
Abstract
Hairy cell leukemia (HCL) is part of the low-grade non-Hodgkin lymphoma family and represents approximately 2% of all leukemias. Treatment with splenectomy and interferon-α historically belonged to the first steps of therapeutic options, achieving partial responses/remissions (PR) in most cases with a median survival between 4 and 6 years in the 1980s. The introduction of the purine analogs (PA) pentostatin and cladribine made HCL a well-treatable disease: overall complete response rates (CRR) range from 76 to 98%, with a median disease-free survival (DFS) of 16 years a normal lifespan can be reached and HCL-related deaths are rare. However, insufficient response to PA with poorer prognosis and relapse rates of 30–40% after 5–10 years of follow-up may require alternative strategies. Minimal residual disease can be detected by additional examinations of bone marrow specimens after treatment with PA. The use of immunotherapeutic monoclonal antibodies (mAB) like rituximab as a single agent or in combination with a PA or more recently clinical trials with recombinant immunotoxins (RIT) show promising results to restrict these problems. Recently, the identification of the possible disease-defining BRAF V600E mutation may allow the development of new therapeutic targets.
Similar content being viewed by others


Search strategy and selection criteria
Data for this review were identified by searches of MEDLINE, PubMed and references from relevant articles using the search terms ‘hairy cell leukemia’ (HCL). Only papers in English and German between 1970 and 2013 were included.
Introduction
Representing approximately 2% of all leukemias, HCL is a chronic B-cell lymphoproliferative disorder.1 In the following, historical and recent treatment options, their results as well as new insights in the pathophysiology and treatment recommendations are reviewed.
Treatment options, results and new insights
Splenic irradiation and splenectomy
Splenomegaly is one of the most common clinical manifestations of HCL (up to 96% of the patients),2 often getting symptomatic in the form of epigastric discomfort or pain.3 Splenectomy was developed as the first effective therapeutic approach in the history of the treatment of HCL and was considered as treatment of choice until 1984,4, 5 resulting in overall response rates (ORR) of 60–100% as documented in eight major reports.5 Achieving a median response duration of 5–20 months1, 6 with an overall survival (OS) rate of 55–70% after 5 years,1, 7, 8 patients with HCL benefit from splenectomy compared with non-splenectomized patients5 and respond better and faster to the following therapy.9 It is recommended to wait at least 6 months after splenectomy before initiating further treatment approaches to reach full benefits of splenectomy.10 Nevertheless, splenectomy does not improve bone marrow infiltration and fibrosis,5 and positive influences of splenectomy with regard to long-term results are reported to be uncertain.1, 10
Despite the fact that more effective chemotherapeutics are available in the treatment of HCL today, splenectomy still remains a valid treatment option in certain cases. HCL with a huge splenomegaly (e.g. >10 cm below costal margin) but low bone marrow involvement can be an indication for splenectomy as first-line treatment.10 In cases of unknown splenomegaly, splenectomy is not only a therapeutic but also a helpful diagnostic instrument to prove the typical transformation of spleen histology.10, 11 Splenectomy can also be an alternative intervention for patients with relapsed or refractory disease.5 Furthermore, HCL can be the reason for rare non-traumatic splenic ruptures.2 In these cases, splenectomy is a necessary treatment method.2 Owing to the fact that HCL is less common in women at childbearing age and thus rare in pregnancy, there are no standardized therapy regimens in this configuration. Then, laparoscopic splenectomy should be considered as a therapeutic option to protect the fetus from toxic effects of chemotherapy, although there are only few data available with regard to the use of chemotherapy during pregnancy.8, 12, 13 In these cases a therapy with cladribine can be initiated after delivery.8
Although splenic irradiation is enumerated together with androgens, lithium, corticosteroids, chlorambucil or cyclophosphamide as unsatisfying treatment approaches before the introduction of interferon-α (IFN-α),14 Lavrenkov et al.3 reported the successful use of palliative splenic irradiation to reduce splenic size and to improve anemia and thrombocytopenia in two patients with symptomatic splenomegaly in HCL.
In conclusion, splenectomy has clearly lost its therapeutic importance after the introduction of IFN-α and later on of purine analogs (PA). Nevertheless, it should still be remembered as an alternative or additional option, especially in symptomatic or refractory/relapsed disease.5
IFN-α
Initially described as an anti-viral agent in 1957, it took almost 30 years until the approval of IFN-α as the first recombinant cytokine for the treatment of malignancies.15 Quesada et al.16 showed that IFN-α can improve and is even able to normalize peripheral blood counts in patients with HCL.5 The main therapeutical effects on patients with HCL are not only the reduction of cytopenia and the elimination of HC from the blood but also the reduction of bone marrow fibrosis.15, 17
Purine analogs
Pentostatin (2′-deoxycoformycin, dcF)
Although early approaches were carried out in refractory CLL,18 pentostatin, a product of Streptomyces antibioticus and a pharmacologic inhibitor of adenosine deaminase (ADA),1, 9 was introduced as the first PA in the therapy of HCL in the 1980s.1, 18, 19 It is not only well tolerated but also leads to durable complete remission rates of >75%.18 With the establishment of PA in the therapy of HCL, prognosis was improved significantly.14, 20
The irreversible inhibition of ADA9 causes an intracellular accumulation of cytotoxic deoxyadenosine triphosphate metabolites inter alia in HC,21 leading to the inhibition of the repair system of spontaneously formed DNA breaks.22 Consequences are the activation of p53 and the release of cytochrome c from mitochondria, initiating apoptosis.22
Efficacy. Table 1 summarizes selected publications of treatment with pentostatin in HCL.23, 24, 25, 26 The main retrospective analysis of several hundred patients resulted in ORR >90% with complete response rates up to 82%.23, 24, 25 The 5- and 10-year OS rates were estimated at 90%23 and 68–81%.18, 23 The long-term follow-up up to 16 years confirmed that pentostatin may not only induce high response rates but also long-lasting responses even in cases of retreatment with pentostatin after relapse.25 Nevertheless, relapse occurs in up to 44% of the patients treated with pentostatin.26
Toxicity. In general, treatment with PA like pentostatin is well tolerated,1, 18, 21 but causes prolonged myelosuppression inducing immunosuppression21, 27 that seems to be less severe, especially at the beginning of the therapy after administration of pentostatin compared with cladribine.18 Pentostatin causes a decrease of T cells9 and B cells.1 While a case of hypereosinophilia with eosinophilic gastroenteritis has been reported,21 more common side effects of pentostatin are neutropenic febrile episodes, skin rash, pruritus, photosensitivity, nausea and vomiting and, in particular, cardiotoxicity causing atrioventricular block, angina pectoris, arrhythmias, extrasystoles or cardiac arrest.1, 9, 21
Cladribine (2-chlorodeoxyadenosine)
Introduced as the second effective PA after pentostatin, cladribine has emerged as the preferred first-line chemotherapeutic agent in HCL.14, 28, 29
Cladribine is resistant against degradation by ADA.30 It accumulates intracellularly and gets polyphosphorylated to its active metabolite cladribine-triphosphate by deoxycytidine kinase.20 Thereby, 2-chlorodeoxyadenosine develops its effectiveness, especially in deoxycytidine kinase-rich cells like lymphocytes or monocytes.20, 30 Recent results suggest that cladribine causes cell type-specific effects.31 Different cytotoxic effects of cladribine-triphosphate are known: direct DNA effects are caused by incorporation of cladribine-triphosphate as a fraudulent base into DNA, leading to a termination of DNA chain elongation and to DNA breakages that cannot be repaired because of the interference of cladribine and DNA repair mechanisms.9, 20, 22 Other effects are the inhibition of DNA-polymerase-β determining DNA synthesis,22 or the inhibition of S-adenylhomocysteine hydrolase, leading to a decrease of DNA methylation.20 Apoptosis gets initiated through caspase system, with cytochrome c and apoptotic protease-activating factor.20 In dendritic cells, apoptosis seems to be induced independent of caspase.31
Efficacy. Cladribine has a proven high effectiveness in HCL. Selected publications are summarized in Table 2.25, 26, 32, 33, 34, 35, 36, 37, 38 A single course of therapy corresponding to a duration of 5–7 days is already able to achieve complete remissions with durable responses: ORR of 87–100%14, 39, 40 and CRR of 85-91%39, 40 have been reported. The 5-year progression-free survival (PFS) rates vary between 72–84% and the OS at 12 years vary between 75–87%.20 The risk of relapse at 4 years amounts to approximately 30%.39
Although the long-term follow-up shows long-lasting remissions, within longer follow-up the number of relapses increases. Then, retreatment with cladribine is a possible approach.30 Admittedly, response rates and duration of response after retreatment with a PA are lower and shorter, respectively.41 Goodman et al.34 reported on 207 assessable patients with a follow-up of at least 7 years. There was an ORR of 100% including 95% CR after only a single cycle of cladribine with a relapse rate of 37% after a median time of 42 months.34 A second cycle of therapy resulted in a response rate of 92% and a relapse rate of 33% during further follow-up.14 Even after a third or fourth treatment cycle patients responded to cladribine despite previous relapses after cladribine.14 The OS at 108 months was 97%, while 3% of the patients had died.14 As the results of Scripps Clinic demonstrate, where after a median time of 29 months 26% relapses were documented, relapses in patients with partial response (PR) are with 64% vs 24% higher than in those who achieved CR.1
Despite these results, ∼10% of the patients are poor responders and ∼5% non-responders to PA.42 In respect to the results of an Italian multicenter clinical trial,43 it seems that the patients who are resistant to cladribine belong to a minor subset of classical HCL characterized by unmutated immunoglobulin heavy-chain variable (IGHV) in immunoglobulin gene analysis and affiliation of the immunoglobulin genes to the VH 4-34 family with higher tumor burden and poorer prognosis, presumably similar to unmutated subsets in other lymphoproliferative disorders like CLL.44, 45 Considering these aspects, IG gene analysis may have an important role in diagnosis and prognosis of HCL.45, 46, 47
Toxicity. Cladribine is generally a well tolerated and highly effective drug.1, 14, 20, 30, 39 Nevertheless, treatment with cladribine often leads to myelosuppression and cytopenias in all three cell lines, potentially arousing severe infections that are in addition to second malignancies common causes of death.20, 29, 30, 48, 49 The severity of side effects depends on duration and extent of disease and therapy.29 There may be a relationship between cladribine treatment and an increased incidence of second malignancies.1 In general, the risk of developing a second malignant disease appears to be higher in patients with HCL with an incidence of 31.9% compared to expected 23.1% in the general population.50, 51
Although fever is documented in 40–69% of the patients, suggesting that a main part of febrile episodes are caused by cytokine release in HC,1 infections occur especially at the beginning of the treatment with high incidences of 30–50%.49 Here, the absolute lymphocyte count at diagnosis can be regarded as a risk factor for the occurrence of severe infections.52 Relatively high rates of febrile and infectious complications are caused by the continuous infusion schedules: grade 3–4 neutropenias were documented in 87% of the 349 patients treated at Scripps Clinic and febrile episodes in 42%.14 The adjunctive application of granulocyte-colony-stimulating factor does not seem to have positive influence on the occurrence of febrile episodes.30 There is a rare risk for opportunistic infections owing to a considerable suppression of CD4- and CD8-positive T lymphocytes by cladribine.30 Occasional cases of various mycobacterial infections, recently tuberculosis meningoencephalitis, have been reported.53 Nephro- and dose-dependent neurotoxicity rather occur infrequently at recommended therapeutical dosage.20 More possible adverse effects are skin rash54 and mild-to-severe gastrointestinal side effects such as nausea, vomiting or diarrhea.1
Fludarabine
Fludarabine is like 2-chlorodeoxyadenosine, a halogenated deoxyadenosine derivative that is primarily a therapeutic option in CLL. Several case reports have been published describing activity of fludarabine in HCL, too.22 Recently, Gerrie et al.41 published a retrospective analysis of 15 patients with relapsed or refractory HCL after different previous treatment attempts including at least one other PA, who were treated with 40 mg/m2 per day fludarabine per os on five consecutive days in combination with the intravenous injection of 375 mg/m2 rituximab, a monoclonal antibody (mAB) that will be discussed below in detail, on day 1 administered every 28 days for four cycles between 2004 and 2010.41 Without assessing the depth of response in most of the cases, the authors report well-tolerated, safe and effective results after a median follow-up of 35 months with 14 progression-free patients, a 5-year PFS of 89%, an OS of 83% and a recurrence rate of 7%.41 These results may offer the possibility of fludarabine as a therapeutic option in relapsed or refractory patients with HCL.
Bendamustine
Bendamustine, actually approved for CLL, has properties of a PA similar to cladribine combined with alkylator activity.55 Multiple mechanisms of action lead to a non-cross-resistance of bendamustine with other cytotoxic agents like PA.55, 56
Kreitman et al.56 reported a case of multiple resistant and refractory classic HCL: a 47-year-old patient did neither respond to cladribine nor splenectomy followed by rituximab and became refractory to pentostatin after repeated cycles over several years so that he was treated with eight cycles of bendamustine 90 mg/m2 on 2 days, every 4–7 weeks, achieving a PR for 6 months. Randomized trials comparing combination therapies of bendamustine and rituximab and pentostatin and rituximab are ongoing.55
Treatment of choice: cladribine or pentostatin?
A superiority of either PA could not be proved yet, thus pentostatin and cladribine, inducing CRR of 76-98%,7 can be still regarded as equally effective.18, 19, 25, 26, 39 Long-term results of the Royal Marsden Hospital demonstrate a median disease-free survival (DFS) of 16 years for both agents, emphasizing that the DFS of complete responders is as five times as long as for partial responders (DFS 20+ years for CR, 4 years for PR, P<0.0001).25 Although there are no satisfactory studies directly comparing the two agents,39 cross-study analyses resulted in 98% ORR with 91% CRR for cladribine and 96% ORR with 81% CRR for pentostatin.30 Moreover, the long-term adverse effects of both agents seem to be comparable.19 The interrupted schedule of pentostatin may lead to less myelosuppression and febrile neutropenia, especially at the beginning of the therapy. A titration of dose and schedule for pentostatin may also allow the treatment of patients suffering from an infection, but what may compensate the disadvantages of months of outpatient therapy with pentostatin.18, 19, 27 On the other hand, the subcutaneous administration of cladribine has a short duration30 and is very convenient for patients and clinicians. Regarding outcome, safety profile and administration regimen, the use of cladribine seems to be more advantageous.29 Else et al.26 showed that all of their patients treated with cladribine are still alive after 10 years compared with 96% of the pentostatin group. Overall, cladribine is used more commonly for initial therapy than pentostatin.27, 29, 39 The important questions about an increased risk of second malignancies and a probably higher risk for late infections due to a treatment with cladribine have to undergo further investigations.19
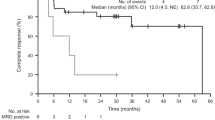
In conclusion, both agents seem to be interchangeable according to the current state of knowledge so that the choice of first-line therapy remains to the therapist, his experiences and the individual settings of the patient, always aiming to achieve CR, as this represents the most significant predictor of prolonged DFS.19, 25 Patients who achieved and kept CR for at least 5 years show a reduced risk of relapse of 25% by 15 years.25 Nevertheless, relapses occur in 30-40% of the cases.18, 41 There is a median relapse-free survival (RFS) <7 years44 and the PFS and DFS curves do not reach a plateau, suggesting that curing HCL with PA is not possible.41, 44 A persistence of HCL after achieving CR with first-line treatment defined as minimal residual disease (MRD) can be detected by different methods like immunophenotypic analysis, immunohistochemical staining or DNA polymerase chain reaction19, 27 varying between values from <50%18, 40 to nearly all of the patients after one cycle of cladribine,14 depending on time and method of bone marrow examination.18
Immunotherapeutic approaches
Clinical trials with immunotherapeutic agents show promising results, especially in the treatment of patients resistant to, or relapsed after, treatment with PA and in the eradication of MRD.1 Owing to the fact that residual HC are strongly positive for CD20, CD22 and CD25, these surface antigens can be used as targets either for unconjugated mAB or for recombinant immunotoxins (RIT), a composite drug combining the binding specificity of the variable fragment of an mAB and the cytotoxic effects of a bacterial toxin.55, 57, 58, 59 Although RIT are more cytotoxic than mAB alone, even to cells with a rare expression of the target sites,60 a problem related to the therapy with RIT is the development of antidrug antibodies, which neutralize drug activity, shorten half-life and may cause life-threatening immune responses in rare cases for which reason therapy has to be terminated in cases of antibody formation.61 Table 3 (refs 62,63,64,65,66,67,68,69,70) and Table 4 (refs 58,71,72,73) summarize selected studies of immunotherapeutic approaches.
Anti-CD20 (rituximab)
CD20 is a non-glycosylated transmembrane protein with calcium channel function, which is probably involved in the regulation and proliferation of normal B cells and highly expressed on a great number of B-lymphoid tumor cells.1, 74, 75 Patients who are assessed positive for MRD after treatment with cladribine show a high expression of CD20 with more than 100 000 sites per cell,55 representing a target for the humanized anti-CD20 mAB rituximab.
A dosage of 375 mg/m2 of rituximab applicated as weekly intravenous infusions is common.10, 76 However, the optimal dosing schedule has not been established yet. There may be six to eight infusions necessary to reach a CR and MRD eradication.70
It should be mentioned that most of the studies reporting on the efficacy of rituximab are based on small study groups, often consisting of not uniformly pretreated patients.19 Although the single use of rituximab in the treatment of HCL achieves CRR of 10–60%,55 the combination of rituximab with a PA, showing synergistic effects, results in higher CRR up to 100%76 with a high rate of MRD eradication up to 92%.42 Kreitman4 suggests that the reduced efficacy of the single use of rituximab compared with PA is based on cellular resistance developments involving the apoptotic pathway, immune mediators and/or limited tumor penetration.4 However, the combination of rituximab and PA is not only effective in newly diagnosed and relapsed cases but also in patients resistant to single use of cladribine and in MRD eradication independently of IGH mutational status.44, 76 An overview of selected trials with rituximab is shown in Table 3.62, 63, 64, 65, 66, 67, 68, 69, 70
Else et al.70 reported in a retrospective review of 18 patients who could be successfully treated with four to eight weekly intravenous infusions of 375 mg/m2 rituximab after one to six courses of either pentostatin (n=12) or cladribine (n=6), achieving an ORR of 100% with 89% durable CR and significant longer DFS after combination therapy compared with first-line treatment with PA.70 Regarding these results, combination therapy with rituximab seems not only to be safe and effective but also beneficial in comparison to single use of a PA.70 A superiority of either PA has not been proved yet, thus the combination drug should be chosen with regard to previous treatment.70
The question whether rituximab should be applicated simultaneously or sequentially to the PA has not been answered yet.69 Although the concurrent therapy of rituximab and a PA in comparison with the sequential administration in patients with CLL seems to result in higher response rates (90% vs 77%), and also in higher rates of toxic events (grade 3–4 neutropenia 74% vs 41%, infusion-related toxicity 20% vs 0%),41, 77, 78 the long-term results for both treatment regimens tend to have a similar outcome.70, 79 With respect to the retrospective review of eight patients by Else et al.,80 a concurrent schedule of rituximab together with a PA in recurrent/refractory HCL may achieve higher and more durable CR but probably with a higher risk of infectious complications.78 Combining a 5-day schedule of 5.6 mg/m2 2-h infusions of cladribine with a sequential course of rituximab of eight weekly infusions of 375 mg/m2 started 1 month later, Ravandi et al.69 achieved a CRR of 100% after completion of the therapy in their phase II study including 36 patients either with HCL or the variant form (n=5), suggesting that this schedule may increase CRR. Kreitman et al.55 prefer the sequential administration of rituximab 6 months after a 5- to 7-day course of cladribine because earlier application may reduce efficacy and increase toxicity with respect to the myelosuppressive effects of cladribine. Moreover, infusional reactions requiring steroids, which may block rituximab, were not observed in the recommended schedule.55
Known side effects of rituximab are reversible grade 3–4 infections including neutropenic fever, cellulitis, herpes zoster dermatitis, grade 1–2 nausea, rash, fatigue, weight loss69 or gastrointestinal bleeding caused by thrombocytopenia.1 Despite significantly low levels of CD4-positive lymphocytes and immunoglobulin G, opportunistic infections are rarely observed in most clinical studies.78 Because of the lower toxicity profile, rituximab may be an alternative option in relapsed patients to avoid the cumulative toxicity of repeated courses of PA.76
Owing to the fact that there is no evident benefit for the eradication of MRD and owing to the high costs for rituximab, the question whether all patients with newly diagnosed HCL should receive a treatment with a combination therapy including rituximab as first-line therapy cannot be answered yet.42, 69, 78 Ravandi78 regards rituximab as an ideal agent for the eradication of MRD with the potential in extending RFS and recommends a combination therapy for patients with a high risk of relapse or refractory disease, such as patients with unmutated IGHV and VH 4-34 gene rearrangement.78 However, standardized indications for a first-line use of rituximab have to be established.42
Anti-CD25
LMB-2 was the first RIT with reported efficacy in HCL.4, 55 It is composed of the variable regions of the anti-CD25 mAB anti-Tac and the truncated form of pseudomonas exotoxin PE38.1, 4, 55 In a phase I trial, four patients with HCL achieved one CR and three PR4, 55 after a total dose of >60 μg/kg per cycle.81 Owing to the fact that RIT may be preferable treatment options for multiple relapsed or refractory patients, including patients with the CD25-negative HCL-V, research is focused on RIT targeting CD22.1, 4, 19, 55
Anti-CD22
CD22 is a B-cell-specific surface antigen of mature B lymphocytes that is neither on normal tissues nor on early progenitor B cells, nor on hematopoietic stem cells,59 but with a median of 40 000 sites per cell strongly expressed on probably all HC.55 Toxic effects of anti-CD22 therapeutics are thus directed substantially against the mature B-cell pool, reducing myelotoxicity.59 Hence, especially neutropenic, immunocompromised or old patients may benefit from therapy with anti-CD22 immunotherapeutics.1 Although initial investigations with unconjugated anti-CD22 mAB did not convince,59 the first developed RIT targeting CD22, BL-22 (CAT-3888), consisting of a single-chain variable fragment of the anti-CD22 RFB4 antibody fused to a truncated form of pseudomonas exotoxin called PE38, showed more successful results60 and its application in HCL developed to the most successful use of RIT.82 The mechanism of action is simplified based on the binding of the variable fragment of the RIT to its target on the cell surface and the initiation of cell death via inhibition of protein synthesis by ADP ribosylation of elongation factor 2 and via activation of caspase system through the bacterial toxin in the cytosol after internalization of the RIT–CD (cluster of determination) complex via the endocytic compartment and the endoplasmic reticulum.59, 60
Using intravenous application schedules with drug administration of 3-40 μg/kg as the maximum-tolerated dose every other day (QOD) for three doses repeating cycle every 21–28 days, Kreitman et al.60 reached CRR of 47–61% in phase I and II trials with BL-22.60 Toxicity profile included the occurrence of a reversible hemolytic uremic syndrome in 13% of the patients in the phase I trial and in 6% of the phase II trial.19 As a median RFS of approximately 7 years of follow-up was documented in these trials, a significant efficacy of BL-22 in relapsed/refractory HCL could be determined.60
To expand the field for the application of anti-CD22 immunotoxins to more common lymphoproliferative disorders with fewer expression of CD22 on cell surface, an RIT with up to a 50-fold higher cytotoxicity owing to a higher affinity to CD22 was developed out of BL-22 by mutagenesis studies, called Moxetumomab pasudotox (HA-22, CAT-8015).60 Kreitman et al.58 pursued a phase I trial with HA-22 including 28 patients with relapsed/refractory HCL who were treated with 5-50 μg/kg every other day for three doses and median four cycles (range 1–16) resulting in an ORR of 86% including 46% durable CR without MRD.58 CR could not be achieved in prior splenectomized patients, suggesting that splenectomy should be avoided in cases where HA-22 is a treatment option.58 Neutralizing antibodies were detected in 5% after one cycle and totally in 38%, indicating that retreatment of most of the patients until maximum response may be feasible without monitoring neutralizing antibodies.58 Although no dose-limiting toxicity occurred, drug-related toxicity was documented in 25–64% including grade 1–2 hypoalbuminemia, aminotransferase elevations, edema, headache, hypotension, nausea and fatigue.58 Considering efficacy and safety of Moxetumomab pasudotox, Kreitman et al.58, 60 and Kreitman and Pastan58, 60 aim further trials to approve the drug for treatment in HCL.58, 60
Anti CD-52 (alemtuzumab)
Alemtuzumab is theoretically useful in HCL because of the common expression of CD52 on HC, but causes prolonged immunosuppression.27
BRAF V600E mutation
Searching for HCL-associated mutations, Tiacci et al.83 discovered the BRAF V600E mutation in HC.83 Therefore, leukemic and normal cells of peripheral blood of an index patient with HCL were analyzed and validated by Sanger sequencing in 47 additional assays of patients with HCL. Although the mutation in BRAF was found in the entire tumor cells of all of the 47 patients with HCL, it did not appear in one of the 195 additional examined samples of patients with other peripheral B-cell lymphomas or leukemias like SMZL.83 In conclusion, BRAF V600E mutation seems to have an important role in the pathogenesis of HCL if it is not even the disease-defining genetic event.83, 84, 85, 86, 87, 88, 89 Although the BRAF V600E mutation is already described with lower frequency in other human cancers, such as melanomas (50%),83, 88 papillary thyroid cancers (40–60%),83, 88 colorectal cancers (8–10%),88 Langerhans cell histiocytosis (57%)83 and more rarely in non-small-cell carcinoma of the lung and ovarian cancers,88 this variant of BRAF mutations is not known in other B-cell disorders.83, 84 It is a missense mutation in the proto-oncogene BRAF, replacing valine to glutamic acid at position 600 of the protein90 and leading to an enhanced constitutive activation of the BRAF kinase, a serine–threonine protein kinase87, 88, 90 that is involved in the RAS-RAF-MEK-ERK and mitogen-activated protein kinase signaling pathways, which regulate cell survival, proliferation and differentiation.83, 91 Tiacci et al.83, 91 could also demonstrate that downstream targets of BRAF kinase like phosphorylated ERK are ubiquitous expressed in HC. Thereby, this mutation can be regarded as the key trigger of MEK-ERK pathway activation in HCL, representing a critical event in the pathogenesis of HCL.84, 88, 91
Although Tiacci’s findings of the presence of the BRAF V600E mutation in HCL and the absence in other B-cell lymphoproliferative disorders were confirmed in further testings,90, 92, 93, 94, 95, 96, 97, 98 Xi et al.99 found wild-type BRAF not only in all his patients with HCL-V or expressing IGHV 4-34 but also in 11 of 53 patients with classic HCL, suggesting that HCL-V, HCL expressing IGHV 4-34 and subgroups of classic HCL differ in terms of pathogenesis.87, 99 Whereas Lennerz et al.100 reported a case of a CD5-positive variant of HCL with BRAF wild type, several cases of BRAF V600E-negative HCL have been reported.101 The lack of BRAF V600E mutation in different HCL cell lines indicates either that these do not seem to be of authentic HCL origin as expected in previous models85, 86 or that they belong to minor subsets of HCL.
Regarding the results of 2019 cases tested for the BRAF V600E mutation in HCL and other hematological malignancies with a Youden index of 0.97 and a specificity of 97.6%, analysis of the BRAF V600E status may be a useful diagnostic method in HCL.100 In this respect, Sanger sequencing,83 high-resolution melting analysis assay,102 allele-specific polymerase chain reaction,103, 104 pyrosequencing-based approaches98, 100, 105 and immunohistochemical markers such as mouse mAB (clone VE1) as an inexpensive alternative to DNA-based methods,106 or the immunostaining for phospho-ERK,91, 107 are under investigation. Regarding these approaches, the diagnostic use of BRAF V600E mutation may simplify the differentiation of HCL to HCL-like disorders, such as SMZL, HCL-V and IGHV 4-34 cases so that the indication for a therapy with PA can be found more easily.84, 87, 88, 106
These new insights of the pathogenesis of HCL may also allow new therapeutic options, especially for patients who need an alternative to PA because of non-sufficient response, persistence of BRAF-V600E at partial remission, relapse or toxic side effects of chemotherapy. The use of active BRAF inhibitors like BRAF V600E inhibitors,39, 83, 84, 91 and/or MEK inhibitors,91 which have already been used very effective in BRAF V600E mutated melanomas,39, 91 could be an alternative to PA. Dietrich et al.108 reported on one patient suffering from HCL refractory to conventional treatments who could be successfully treated with the BRAF inhibitor vemurafenib: starting at low doses of 240 mg two times daily followed by a slow dose escalation, CR could be achieved on day 43 of 56 days of treatment, supporting the hypothesis that BRAF V600E represents the disease-causing event in HCL and offers a promising therapeutic target.108 Prolonged remission at 6 months after treatment without evidence for MRD has been documented.109 More off-trial use of low-dose vemurafenib in HCL resulting in one complete110 and one partial111 remission has been reported. Recently, Simalikalactone E, an extract of an Amazonian antimalarial agent, has been identified as a potent BRAF V600E inhibitor.112 BRAF inhibitors are specifically directed against BRAF mutated cells causing apoptosis by interruption of MEK-ERK signaling clearing CD20-positive HC whilst probably sparing normal hematopoiesis.109 However, it has to be mentioned that BRAF inhibitor-induced remissions are often short lived.87
In conclusion, the role of BRAF V600E mutation in pathogenesis, diagnosis and therapy in HCL has to be defined in further investigations.
Alternative approaches
Kreitman4 and Jones et al.113 reported a case of a patient with multiple relapsed HCL refractory to PA and rituximab who could reach CR after treatment with the non-selective cyclin-dependent kinase inhibitor flavopiridol as part of a phase II study for the treatment of indolent B-cell malignancies including HCL.4, 113
Phase I and II trials targeting the B-cell receptor signaling pathways have already been tested in non-Hodgkin lymphoma such as CLL or mantle cell lymphoma. Sivina et al.114 showed that Bruton’s tyrosine kinase inhibitors such as ibrutinib have the potential to block B-cell receptor signaling, proliferation and survival in primary HCL and HCL cell lines in vitro.114 Further investigations are needed to prove the therapeutical potential of these agents in HCL.
Kiyasu et al.115 reported on a patient suffering from HCL who was refractory to several treatment approaches inter alia to a combination therapy of cladribine and rituximab but who could be successfully treated with a combination of allogeneic hematopoietic stem cell transplantation followed by four weekly doses of 375 mg/m2 rituximab.115 Regarding a case report of Zinzani et al.,116 allogeneic stem cell transplantation may be an alternative strategy in HCL patients resistant to chemoimmunotherapy.116 Further experiences with stem cell transplantation in HCL are limited to few case reports.116, 117
Treatment recommendations
Initial treatment
When there are no contraindications, PA are still recommended as treatment of choice,10, 27, 39 yielding CRR of 75–100%.76 Although a superiority in the efficacy of the two PA has not been proved yet,10, 39 cladribine is used more often as first-line agent than pentostatin because of its advantageous administration and safety profile.27 Conclusive recommendations for the dosage of cladribine are lacking. The most favored dosing schemes are the 7-day continuous infusion schedule of 0.1 mg/kg per day,19, 39 or the administration of short intravenous infusions of 0.14 mg/kg per day for five consecutive days.6 The alternative subcutaneous application of cladribine seems to be the most cost-effective option10 and is already used as treatment of choice.28
If possible, the application of PA should be delayed in patients with an ongoing infection owing to the possible prolonged myelosuppression aggravated, especially through cladribine.19 An alternative strategy in this situation is the previous administration of IFN-α to increase granulocyte levels for a better anti-infective response of antibiotic or anti-fungal therapy.19, 28 Induction therapy can be started with 3 × 106 IU daily until maximum response followed by a maintenance therapy with thrice weekly subcutaneous injections of the same dose.10 A reduced regimen only consisting of thrice weekly application of IFN-α is recommended for very cytopenic patients.10 If there are contraindications against PA and IFN-α, which often can be difficult to handle in older patients because of flu-like symptoms, fatigue, depression, immune disturbances or cardiac arrhythmia, rituximab may be an alternative as first-line strategy.76
Torrey et al.30 recommend the use of pentostatin, especially in patients who relapse within 12 months after a first treatment approach with cladribine. The most common regimen for pentostatin is based on an intravenous dose of 4 mg/m2 every 14 days until CR plus one or two consolidating injections if applicable.10, 19 In cases of severe neutropenias or ongoing infections, there is the possibility for a delayed application.19, 27 Contraindications for a therapy with the renal cleared pentostatin are a serum creatinine >1.5 mg/dl19 or a clearance <60 ml/min.10 Nevertheless, at a clearance of 40–60 ml/min,10 the dosage of pentostatin can be reduced to 2 mg/m2.10, 19 In every case, renal function has to be observed during therapy and 1.5 l fluid hydration with each dose should be administered.10, 19 In patients with huge splenomegaly10 or in pregnancy,12, 13 splenectomy may be considered as first-line strategy. If treatment is necessary, alternative therapeutic options in pregnant HCL patients are IFN-α8, 10, 13 or mAB (rituximab),8 depending on individual multitude factors. Figure 1 gives an overview of the management of HCL.

Management of hairy cell leukemia.
After normalization of blood counts, a bone marrow examination 4–6 months after therapy with cladribine or after eight to nine courses of pentostatin is recommended to evaluate response.10 Grever27 recommends a careful follow-up of the patient within the first year after achieving CR including monthly to quarterly intervals depending on blood values. Before starting (re)treatment at suspected relapse, a bone marrow specimen should be examinated.27
Treatment of relapsed, resistant and residual disease
There is no international consensual agreement for a standardized procedure for relapsed, refractory or patients with residual disease in HCL, yet.76 The most commonly used options in these situations include repeated cycles either with the same or the alternative PA, IFN-α, splenectomy, combination therapies with rituximab or clinical trials with RIT.
Naik and Saven39 recommend repeated cycles with cladribine in late relapses with RFS intervals >12-18 months and limit the use of pentostatin for early relapses after cladribine within 6 months. After first-line treatment with either cladribine or pentostatin, the administration of the alternative PA is recommended in early relapses within 2 years after the first treatment cycle10, 19 and a repeated cycle of the same PA for relapses that appear after 2 years or in cases of residual disease.10 Subsequent cycles of PA result in lower response rates (70–90%) with shorter duration and may induce the risk for prolonged CD4 lymphopenia, bone marrow hypoplasia with cytopenia and opportunistic infections owing to the prolonged immuno- and myelosuppression.76
To avoid the accumulation of such adverse effects of further treatment with PA, combination or consolidation chemotherapy with rituximab can be pursued as an alternative strategy.19, 76 By default, 375 mg/m2 rituximab are applicated as a weekly intravenous infusion for six to eight doses in total either concurrently or sequentially to PA.10 In cases of refractory HCL rituximab alone or in combination with a PA, the participation in a clinical trial with RIT,42 or splenectomy or splenic irradiation,1 may be successful possibilities. In regard to the controversial role of MRD, the eradication of MRD is not recommended generally, but only as part of clinical trials.10
Supportive care
Before the initiation of chemotherapy, all sources of infection should be rehabilitated.9 Pneumocystis jirovecii prophylaxis with 960 mg co-trimoxazol three times weekly, especially when CD4-positive T-cell counts decrease <200/μl,9, 10 and herpes prophylaxis with 200 mg acyclovir three times daily, especially for patients with prior herpetic infections, is recommended for lymphopenic patients at least until counts ⩾1 × 109/l.10
Saven et al.39 do neither recommend a routine use of granulocyte-colony-stimulating factor based on their findings in a phase II study investigating the influence of filgrastim on cladribine-induced severe neutropenia nor prophylactic erythropoietin-stimulating agents because of the rareness of severe cladribine-induced anemia. However, filgrastim may be an option in patients with previous life-threatening infections or with severe neutropenia with high risk for infection.19
If transfusions of blood products are necessary in patients once treated with pentostatin or cladribine, all of the products have to be irradiated to reduce the occurrence of transfusion-related graft-versus-host-disease.1, 10
References
Mey U, Strehl J, Gorschlüter M, Ziske C, Glasmacher A, Pralle H et al. Advances in the treatment of hairy-cell leukaemia. Lancet Oncol 2003; 4: 86–94.
Gedik E, Girgin S, Aldemir M, Keles C, Tuncer M, Aktas A . Non-traumatic splenic rupture: report of seven cases and review of the literature. Gastroenterology 2008; 14: 6711–6716.
Lavrenkov K, Krepel-Volsky S, Levi I, Ariad S . Low dose palliative radiotherapy for splenomegaly in hematologic disorders. Leuk Lymphoma 2011; 53: 430–434.
Kreitman RJ . Immunoconjugates and new molecular targets in hairy cell leukemia. Hematology Am Soc Hematol Educ Program 2012; 2012: 660–666.
Habermann TM, Rai K . Historical treatments of in hairy cell leukemia, splenectomy and interferon: past and current uses. Leuk Lymphoma 2011; 52: 18–20.
Golomb HM . Fifty years of hairy cell leukemia treatments. Leuk Lymphoma 2011; 52: 3–5.
Zinzani PL, Pellegrini C, Stefoni V, Derenzini E, Gandolfi L, Broccoli A et al. Hairy cell leukemia: evaluation of the long-term outcome in 121 patients. Cancer 2010; 116: 4788–4792.
Daver N, Nazha A, Kantarjian H, Haltom R, Ravandi F . Treatment of hairy cell leukemia during pregnancy: are purine analogues and rituximab viable therapeutic options. Clin lymphoma, myeloma leuk 2012; 13: 86–89.
Barth J, Rummel MJ . Haarzellleukämie: teil 1: krankheitsbild und allgemeine therapie. Med Monatsschr Pharm 2009; 32: 249–251.
Jones G, Parry-Jones N, Wilkins B, Else M, Catovsky D . Revised guidelines for the diagnosis and management of hairy cell leukaemia and hairy cell leukaemia variant*. Br J Haematol 2012; 156: 186–195.
Gujral S, Lad P, Subramanian PG, Shet TM, Epari S, Laskar S et al. Histopathological audit of splenectomies received at a cancer hospital. Indian J Pathol Microbiol 2011; 54: 487–496.
Adeniji BA, Fallas M, Incerpi M, Hamburg S, Katz R, Ogunyemi D . Laparoscopic splenectomy for hairy cell leukemia in pregnancy. Case Rep Med 2010; 2010, Article ID 136823 (e-pub).
Rizack T, Mega A, Legare R, Castillo J . Management of hematological malignancies during pregnancy. Am J Hematol 2009; 84: 830–841.
Lauria F, Cencini E, Forconi F . Alternative methods of cladribine administration. Leuk Lymphoma 2011; 52: 34–37.
Bekisz J, Baron S, Balinsky C, Morrow A, Zoon KC . Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals 2010; 3: 994–1015.
Quesada JR, Reuben J, Manning JT, Hersh EM, Gutterman JU . Alpha interferon for induction of remission in hairy-cell leukemia. N Engl J Med 1984; 310: 15–18.
Habermann TM, Andersen JW, Cassileth PA, Bennett JM, Oken MM . Sequential administration of recombinant interferon alpha and deoxycoformycin in the treatment of hairy cell leukaemia. Br J Haematol 1992; 80: 466–471.
Grever MR . Hairy cell leukemia: a successful model for experimental therapeutics—pentostatin and new ideas. Leuk Lymphoma 2011; 52: 25–28.
Grever MR, Lozanski G . Modern strategies for hairy cell leukemia. J Clin Oncol 2011; 29: 583–590.
Spurgeon S, Yu M, Phillips JD, Epner EM . Cladribine: not just another purine analogue? Exp Opin Invest Drugs 2009; 18: 1169–1181.
Shouval R, Duffield A, Gocke C, Lee L, Brodsky RA . Pentostatin-induced hypereosinophilia with eosinophilic gastroenteritis. Leuk Lymphoma 2010; 51: 1567–1569.
Johnston JB . Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk Lymphoma 2011; 52: 43–45.
Flinn IW, Kopecky KJ, Foucar MK, Head D, Bennett JM, Hutchison R et al. Long-term follow-up of remission duration, mortality, and second malignancies in hairy cell leukemia patients treated with pentostatin. Blood 2000; 96: 2981–2986.
Maloisel F, Benboubker L, Gardembas M, Coiffier B, Divine M, Sebban C et al. Long-term outcome with pentostatin treatment in hairy cell leukemia patients. A French retrospective study of 238 patients. Leukemia 2003; 17: 45–51.
Dearden CE, Else M, Catovsky D . Long-term results for pentostatin and cladribine treatment of hairy cell leukemia. Leuk Lymphoma 2011; 52: 21–24.
Else M, Dearden CE, Matutes E, Garcia-Talavera J, Rohatiner AZS, Johnson SAN et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol 2009; 145: 733–740.
Grever MR . How I treat hairy cell leukemia. Blood 2010; 115: 21–28.
Austein T, Schmitt A . 50-jähriger Mann mit Gewichtsverlust und Splenomegalie. Dtsch Med Wochenschr (1946) 2009; 134: 251–252.
Huynh E, Sigal D, Saven A . Cladribine in the treatment of hairy cell leukemia: initial and subsequent results. Leuk Lymphoma 2009; 50: 12–17.
Torrey ML, Sigal DS, Saven A . Development of cladribine at Scripps for hairy cell leukemia and current results. Leuk Lymphoma 2011; 52: 29–33.
Singh V, Chittappen KP, Gudi V, Bénardais K, Voss EV, Stangel M . 2-chlorodeoxyadenosine (cladribine) induces apoptosis in human monocyte-derived dendritic cells. Clin Exp Immunol 2013; 173: 288–297.
Cheson BD, Sorensen JM, Vena DA, Montello MJ, Barrett JA, Damasio E et al. Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine via the Group C protocol mechanism of the National Cancer Institute: a report of 979 patients. J Clin Oncol 1998; 16: 3007–3015.
Saven A, Burian C, Koziol JA, Piro LD . Long-term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood 1998; 92: 1918–1926.
Goodman GR, Burian C, Koziol JA, Saven A . Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol 2003; 21: 891–896.
Jehn U, Bartl R, Dietzfelbinger H, Haferlach T, Heinemann V . An update: 12-year follow-up of patients with hairy cell leukemia following treatment with 2-chlorodeoxyadenosine. Leukemia 2004; 18: 1476–1481.
Chadha P, Rademaker AW, Mendiratta P, Kim B, Evanchuk DM, Hakimian D et al. Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CdA): long-term follow-up of the Northwestern University experience. Blood 2005; 106: 241–246.
Robak T, Jamroziak K, Gora-Tybor J, Blonski JZ, Kasznicki M, Dwilewicz-Trojaczek J et al. Cladribine in a weekly versus daily schedule for untreated active hairy cell leukemia: final report from the Polish Adult Leukemia Group (PALG) of a prospective, randomized, multicenter trial. Blood 2007; 109: 3672–3675.
Zenhäusern R, Schmitz SH, Solenthaler M, Heim D, Meyer-Monard S, Hess U et al. Randomized trial of daily versus weekly administration of 2-chlorodeoxyadenosine in patients with hairy cell leukemia: a multicenter phase III trial (SAKK 32/98). Leuk Lymphoma 2009; 50: 1501–1511.
Naik RR, Saven A . My treatment approach to hairy cell leukemia. Mayo Clin Proc 2012; 87: 67–76.
Sigal DS, Sharpe R, Burian C, Saven A . Very long-term eradication of minimal residual disease in patients with hairy cell leukemia after a single course of cladribine. Blood 2010; 115: 1893–1896.
Gerrie AS, Zypchen LN, Connors JM . Fludarabine and rituximab for relapsed or refractory hairy cell leukemia. Blood 2012; 119: 1988–1991.
Lauria F, Forconi F . Combination therapies to improve the long-term outcome in hairy cell leukemia. Leuk Lymphoma 2009; 50: 18–22.
Forconi F, Sozzi E, Cencini E, Zaja F, Intermesoli T, Stelitano C et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009; 114: 4696–4702.
Forconi F . Hairy cell leukaemia: biological and clinical overview from immunogenetic insights. Hematol Oncol 2011; 29: 55–66.
Caligaris-Cappio F . IG genes and hairy cell leukemia. Blood 2009; 114: 4610–4611.
Forconi F, Cencini E, Sozzi E, Sicuranza A, Raspadori D, Lauria F . Insight into the behavior of hairy cell leukemia by immunogenetic analysis. Leuk Lymphoma 2011; 52: 103–107.
Forconi F, Cencini E, Sicuranza A, Sozzi E, Lauria F . Molecular insight into the biology and clinical course of hairy cell leukemia utilizing immunoglobulin gene analysis. Leuk Lymphoma 2011; 52: 15–23.
Juliusson G, Samuelsson H . Hairy cell leukemia: epidemiology, pharmacokinetics of cladribine, and long-term follow-up of subcutaneous therapy. Leuk Lymphoma 2011; 52: 46–49.
Kraut E . Infectious complications in hairy cell leukemia. Leuk Lymphoma 2011; 52: 50–52.
Cannon T, Mobarek D, Wegge J, Tabbara IA . Hairy cell leukemia: current concepts. Cancer invest 2008; 26: 860–865.
Hisada M, Chen BE, Jaffe ES, Travis LB . Second cancer incidence and cause-specific mortality among 3104 patients with hairy cell leukemia: a population-based study. J Natl Cancer Inst 2007; 99: 215–222.
Damaj G, Kuhnowski F, Marolleau J, Bauters F, Leleu X, Yakoub-Agha I . Risk factors for severe infection in patients with hairy cell leukemia: a long-term study of 73 patients. Eur J Haematol 2009; 83: 246–250.
Girardi K, Paviglianiti A, Cirillo M, Bianchi A, Gherardi G, Annibali O et al. Tuberculous meningoencephalitis in a patient with hairy cell leukemia in complete remission. J Clin Exp Hematopathol 2012; 52: 31–34.
Ganzel C, Gatt ME, Maly A, Ben-Yehuda D, Goldschmidt N . High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma 2012; 53: 1169–1173.
Kreitman RJ, Arons E, Stetler-Stevenson M, Fitzgerald DJP, Wilson WH, Pastan I . Recombinant immunotoxins and other therapies for relapsed/refractory hairy cell leukemia. Leuk Lymphoma 2011; 52: 82–86.
Kreitman RJ, Arons E, Stetler-Stevenson M, Miller KB . Response of hairy cell leukemia to bendamustine. Leuk Lymphoma 2011; 52: 1153–1156.
Fitzgerald DJ, Moskatel E, Ben-Josef G, Traini R, Tendler T, Sharma A et al. Enhancing immunotoxin cell-killing activity via combination therapy with ABT-737. Leuk Lymphoma 2011; 52: 79–81.
Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M et al. Phase I Trial of Anti-CD22 Recombinant Immunotoxin Moxetumomab Pasudotox (CAT-8015 or HA22) in Patients With Hairy Cell Leukemia. J Clin Oncol 2012; 30: 1822–1828.
Park JH, Levine RL . Targeted immunotherapy for hairy cell leukemia. J Clin Oncol 2012; 30: 1888–1890.
Kreitman RJ, Pastan I . Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011; 17: 6398–6405.
Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci USA 2011; 108: 5742–5747.
Hagberg H, Lundholm L . Rituximab, a chimaeric anti-CD20 monoclonal antibody, in the treatment of hairy cell leukaemia. Br J Haematol 2001; 115: 609–611.
Lauria F, Lenoci M, Annino L, Raspadori D, Marotta G, Bocchia M et al. Efficacy of anti-CD20 monoclonal antibodies (Mabthera) in patients with progressed hairy cell leukemia. Haematologica 2001; 86: 1046–1050.
Nieva J, Bethel K, Saven A . Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood 2003; 102: 810–813.
Thomas DA, O'Brien S, Bueso-Ramos C, Faderl S, Keating MJ, Giles FJ et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003; 102: 3906–3911.
Forconi F, Toraldo F, Sozzi E, Amato T, Raspadori D, Lauria F . Complete molecular remission induced by concomitant cladribine—rituximab treatment in a case of multi-resistant hairy cell leukemia. Leuk Lymphoma 2007; 48: 2441–2443.
Cervetti G, Galimberti S, Andreazzoli F, Fazzi R, Cecconi N, Caracciolo F et al. Rituximab as treatment for minimal residual disease in hairy cell leukaemia: extended follow-up. Br J Haematol 2008; 143: 296–298.
Zenhäusern R, Simcock M, Gratwohl A, Hess U, Bargetzi M, Tobler A . Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008; 93: 1426–1428.
Ravandi F, O'Brien S, Jorgensen J, Pierce S, Faderl S, Ferrajoli A et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011; 118: 3818–3823.
Else M, Dearden CE, Matutes E, Forconi F, Lauria F, Ahmad H et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011; 52: 75–78.
Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol 2000; 18: 1622–1636.
Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, Fitzgerald DJP, Wilson WH et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol 2005; 23: 6719–6729.
Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJP, Wilson WH et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol 2009; 27: 2983–2990.
Leung K Cy5.5-Rituximab. Molecular Imaging and Contrast Agent Database (MICAD) [Internet] 10 March 2009 [updated 4 June 2009]. National Center for Biotechnology Information (US): Bethesda, MD, USA; 2004–2013.
Fey MF . Rekombinante Proteine als Therapeutika in derklinischen Onkologie. Ther Umsch 2011; 68: 618–630.
Malfuson JV, Fagot T, Konopacki J, Souleau B, Cremades S, Revel Tde . Which role for rituximab in hairy cell leukemia? Reflections on six cases. Acta haematol 2010; 123: 110–116.
Byrd JC, Peterson BL, Morrison VA, Park K, Jacobson R, Hoke E et al. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003; 101: 6–14.
Ravandi F . Chemo-immunotherapy for hairy cell leukemia. Leuk Lymphoma 2011; 52: 72–74.
Byrd JC, Rai K, Peterson BL, Appelbaum FR, Morrison VA, Kolitz JE et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005; 105: 49–53.
Else M, Osuji N, Forconi F, Dearden C, Del Giudice I, Matutes E et al. The role of rituximab in combination with pentostatin or cladribine for the treatment of recurrent/refractory hairy cell leukemia. Cancer 2007; 110: 2240–2247.
Kreitman RJ . Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. BioDrugs 2009; 23: 1–13.
Kreitman RJ, Fitzgerald DJP, Pastan I . Approach to the patient after relapse of hairy cell leukemia. Leuk Lymphoma 2009; 50: 32–37.
Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364: 2305–2315.
Tiacci E, Schiavoni G, Forconi F, Santi A, Trentin L, Ambrosetti A et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood 2011; 119: 192–195.
Tiacci E, Pucciarini A, Bigerna B, Pettirossi V, Strozzini F, Martelli MP et al. Absence of BRAF-V600E in the human cell lines BONNA-12, ESKOL, HAIR-M, and HC-1 questions their origin from hairy cell leukemia. Blood 2012; 119: 5332–5333.
Weston-Bell NJ, Hendriks D, Sugiyarto G, Bos NA, Kluin-Nelemans HC, Forconi F et al. Hairy cell leukemia cell lines expressing annexin A1 and displaying B-cell receptor signals characteristic of primary tumor cells lack the signature BRAF mutation to reveal unrepresentative origins. Leukemia 2013; 27: 241–245.
Burger JA . BRAF mutation: supporting diversity in HCL. Blood 2012; 119: 3193–3194.
Tadmor T, Tiacci E, Falini B, Polliack A . The BRAF-V600E mutation in hematological malignancies: a new player in hairy cell leukemia and Langerhans cell histiocytosis. Leuk Lymphoma 2012; 53: 2339–2340.
Rinaldi A, Kwee I, Young KH, Zucca E, Gaidano G, Forconi F et al. Genome-wide high resolution DNA profiling of hairy cell leukaemia. Br J Haematol 2013; 162: 566–569.
Ping N, Wang Q, Wang Q, Dong S, Wu L, Xue Y et al. Absence of BRAF V600E mutation in hematologic malignancies excluding hairy-cell leukemia. Leuk Lymphoma 2012; 53: 2498–2499.
Tiacci E, Schiavoni G, Martelli MP, Boveri E, Pacini R, Tabarrini A et al. Constant activation of the RAF-MEK-ERK pathway as a diagnostic and therapeutic target in hairy cell leukemia. Haematologica 2013; 98: 635–639.
Takahashi Y, Mori J, Kami M . BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 365, 960-961; author reply 961–962.
Pardanani A, Tefferi A . BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 365, 961; author reply 961–962.
Auer RL, Cotter FE . A defining moment for hairy cell leukaemia. Br J Haematol 2011; 155: 607–608.
Blombery P, Wong SQ, Hewitt CA, Dobrovic A, Maxwell EL, Juneja S et al. Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica 2011; 97: 780–783.
Trifa AP, Popp RA, Cucuianu A, Coadă CA, Urian LG, Militaru MS et al. Absence of BRAF V600E mutation in a cohort of 402 patients with various chronic and acute myeloid neoplasms. Leuk Lymphoma 2012; 53: 2496–2497.
Berghoff AS, Capper D, Preusser M . Lack of BRAF V600E protein expression in primary central nervous system lymphoma. Appl Immunohistochem Mol Morphol 2013; 21: 351–353.
Laurini JA, Aoun P, Iqbal J, Chan W, Greiner TC . Investigation of the BRAF V600E mutation by pyrosequencing in lymphoproliferative disorders. Am J Clin Pathol 2012; 138: 877–883.
Xi L, Arons E, Navarro W, Calvo KR, Stetler-Stevenson M, Raffeld M et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012; 119: 3330–3332.
Lennerz JK, Klaus BM, Marienfeld RB, Möller P . Pyrosequencing of BRAF V600E in routine samples of hairy cell leukaemia identifies CD5+ variant hairy cell leukaemia that lacks V600E. Br J Haematol 2012; 157: 267–269.
Langabeer SE, O'Brien D, McElligott AM, Lavin M, Browne PV . BRAF V600E-Negative Hairy Cell Leukaemia. Case Rep Hematol 2013; 2013: 513049.
Boyd EM, Bench AJ, van Veer MB 't, Wright P, Bloxham DM, Follows GA et al. High resolution melting analysis for detection of BRAF exon 15 mutations in hairy cell leukaemia and other lymphoid malignancies. Br J Haematol 2011; 155: 609–612.
Arcaini L, Zibellini S, Boveri E, Riboni R, Rattotti S, Varettoni M et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood 2011; 119: 188–191.
Szankasi P, Reading NS, Vaughn CP, Prchal JT, Bahler DW, Kelley TWA . Quantitative Allele-Specific PCR Test for the BRAF V600E Mutation Using a Single Heterozygous Control Plasmid for Quantitation: A Model for qPCR Testing without Standard Curves. J Mol Diagn 2013; 15: 248–254.
Verma S, Greaves WO, Ravandi F, Reddy N, Bueso-Ramos CE, O'Brien S et al. Rapid detection and quantitation of BRAF mutations in hairy cell leukemia using a sensitive pyrosequencing assay. Am J Clin Pathol 2012; 138: 153–156.
Andrulis M, Penzel R, Weichert W, Deimling A von, Capper D . Application of a BRAF V600E mutation-specific antibody for the diagnosis of hairy cell leukemia. Am J Sugr Pathol 2012; 36: 1796–1800.
Warden DW, Ondrejka S, Lin J, Durkin L, Bodo J, Hsi ED . Phospho-ERKTHR202/Tyr214 Is Overexpressed in Hairy Cell Leukemia and Is a Useful Diagnostic Marker in Bone Marrow Trephine Sections. Am J Sugr Pathol 2013; 37: 305–308.
Dietrich S, Glimm H, Andrulis M, Kalle C, von, Ho AD, Zenz T . BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012; 366: 2038–2040.
Dietrich S, Hüllein J, Hundemer M, Lehners N, Jethwa A, Capper D et al. Continued Response Off Treatment After BRAF Inhibition in Refractory Hairy Cell Leukemia. J Clin Oncol 2013; 31: e300–e303.
Peyrade F, Re D, Ginet C, Gastaud L, Allegra M, Ballotti R et al. Low-dose vemurafenib induces complete remission in a case of hairy-cell leukemia with a V600E mutation. Haematologica 2013; 98: e20–e22.
Follows GA, Sims H, Bloxham DM, Zenz T, Hopper MA, Liu H et al. Rapid response of biallelic BRAF V600E mutated hairy cell leukaemia to low dose vemurafenib. Br J Haematol 2012; 161: 150–153.
Robert G, Jullian V, Jacquel A, Ginet C, Dufies M, Torino S et al. Simalikalactone E (SkE), a new weapon in the armamentarium of drugs targeting cancers that exhibit constitutive activation of the ERK pathway. Oncotarget 2012; 3: 1688–1699.
Jones JA, Kraut EH, Deam D, Byrd JC, Grever MR . Hematologic improvement after flavopiridol treatment of pentostatin and rituximab refractory hairy cell leukemia. Leuk Lymphoma 2012; 53: 490–491.
Sivina M, Kreitman RJ, Arons E, Buggy JJ, Ravandi F, Burger JA . Bruton's tyrosine kinase (BTK) inhibitor Ibrutinib (PCI-32765) blocks hairy cell leukemia (HCL) survival, proliferation, and BCR signaling: a new therapeutic approach for HCL. American Society of Hematology 2012, CLL – Therapy, excluding Transplantation: Poster I: 1802.
Kiyasu J, Shiratsuchi M, Ohtsuka R, Sada E, Idutsu K, Nagasawa E et al. Achievement of complete remission of refractory hairy cell leukemia by rituximab progressing after allogeneic hematopoietic stem cell transplantation. In Hematol 2009; 89: 403–405.
Zinzani PL, Bonifazi F, Pellegrini C, Casadei B, Argnani L, Motta MR et al. Hairy cell leukemia: allogeneic transplantation could be an optimal option in selected patients. Clin lymphoma, myeloma & leukemia 2012; 12: 287–289.
Cheever MA, Fefer A, Greenberg PD, Appelbaum F, Armitage JO, Buckner CD et al. Treatment of hairy-cell leukemia with chemoradiotherapy and identical-twin bone-marrow transplantation. N Engl J Med 1982; 307: 479–481.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Maevis, V., Mey, U., Schmidt-Wolf, G. et al. Hairy cell leukemia: short review, today’s recommendations and outlook. Blood Cancer Journal 4, e184 (2014). https://doi.org/10.1038/bcj.2014.3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2014.3
Keywords
This article is cited by
-
Adverse event rates and economic burden associated with purine nucleoside analogs in patients with hairy cell leukemia: a US population-retrospective claims analysis
Orphanet Journal of Rare Diseases (2020)
-
sIL2R ratio as early marker for response in hairy cell leukemia and the prognostic relevance of IL28B genotype to interferon-α therapy
Annals of Hematology (2017)
-
Superior efficacy of rituximab-based chemoimmunotherapy as an initial therapy in newly diagnosed patients with B cell indolent lymphomas: long-term results from a single center in China
BMC Cancer (2015)
-
Rituximab as frontline monotherapy in untreated hairy cell leukemia patients
Annals of Hematology (2015)
