1975–1982.
Acknowledgements
This work was supported in part by Funds from Enid A. Haupt Endowed Chair, Kids Walk for Kids with Cancer NYC, Katie Find a Cure Foundation and the Robert Steel Foundation. We thank Dr Irene Cheung, Dr. Mahiuddin Ahmed and Brian Santich for reviewing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MSKCC and NK Cheung have financial interest in Y-mabs Therapeutics Inc.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hoseini, S., Cheung, N. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer Journal 7, e522 (2017). https://doi.org/10.1038/bcj.2017.2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2017.2
This article is cited by
-
Update on Small Molecule Targeted Therapies for Acute Myeloid Leukemia
Current Treatment Options in Oncology (2023)
-
Treatment outcomes of pediatric acute myeloid leukemia: a retrospective analysis from 1996 to 2019 in Taiwan
Scientific Reports (2021)
-
Graphene Quantum Dots Decorated Gold-Polyaniline Nanowire for Impedimetric Detection of Carcinoembryonic Antigen
Scientific Reports (2019)
-
Self-nanoemulsifying system improves oral absorption and enhances anti-acute myeloid leukemia activity of berberine
Journal of Nanobiotechnology (2018)
-
Recent developments in immunotherapy of acute myeloid leukemia
Journal of Hematology & Oncology (2017)
