
Abstract
T- and natural killer (NK)-cell lymphomas are challenging childhood neoplasms. These cancers have varying presentations, vast molecular heterogeneity, and several are quite unusual in the West, creating diagnostic challenges. Over 20 distinct T- and NK-cell neoplasms are recognized by the 2008 World Health Organization classification, demonstrating the diversity and potential complexity of these cases. In pediatric populations, selection of optimal therapy poses an additional quandary, as most of these malignancies have not been studied in large randomized clinical trials. Despite their rarity, exciting molecular discoveries are yielding insights into these clinicopathologic entities, improving the accuracy of our diagnoses of these cancers, and expanding our ability to effectively treat them, including the use of new targeted therapies. Here, we summarize this fascinating group of lymphomas, with particular attention to the three most common subtypes: T-lymphoblastic lymphoma, anaplastic large cell lymphoma, and peripheral T-cell lymphoma-not otherwise specified. We highlight recent findings regarding their molecular etiologies, new biologic markers, and cutting-edge therapeutic strategies applied to this intriguing class of neoplasms.
Similar content being viewed by others

Introduction
T-cell lymphomas encompass several hematological cancers in children and adolescents. Arising in cells of the innate and adaptive immune systems, T- and natural killer (NK)-cell neoplasms comprise over 20 distinct entities in the current World Health Organization (WHO) schema (Table 1 and Figure 1).1, 2 The most common types in pediatric patients are T-cell lymphoblastic lymphoma (T-LBL) and anaplastic large cell lymphoma (ALCL). Of the other rarely occurring pediatric T-cell neoplasms, peripheral T-cell lymphoma-not otherwise specified (PTCL-NOS) is seen most frequently. Most of these lymphomas are aggressive diseases3 that present formidable diagnostic and therapeutic challenges.4 In this review, we update the biologic features of several T-cell lymphomas in children and adolescents, with a focus on their molecular pathology and its implications for novel therapeutics.

Montage of pathology images for several different T- and NK- cell lymphomas in children and adolescents. (a) T-LBL showing diffuse infiltration of T-lymphoblasts with fine nuclear chromatin and scarce cytoplasm. Blasts have inconspicuous nucleoli and irregular nuclear contours. × 200 magnification, hematoxylin and eosin (H&E) stain. (b) ALCL, ALK+, showing large neoplastic cells with marked pleomorphism and abundant cytoplasm. Characteristic horseshoe-shaped cells are seen. Cells stained strongly with CD30 and ALK antibodies demonstrate a mature T-cell immunophenotype. × 400 magnification, H&E. (c) PTCL, NOS with predominantly large cell morphology. Neoplastic cells are large with abundant cytoplasm and variably prominent nucleoli. Immunophenotyping reveals a mature T-cell phenotype, but cells lack expression of CD30 or ALK. × 400 magnification, H&E. (d) Hepatosplenic γ/δ T-cell lymphoma. Small-to-intermediate neoplastic cells have infiltrated liver sinusoids. These cells show round nuclear contours, clumped chromatin without prominent nucleoli, and abundant clear cytoplasm. × 100 magnification, H&E. (e) SPTCL demonstrating invasion of malignant cells into fat. Small-to-intermediate size neoplastic cells with moderate cytologic atypia and irregular nuclear contours surround fat lobules. × 400 magnification, H&E. (f) Primary cutaneous γ/δ T-cell lymphoma that presented with skin nodules and plaques with extensive dermal involvement and extension into panniculitic fat. Medium-to-large neoplastic cells have occasional prominent nucleoli. Neoplastic cells lacked CD4 and CD8 expression and were EBV negative. × 200 magnification, H&E. (g) T-cell post-transplant lymphoproliferative disorder in a patient with liver transplant 2 years prior. Clonal T cells have invaded the intestine and are EBV positive. × 400 magnification, H&E. (h) Extranodal T/NK-cell lymphoma of the nasal cavity showing angiocentric pattern with vascular invasion. Neoplastic cells are small-to-intermediate in size with abundant cytoplasm. Irregular nuclei show clumped hyperchromatic chromatin. Cells are CD8 and EBV positive. × 200 magnification, H&E. (i) AITL with diffuse nodal effacement and prominent arborizing vessels. Neoplastic cells are intermediate size and show variable clear cytoplasm. × 200 magnification, H&E.
T-LBL in children and adolescents
After Burkitt lymphoma, LBL is the second most common pediatric non-Hodgkin lymphoma (NHL). In 80–90% of LBL, disease is T-cell lineage, unlike acute lymphoblastic leukemia (ALL) where precursor B-cell is typical. Thus, T-LBL is the most common pediatric T-cell lymphoma.5, 6, 7 Historically, T-LBL and T-ALL have been considered variant clinical manifestations of the same disease, but genomic and gene expression studies reveal molecular differences between T-LBL and T-ALL.8, 9 Clinical features also subtly differentiate these cancers, with T-LBL tending to have earlier and local relapses, and T-ALL more often having central nervous system (CNS) involvement at diagnosis.10, 11, 12
Children with T-LBL frequently present with supra-diaphragmatic mass. Overlapping symptoms in T-LBL and T-ALL derive from their shared propensity to manifest with hepatic, splenic, and nodal enlargement, as well as with mediastinal mass. Serious sequelae from these clinical features can occur, including obstructive airway compromise and superior vena cava syndrome; both may require emergent glucocorticoid and/or radiation therapy.13 T-LBL is often advanced at diagnosis (stage III–IV), unlike B-cell LBL, which primarily is a localized disease of skin, bone, or lymph node. Accordingly, stage I and II T-LBLs are relatively rare. As no molecular features can yet reliably differentiate LBL from ALL, the clinical finding of ⩾25% of marrow infiltration by malignant lymphoblasts continues to define ALL, while 5–25% marrow involvement is regarded as stage IV LBL.14
Current treatments for pediatric T-LBL
Children with limited disease (stage I–II) T-LBL do well, with long-term overall survival (OS) of 85–90%.5, 6, 7, 14 Disease-free survival is lower at 63–73%, but relapsed patients show good responses to salvage therapy.15 As in advanced-stage T-LBL, ALL protocols are the basis of treatment for T-LBL patients with localized disease.16 These patients are treated without local surgery or radiation, and do not receive craniospinal radiation therapy (CRT) prophylaxis. The following discussion will focus on therapies for advanced (stage III–IV) T-LBL patients.
Advanced pediatric T-LBL outcomes improved dramatically on the 10-drug LSA2L2 regimen.17 Most LBL therapies can be traced to this protocol, with nearly all current strategies comprised of induction, consolidation, re-intensification, and maintenance phases. The specific timing and doses of some agents may vary, but overall, treatments since 2000 have achieved 70–90% event-free survival (EFS) with 12- to 24-month regimens (Table 2).
Many treatment protocols advanced LBL therapy over the past 15 years; highlights of these studies are briefly summarized: The German Berlin–Frankfurt–Munster (BFM)-90 regimen administered CRT to all advanced patients, independent of CNS involvement. Although EFS was 90% in stage III–IV patients, long-term effects prompted ensuing studies to apply CRT to only CNS-positive patients.11 The Italian lymphoma non-Hodgkin (LNH)-92 protocol reported only 69% EFS in advanced T-LBL, but this regimen lacked re-intensification, demonstrating the value of this phase.18 The BFM-95 study queried whether systemic methotrexate (MTX) and intrathecal (IT) chemotherapy could safely replace CRT as prophylaxis for CNS recurrence. Although its 84% EFS was lower than the 90% EFS of BFM-90, comparison with other earlier BFM studies determined that omitting CRT was not inferior to prior regimens that applied CRT to CNS-negative patients.19 The Children's Oncology Group (COG) used an intense and truncated (12-month) multi-agent protocol for advanced T-LBL. This yielded a 78% EFS, similar to longer ALL-based regimens.20 Early reports from the current COG study testing high-dose MTX and early intensification suggest that neither intervention improves EFS.21
Two studies have explored completely omitting CRT in advanced disease, including CNS-positive patients.22, 23 A European Organization for Research and Treatment of Cancer (EORTC) trial showed omission of CRT did not raise CNS relapse rates, with 78% 6-year EFS. Although only three CNS-positive patients participated in this trial, the overall low rate of CNS relapse validates that intrathecal treatments can safely replace CRT in stage III–IV, CNS-negative patients. This study also identified response to a prednisone-only prephase as an important prognostic factor. Patients in complete remission (CR) after prephase (16/121 patients) had 100% EFS, but only 14% EFS was seen in prednisone-resistant T-LBL.22 St Jude's NHL13 trial also eliminated CRT. Like their institutional ALL regimen, NHL13 used high-dose MTX every 8 weeks and added a re-induction phase during maintenance. This study had an impressive 83% EFS in advanced T-LBL, despite its lack of CRT.23
Therapies for relapsed pediatric T-LBL
Historically, children with refractory or relapsed LBL have poor prognoses, with 10% 5-year OS.24 Most relapses occur within 2 years of diagnosis (Figure 2).10 Successful outcomes have been reported in recurrent T-LBL using intense re-induction chemotherapy followed by either autologous (auto-) or allogeneic stem cell transplant (allo-SCT), but these series each contained 10 or fewer T-LBL patients, so reliable survival figures are not available.25, 26, 27 Modestly effective re-induction regimens include ICE (ifosfamide, carboplatin, and etoposide; 92% response rate seen in a 40 patient cohort containing 24 relapsed NHL) and DECAL (dexamethasone, etoposide, cisplatin, high-dose cytarabine and L-asparaginase; 50% response rate in a 58 patient NHL cohort, including 26 LBL cases).28, 29

Time and site of disease recurrence in children with relapsed lymphoblastic lymphoma. I, patients with T-LBL; X, patients with precursor B-cell lymphoblastic lymphoma; BM, bone marrow. (*) Patient was treated on a high-risk arm and experienced relapse during an intensive phase of treatment 11 months after start of therapy. Reprinted from Burkhardtet al.10
Chemo-sensitive relapses have better outcome after auto or allo-SCT.28, 30 Relapses after allo-SCT were less common in a group of 204 LBL patients (most of whom had T-LBL), but higher transplant-related mortality was felt to offset any survival benefit.31 However, this remains an open question. In one relapsed T-LBL BFM cohort, OS was quite low (14%; 4/28 patients), but all 4 long-term survivors received allo-SCT. Yet, because only 2 patients received auto-SCT in this study (9 received allo-SCT and 17 chemotherapy-only), these data do not convincingly favor allo- over auto-SCT.10 However, other studies imply that graft vs lymphoma effect after allo-SCT may help prevent relapses. Specifically, one series of 53 refractory or recurrent LBL patients showed much higher EFS after allo- compared with auto-SCT (40% vs 4%).32 A second study of 48 refractory or relapsed LBL patients (32 with T-LBL) also demonstrated higher EFS and OS in the allo-SCT group.33 Although neither study examined T-LBL exclusively, their large sample sizes and the preponderance of T-cell disease within LBL both suggest that results are likely generalizable to T-LBL.
Future therapies for pediatric T-LBL
To date, new therapies for T-LBL are limited. The novel purine analog nelarabine shows promise in relapsed and refractory T-ALL, but not T-LBL.34 Impetus for the use of nelarabine in T-cell cancer was suggested by the finding that purine nucleoside phosphorylase-deficient patients develop T-cell cytopenias because of toxic deoxyguanosine triphosphate levels in T cells. As a deoxyguanosine triphosphate derivative, nelarabine resists purine nucleoside phosphorylase-mediated degradation and shows marked T-lymphocyte toxicity. In a phase II COG study, T-ALL patients in first relapse had a 55% response rate, and 48% of patients achieved CR. Unfortunately, nelarabine lacked similar efficacy in T-LBL, and 18% of patients exhibited severe (⩾ grade 3) CNS toxicity, such as peripheral neuropathy, hallucinations, and seizures.34 Other novel agents targeting frequent NOTCH1 and mammalian target of rapamycin (mTOR) kinase upregulation in T-LBL are being investigated in vitro. Finally, phase I studies are also testing another purine nucleoside phosphorylase inhibitor, forodesine, in T-cell malignancy. However, these trials investigate adults with PTCL rather than pediatric T-LBL.35
As salvage rates for relapsed T-LBL are dismal and promising novel agents do not exist, delineating prognostic factors that predict relapse would be useful in stratifying high-risk patients for intensified upfront treatment regimens. Currently, such factors are not well established in pediatric T-LBL. Aside from the striking EORTC result where response to a 1-week prednisone pre-phase predicted opposite extremes in EFS, other clinical prognostic factors are lacking. Assessment of treatment responses via laboratory testing, molecular studies, or radiographically (such as 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography) are all tenable options to identify high-risk patients early-on.36
The paucity of cytogenetic and molecular features linked to T-LBL clinical response presents intriguing opportunities for study. One recent discovery is the finding that chromosome 6q loss of heterozygosity in T-LBL predicts a higher risk of relapse, but other cryptic karyotypic features may also exist.8 Meanwhile, our assessment of low-level minimal disseminated disease (MDD) and minimal residual disease is also improving, with T-cell receptor (TCR) PCR assays able to quantify these reliably.37 Flow cytometry can also detect occult disease, and these two techniques have been compared with respect to their detection and quantification of MDD and minimal residual disease in T-LBL.38 In a separate study using a 0.01% MDD threshold, two-thirds of patients had MDD-positive marrow at diagnosis (Figure 3a). Critically, patients with high marrow MDD had much lower 2-year EFS (52%) than patients with <5% MDD (EFS=89%; Figure 3b). These data clearly demonstrate poor outcomes in stage III patients with higher MDD.39 However, prior BFM studies have shown no prognostic difference between stage III and IV T-LBL patients.19 Similarly, stage IV patients with gross marrow disease (6–24% blasts) treated with COG-based therapy greatly exceed the poor 2-year EFS seen in these high-MDD stage III patients.20 This curious contradiction alludes to important questions that remain in T-LBL biology. Further study of the aforementioned predictors and discovery of new prognostic features will permit prospective studies to determine which at-risk groups may benefit from intensified treatments, like allo-SCT, as first-line approaches.

Prevalence, degree, and impact of marrow involvement in children with T-cell lymphoblastic lymphoma (T-LL). (a) Percentage of T-LL cells in bone marrow at diagnosis as detected by flow cytometry, according to disease stage based on conventional criteria. Horizontal bars indicate median value for each group. (b) Event-free survival stratified by level of T-LL cells in bone marrow at diagnosis as measured by flow cytometry: <5% and ⩾5% T-LL cells. Reprinted from Coustan-Smith et al.39
For instance, a new T-ALL type was recently described with markedly poor prognosis, raising the question of whether this group might also be relevant to T-LBL.40 This new type, dubbed early T-cell precursor ALL (ETP-ALL), has distinct biology from classic precursor T-ALL. ETP-ALL retains stem-cell features and has high genomic instability. In a cohort of over 200 T-ALL cases, over 12% met ETP-ALL criteria. These patients had higher minimal residual disease during induction (Figure 4a) and increased rates of induction failure and relapse (Figure 4b).40 To optimally treat these patients, St Jude's now performs allo-SCT in first remission for ETP-ALL. Analyses of T-LBL specimens are needed to determine if ‘ETP-LBL’ exists, and if so, whether a similar therapeutic strategy might be warranted.

(a) Prevalence of minimal residual disease (MRD) during the early phases of therapy for patients with early T-precursor (ETP) vs standard T-ALL. MRD levels were measured by flow cytometry. Horizontal bars indicate median values, if above 0.01%. (b) Kaplan–Meier plots showing cumulative incidence of remission failure or hematological relapse in patients with standard T-lymphoblastic leukemia (T-ALL; red) vs early T-precursor (ETP-ALL; blue) treated on St Jude protocols. Curves start at time of diagnosis. Outcome estimates at 10 years of follow-up are shown; P-values are from the log-rank test. Reprinted from Coustan-Smith et al.40
Molecular basis of T-cell lymphoblastic neoplasia and therapeutic implications
Patients with T-LBL and T-ALL present similar challenges, so understanding the molecular origins of these diseases is vital for efforts to develop better treatment outcomes. It is generally accepted that malignant transformation is a multistep process of genetic mutations that arrest differentiation and allow uncontrolled cell growth.41, 42 In T-lymphoblastic cancers, these genetic events have been queried extensively.43, 44 T-LBL and T-ALL share some cytogenetic and molecular features, implying these diseases are closely related neoplasms.41 As most analyses of T-lymphoblastic neoplasia have studied T-ALL, not all of this impressive body of work may apply to T-LBL. However, efforts have been undertaken to compare the genetic and genomic origins of these related diseases, and they reveal both shared features and potentially important differences.45, 46
Juxtaposition of the TCRβ or TCRαδ enhancers with proto-oncogenes via chromosomal translocations can mis-activate many transcription factors or repressors.47 Such TCR-driven translocations are seen in about 33% of T-ALL cases, and dysregulate basic helix-loop-helix and homeobox transcription factors, LIM-only domain transcriptional regulators, or other oncogenes like NOTCH1 and MYB. Non-TCR translocations are also described which create oncogenic fusion proteins such as MLL-ENL, CALM-AF10, and SET-NUP214.48, 49
Expression profiling in T-ALL supports the paradigm that oncogenic transcription factors can disrupt the normal pathways governing cell proliferation, differentiation, and survival during T-cell development.50, 51, 52 As specific translocations and expression patterns are associated with T-cell developmental arrest at distinct stages, it is believed that these genetic events can define different molecular subtypes of T-ALL.44
However, two other common genetic lesions are present across many T-ALL subtypes. The most prevalent genetic abnormality in T-ALL is inactivation of CDKN2A and CDKN2B at chromosome 9p21. Deletions at this locus occur in >70% of T-ALL.53 Another frequent aberration is activating mutation(s) in the NOTCH1 proto-oncogene, which are seen in over half of T-ALL cases.54 Other rare but recurrent genetic lesions in T-ALL include mutations activating JAK1, FLT3, and RAS,55, 56, 57 or inactivating the tumor suppressors NF1, PTEN, and WT1.58, 59, 60 Mutations in these genes are infrequent, so it is not yet clear whether they occur in several T-ALL types like CDKN2A/N2B and NOTCH1.
NOTCH1 activation is important to T-ALL pathogenesis. NOTCH1 normally promotes T-cell lineage commitment by lymphoid progenitors,61 and blocking its signaling in progenitors impedes T-cell development, instead favoring the B-cell lineage.62, 63 A role for NOTCH1 in T-ALL was first suspected because of a rare translocation, t(7;9)(q34;q34.3). This rearrangement coupled the TCRβ locus to ICN1, the IntraCellular portion of NOTCH1, a constitutively active form of the protein.64 NOTCH1's role was further substantiated by animal models where NOTCH1 and ICN1 can induce T-ALL and T-LBL in vivo.65, 66
Scrutiny of the NOTCH1 pathway has led to a novel therapeutic strategy. Cell surface NOTCH1 undergoes proteolysis to generate active ICN1, including a cleavage by the enzyme gamma-secretase. Consequently, gamma-secretase inhibitors (GSIs) inhibit NOTCH1 signaling (Figure 5). GSIs block growth in some T-ALL cell lines and cause cell cycle arrest and apoptosis of primary T-ALL cells.54, 67 Added to glucocorticoids, GSIs show potent anti-T-ALL effects in vivo, and also diminish the gastrointestinal toxicity seen with GSIs alone. Combining GSIs with glucocorticoids can also induce apoptosis in previously glucocorticoid-resistant T-ALL.68 Unfortunately, loss of the tumor-suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) confers GSI resistance to T-ALL cell lines.59 However, as PTEN-null T cells are highly sensitive to AKT inhibitors, combined NOTCH1 and phosphatidylinositol 3-kinase (PI3K)/AKT therapies are also being investigated in T-ALL.59, 69

Targeted treatment strategies for T-LBL. Glucocorticoid binding causes nuclear translocation of cytoplasmic receptors, which then bind glucocorticoid response elements (GREs) to promote transcription of pro-apoptotic genes, leading to cell death. Other drugs block pathways needed for growth and survival of malignant T-cell lymphoblasts. GSIs prevent release of intracellular notch (ICN1) from membrane-tethered heterodimeric NOTCH1 protein. Combined glucocorticoid and GSI therapies show synergy, but pten deletions can subvert reliance upon activated NOTCH1. Inhibitors of phosphatidylinositol 3-kinase (PI3K), AKT, and mTOR counteract this, thereby restoring GSI-sensitivity. One of the activities of ICN1 is to augment NF-κB signaling. Blocking proteosomal degradation can stabilize inhibitors of NF-κB (IκB), allowing Bortezomib to cooperate with GSI as well (figure design by Yaya Chu, New York Medical College).
Targeting of the mTOR pathway is similarly being explored, as simultaneous GS- and mTOR-inhibition shows synergy in T-ALL.70, 71 Similarly, because NOTCH1 regulates nuclear factor kappaB (NF-κB) signaling, GSIs synergize with bortezomib, which inhibits NF-κB (Figure 5).72 Another target of NOTCH1 is CCR7, a chemokine receptor that mediates CNS infiltration by T-ALL.73 As the CNS represents a relatively common site of relapse, GSIs may be able to prevent CNS recurrence. Although promising, clearly, it remains to be seen whether these ideas will improve T-ALL outcomes, and if they can be extrapolated to T-LBL treatment.
CDKN2A/N2B and NOTCH1 mutations are prevalent in T-ALL, but not universal. Thus, they fall short of an absolute molecular explanation for all T-lymphoblast transformation. Given the vast diversity of additional aberrations already described, it is likely that other genetic lesions in T-cell malignancy are yet to be identified. Certainly, future discoveries may generate further therapeutic options. Notably, additional animal models of T-LBL offer promising new tools to expand our knowledge of the molecular basis of this disease.66, 74, 75
ALCL in children and adolescents
ALCL comprises about 10% of pediatric NHL. It was recognized as a distinct disease in the 1980s,76 and much of its biology is now understood.77 ALCL has two presentations: systemic ALCL and primary cutaneous ALCL occurring exclusively in skin.78 Categorized as a mature T-cell lymphoma, most ALCLs have TCR gene rearrangements even if they lack T-cell antigen expression.79 In children and adolescents, systemic ALCL is associated with anaplastic lymphoma kinase (ALK) gene translocations, denoting the disease as ALK+. The most common translocation creates an NPM–ALK fusion gene that joins the nucleophosmin gene upstream to ALK. Less common translocations fusing ALK downstream of other partners are also described (Table 3).80 Both expression profiling and genomic analyses of ALCL suggest that ALK+ and ALK− lymphomas actually represent distinct clinical entities.81, 82 Accordingly, ALK+ and ALK− lymphomas are treated differently and have differing prognoses. Apart from ALK− cutaneous ALCL, which can be successfully managed by surgery alone, ALK+ disease has better outcomes, and this distinction stratifies patients to receive less-intensified therapy.
ALCL is characterized histologically by large, pleomorphic, and multinucleate ‘hallmark cells,’ with horseshoe nuclei, abundant cytoplasm, and peri-nuclear eosinophilia.83 Hallmark cells may resemble Reed–Sternberg cells of Hodgkin lymphoma (HL), but have less conspicuous nucleoli. Morphologic ALCL variants include the common variant (75%) composed chiefly of hallmark cells, the lymphohistiocytic variant (10%) with benign histiocytes admixed with neoplastic cells, and a small cell variant (10%) of primarily small neoplastic cells and only scattered hallmark cells.84 Other subtypes (<5%) include sarcomatoid, signet ring, neutrophil-rich, and giant cell variants.79
Pediatric and adult ALCL have several differences. Over 90% of childhood ALCL is ALK+,79 compared with only 60% of adult ALCL.85 Also, primary cutaneous ALCL is nearly always ALK−, and rare in children.80 Children typically present with advanced stage disease. A compendium of 225 childhood ALCL patients treated on BFM, SFOP, or UKCCSG protocols revealed that 74% were stage III–IV by St Jude criteria and 75% were Ann Arbor stage III or IV. Extranodal disease was common (68%), with skin (26%), bone (14%), and soft-tissues (15%) all relatively frequently involved.86 B symptoms were seen in 54%, consistent with prior reports.86, 87 Despite these aggressive features, both marrow and CNS invasion are uncommon in pediatric ALCL, occurring in <10% and <5% of cases, respectively.85, 86, 88
Current treatments for pediatric ALCL
The optimal approach for limited disease is not established, as both B-NHL and T-ALL regimens show similar efficacy. EFS may be as high as 100% for children with localized ALCL (stage I–II resected) as shown in the NHL-BFM-90 trial using 2 months of combined chemotherapy.89 Previously, St Jude's reported 75% EFS in children with localized CD30+ large cell lymphoma (presumably ALCL) treated with three cycles of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), either with or without maintenance therapy.90
Advanced pediatric ALCL treatment has evolved over the past two decades. Different strategies have achieved 65–75% EFS with either B-NHL protocols or LSA2L2-type therapies (Table 4).89, 91, 92, 93, 94, 95 Other trials have used a doxorubicin, prednisone, and vincristine (APO) backbone. Pediatric Oncology Group (POG)-9315 added intermediate-dose MTX and high-dose cytarabine to APO, but this did not improve efficacy, with MTX and cytarabine causing greater toxicity. Children on the APO-only arm had 75% 2-year EFS.92 COG is evaluating the addition of weekly vinblastine to APO. This stems from encouraging French data in relapsed ALCL, where 10/12 patients responded to weekly vinblastine.96 Unfortunately, a recent COG report showed merely worse myelosuppression with vinblastine, with an equivalent 77% 2-year EFS.97
The European intergroup ALCL99 trial, which is based on the BFM B-NHL treatment strategy, employed a less toxic regimen with high-dose MTX and no IT therapy. They reported 74% 2-year EFS and diminished toxicity.98 Until ALCL99, CNS-negative ALCL treatment usually included IT prophylaxis. With their recent success in preventing CNS relapse without IT treatment, perhaps reducing the number of high-dose MTX administrations should also be considered. For rare ALCL patients with CNS disease, CRT doses of 18–24 Gy, in addition to high-dose MTX and/or cytarabine plus IT chemotherapy, has been used.89, 91
Several prognostic factors have been identified in children with ALCL. Clinical features associated with poor outcome include visceral organ (liver, lung, or spleen) or mediastinal involvement, elevated lactate dehydrogenase (LDH), and diffuse skin disease.86, 91, 99 In addition, correlation between ALCL biology and outcome is now possible via PCR-based MDD detection of the NPM-ALK transcript. Higher levels of this mRNA in marrow predicted a 71% chance of relapse, in contrast to 18% in MDD-negative patients.100 Meanwhile, detection of endogenous anti-ALK antibodies are inversely correlated to ALCL dissemination and risk of relapse in ALK+ disease, implying that ALK may be a potential immunotherapeutic target.101, 102 Additional results from the recent ALCL99 trial reveal that two less common ALCL variants, small cell and lymphohistiocytic, have a high risk of treatment failure, independent of clinical risk factors.103 Further advances in our ability to identify patients at high risk for relapse will enable appropriate application of intensified front-line therapies to potentially improve outcomes.
Relapsed ALCL is quite different from other pediatric NHL subtypes. Chemosensitivity of recurrent disease is a hallmark of childhood ALCL, rendering salvage strategies generally effective.104, 105 Clinical behavior after relapse varies from aggressive to indolent, with a waxing and waning course.106 Historically, ALCL has been considered prone to late relapse, like other lymphomas seen in adult patients.107 However, a recent report describing 74 children and adolescents with recurrent ALCL revealed a median time of only 7.1 months between diagnosis and relapse. Of 58 patients who achieved remission after front-line therapy, only 18 recurred >1 year from original diagnosis.108 The prognostic significance of early vs late ALCL relapse remains an open question, with recurrences >1 year after diagnosis perhaps having superior outcome,96 but this finding has not been consistent in all cohorts.109 Late relapse again showed trends of better EFS and OS in the most recent report,108 but because these studies all compile data from patient groups who received differing initial treatments and relapse therapies, no definitive answer is yet available.
In a series of three French clinical trials over two decades, relapse therapies varied widely from single-agent vinblastine to multi-agent treatment to fully ablative chemotherapy with auto- or allo-SCT. Early relapses and failures with intensive initial regimens carry higher risk for treatment failure. Three-year disease-free survival did not differ in patients who received ablative SCT in CR2 vs chemotherapy alone.96, 110 However, recent studies using allo-SCT in relapsed and refractory ALCL are promising, with 75% 3-year EFS seen in one 20 patient cohort.111, 112
Progress in ALCL treatment lies in both new targeted agents and optimizing risk stratification to identify patients who may benefit from front-line allo-SCT. The CD30 antigen is one compelling novel therapeutic target. Present in nearly all cases of childhood and adolescent ALCL, CD30 expression is controlled by ALK (Figure 6). SGN-35, an anti-CD30 monoclonal antibody, has shown even greater success than its predecessor SGN-30.113, 114 Phase I trials have examined SGN-35 monotherapy in patients with refractory/recurrent CD30+ ALCL and HL.115 Dose-limiting toxicities (thrombocytopenia, hyperglycemia, and febrile neutropenia) were used to derive a treatment dose of 1.8 mg/kg given every 3 weeks. At this dose, SGN-35 was generally well tolerated, with patients exhibiting primarily grade 1 or 2 adverse effects consisting of fatigue, nausea, neutropenia, and peripheral neuropathy (33% of patients) as well as fever, headache, back pain, and cough (25% of patients).115 At higher doses, one trial showed 7/8 patients achieving CR. In a second trial, also at higher SGN-35 doses, 7/28 patients achieved CR with an overall response rate (ORR) of 46%.116, 117 These responses to monotherapy in heavily pre-treated patients offer exciting hope that adding SGN-35 to combination therapy will yield even better results in front-line therapy.

Model of ALK and CD30 signaling. In ALCL, the NPM–ALK fusion protein governs CD30 expression by phosphorylating signal transducer and activator of transcription 3 (STAT3). NPM–ALK also regulates extracellular signal-regulated kinase 1 (ERK1)- and ERK2-mediated activation of JUNB protein. Phosphorylated STAT3 and activated AP1 complexes containing JUNB then cooperate to enhance CD30 transcription. NPM–ALK impedes CD30 signaling and NFκB activation by sequestering tumor necrosis factor receptor-associated factor 2 (TRAF2) away from CD30. This occurs via dimerization of NPM-ALK with wild-type (WT) NPM. CD30 engagement normally causes TRAF2 degradation. Engagement of CD30 on ALCL cells results in activation of both the canonical and alternative NFκB pathways, which causes apoptosis and p21-mediated cell-cycle arrest. A clinical trial is testing the use of anti-CD30 antibodies (red arrow) in ALCL109. Reprinted from Chiarle et al.77
Development of other novel therapeutics has been fostered by advances in understanding of ALCL biology. Targets include the ALK protein itself, via small molecule and antibody-mediated inhibition. The ALK inhibitor NVP-TAE684 both prevented tumors and promoted pre-induced tumor regression in separate mouse models.118 Another kinase inhibitor, the dual MET/ALK inhibitor Crizotinib, also shows promise in ALK+ ALCL. Spurred on by the finding that some non-small cell lung carcinoma patients have a related translocation involving the ALK gene (EML4-ALK), Crizotinib is being developed as a targeted agent for cancers driven by constitutive ALK activity and was recently approved by the FDA in patients with lung cancer with the presence of an ALK mutation. Exciting results with this medication in refractory and relapsed ALK+ ALCL suggest this strategy holds great clinical promise.119
These data with ALK inhibitors agree with findings that circulating anti-ALK antibodies predict lower relapse rates, and have led to vaccination studies using the ALK antigen.120 Other investigations have focused on different aspects of ALCL biology. Although somatic mutations in ALCL are rare, sequencing identified 6 different mutations in 12/44 (27%) patients with ALK+ ALCL. Perforin (PRF1) mutations were particularly prevalent in ALCL, compared with control subjects where the A91V mutation was infrequently seen (P<0.01).121 PRF1 mutations occur in other NHLs, and may impair cytotoxicity because of abnormal PRF1 conformation caused by the A91V mutation.122 Additionally, amplification of the sonic hedgehog (SHH) gene in a subset of ALK+ ALCL123 has been shown to lead to dysregulated SHH signaling.
Proteomic analyses of ALCL complement these landmark genomic studies and have been instrumental in understanding the ALK protein network.124, 125 Proteomic signatures from cells expressing the NPM–ALK fusion show alterations in cellular processes including proliferation, ribosome synthesis, survival, apoptosis, angiogenesis, and cyto-architectural organization.124 Further studies revealed loss of cell adhesion caused by NPM-ALK expression in a kinase-dependent manner, as well as sensitivity of ALK+ ALCL to inhibition of the RAS, extracellular signal-regulated kinase (ERK) and FK506 binding protein-rapamycin-associated protein (FRAP) mTOR signaling pathways.124 Understanding the myriad effects of NPM-ALK on these cellular pathways offers key insights into ALK+ ALCL biology.
Several potential therapeutic targets in ALCL have also been identified by proteomic studies. Constitutive CD25 expression by pediatric ALCL has led to in vitro investigations of the anti-CD25 agent, denileukin diftitox.126, 127 Identification of other downstream pathways interconnected with ALK has led to studies examining disruption of ALK-associated pathways. Heat shock protein 90 (Hsp-90) and the PI3K/Akt pathway both have interactions with the ALK protein network. In vitro studies targeting Hsp-90 cause increased degradation of NPM-ALK and apoptosis of ALCL cell lines,128 while PI3K/Akt-null mice injected with NPM-ALK+ cells have impaired tumor formation.129 These advances in the genomics and proteomics of ALCL have enabled new therapeutic approaches. Combining new and less-toxic therapies offers exciting opportunities to enhance the efficacy and safety of therapies for future ALCL patients.
PTCL-NOS in children and adolescents
PTCLs are a heterogeneous group of cancers arising from mature T and NK cells. These diseases include all forms of T-cell lymphoma except T-lymphoblastic disease. PTCL-NOS is the second most common PTCL after ALCL. In children, PTCL-NOS accounts for about 1% of NHL cases.130 In contrast, it comprises about 4% of adult NHL.131
The biology of PTCL-NOS shows great diversity. Identifying the normal cells that correspond to the cellular origin of different PTCL-NOS diseases has been challenging. Several pan-T-cell antigens, as well as cytotoxic and activated T-cell markers, are seen across different T-cell developmental stages.132 Although several cytogenetic abnormalities have been observed in PTCL-NOS, a specific schema of clinically useful classification has not been established.133 In fact, ALK translocations in ALCL represent the only recurrent genetic aberration in PTCL.134 Furthermore, apart from the frequent clonal TCR rearrangements seen in PTCL-NOS, other molecular changes vary and display no consistent pattern.3
Recent studies in PTCL-NOS biology have led to exciting findings. Genomic analyses of tumor specimens have linked reduced expression of NF-κB genes to shortened survival.135 Another report used expression profiling to stratify PTCL-NOS into three subgroups: one notable for cyclin D2 expression, another with NF-κB1 and BCL-2 overexpression, and a third with high expression of genes in the interferon/Janus kinase/signal transducers and activators of transcription pathway.136 Although further studies are needed to confirm these results and deepen our understanding as pertains to lymphomagenesis, they represent potentially important biological observations in PTCL-NOS.
The clinical presentations of PTCL-NOS can be as varied as its biological characteristics. Patients often exhibit generalized lymphadenopathy (frequently cervical) or extranodal disease of the liver, spleen, skin, and marrow. Most patients have advanced stage disease with high LDH at diagnosis, as well as B-symptoms like fever, night sweats, and/or weight loss. These features derive from increased cytokine production by malignant T cells, and can even cause the hyper-inflammatory signs of hemophagocytic syndrome.3
Treatment strategies for PTCL, including PTCL-NOS, are similarly varied. It has been challenging to develop and establish effective regimens because clinical experience in pediatric PTCL is sparse and individual studies are hampered by too few patients. Overall, while a few PTCL types require unique approaches (for example, adult T-cell leukemia/lymphoma (ATLL) and Epstein–Barr virus (EBV)-positive T-cell lymphoproliferative disease (T-LPD) of childhood, ALCL, and some cutaneous entities), other PTCL patients receive similar therapies irrespective of subtype.
The largest pediatric PTCL cohorts are studies from the USA and the UK. The COG analyzed 20 pediatric patients over a 9-year period. This cohort had 12 PTCL-NOS patients; other diagnoses included extranodal NK/T-cell lymphoma nasal type, subcutaneous panniculitis-like T-cell lymphoma (SPTCL), and enteropathy-type T-cell lymphoma. Treatments varied based upon clinical staging. Advanced stage III–IV disease received a regimen of doxorubicin, prednisone, vincristine, mercaptopurine, and MTX ± alternating therapy with high-dose cytarabine and intermediate-dose MTX. Localized stage I–II patients were treated with CHOP. Patients with localized disease fared well; only 2 relapsed and 9/10 survived. In advanced disease cases, 6/10 relapsed and only 5 survived. These results are markedly better than typically seen in adults with PTCL. However, while CHOP seems adequate for localized cases, the 50% OS of advanced stage patients leaves room for improvement.137
The UK experience was similar. This study retrospectively analyzed 25 PTCL cases in children and adolescents over 20 years. A similar distribution of PTCL subtypes was seen with 68% categorized as PTCL-NOS. Remaining diagnoses included angioimmunoblastic T-cell lymphoma (AITL), angiocentric PTCL, and SPTCL. Patients received either B-NHL CHOP-like regimens or T-ALL therapy. In children with PTCL-NOS, 9/12 survived after T-ALL therapy, but 4/5 died following B-NHL treatment. Like the COG study, when analyzed by extent of disease, most patients (9/12) with local stage I–II disease survived, but only 6/12 advanced stage patients survived. The authors concluded that T-ALL therapy was appropriate for children with PTCL-NOS, but prognoses for children with advanced stage disease remain guarded.130
Adults with PTCL generally have inferior outcome. Five-year OS in adult studies ranges from 25 to 45% with many treatment strategies used.3, 138, 139, 140 Chemotherapy regimens for adults frequently include CHOP-like therapy, and incorporation of cytarabine, cisplatin, and etoposide have not bolstered survival rates. Owing to poor outcomes, high-dose chemotherapy followed by auto- or allo-SCT has also been explored. High-dose chemotherapy with auto-SCT has been attempted in both the initial diagnosis and relapse setting. In patients with refractory or recurrent disease, OS after auto-SCT is about 33%.141 Some studies report slightly higher OS rates of 39–48%, but these cohorts included ALCL patients, who typically do well with auto-SCT salvage therapy. When separated out, non-ALCL PTCL cases had OS closer to 30% in these same studies.142 Attempts with front-line auto-SCT in newly diagnosed PTCL have yielded only marginally better results. A large Italian study in high-risk PTCL patients who received up-front auto-SCT reported long-term OS rates of 39%. OS was hampered by the fact that many patients progressed before auto-SCT and never received transplant. But, in patients who did receive high-dose chemo and auto-SCT, 12-year disease-free survival rate was improved to 55%.143
Allo-SCT has also been used in high-risk PTCL. This strategy provides the advantages of infusing a known lymphoma-free graft, as well as potential graft-vs-lymphoma effect. Although patients receiving allo-SCT have proven lower relapse risks (compared with auto-SCT), historically high rates of transplant-related mortality in fully ablative conditioning regimens have offset any survival advantage.144 Better success has been achieved using allo-SCT with reduced intensity conditioning regimens. In a pilot study of 17 patients with refractory and recurrent disease (eight after failed auto-SCT), 3-year progression-free survival was 64%, and only one patient experienced transplant-related mortality.145 These results offer hope for this notoriously recalcitrant disease.
Establishing prognostic criteria for high-risk PTCL is important to identify patients who may be candidates for intensified treatment. As mentioned above, pediatric studies show patients with advanced stage III–IV disease have markedly worse outcomes than localized stage I–II disease.130, 137 In adults, the International Prognostic Index (IPI) provides risk stratification. The IPI incorporates features such as stage, LDH level, patient age, multifocal extranodal disease involvement, and performance status. Its validity in adult PTCL-NOS patients was verified in a Canadian study of 117 patients. Those with an IPI score of 0–1 (30% of the cohort) had 64% 5-year OS, while those with IPI >2 (70% of patients) had OS of only 30%.140 A Prognostic Index for PTCL-NOS (PIT) model was established based on IPI principles and results from a series of nearly 400 patients. Four variables were used to stratify prognostic groups: age, performance, LDH, and marrow involvement. Five-year OS rates by PIT score were: 0, 62%; 1, 53%; 2, 33%; and >2, 18%.146 In applying this schema to pediatric patients, advanced disease markers like high LDH and marrow involvement would portend worse prognoses.
Considering the poor outcomes of high-risk PTCL patients, new therapeutic strategies are desperately needed. Agents being investigated include compounds from various pharmacological classes including nucleoside analogs, antifolates, histone deacetylase and proteasome inhibitors, anti-angiogenesis agents, and monoclonal antibodies. Of nucleoside analogs, pentostatin and gemcitabine have been most promising. A single institution study of 10 patients demonstrated 60% ORR to gemcitabine with refractory/recurrent disease.147 Nelarabine, on the other hand, achieved only 11% ORR and showed marked toxicity.148 The histone deacetylase-inhibitor depsipeptide showed a 26% ORR in a phase II trial,149 and the anti-angiogenic bevacizumab has induced CR in case reports of refractory/recurrent AITL.150, 151 Pralatrexate, an anti-folate, has shown promise in phase I–II trials with 47% ORR in 26 patients with T-cell lymphoma, many with PTCL-NOS.152 This work has expanded to a multicenter trial, and interim data show a 29% ORR in 65 patients, with 11% of patients achieving CR.153 Combining liposomal doxorubicin with the proteasome inhibitor bortezomib has proven safe and effective in other advanced hematological malignancies.154 Proteasome inhibitors promote apoptosis and other anti-proliferative properties by inhibiting the NF-κB pathway. Bortezomib thus is an attractive candidate drug for PTCL-NOS diseases with demonstrated overexpression of NF-κB1 target genes.136 Bortezomib has also been studied in refractory and relapsed cutaneous T-cell lymphomas, where 67% ORR was seen with single-agent therapy. Although most cases in that trial were mycosis fungoides, one of two PTCL-NOS patients also responded.155 Current efforts add bortezomib to first-line PTCL agents,156 with in vitro testing of cutaneous PTCL demonstrating synergy between bortezomib and the histone deacetylase inhibitor suberoylanilide hydroxamic acid.157
Monoclonal antibody therapies have gained considerable momentum in the treatment of pediatric lymphomas over the past decade. These successes have fostered interest evaluating them in PTCL as well. Alemtuzumab is a humanized monoclonal antibody targeting CD52, an antigen expressed on most lymphocytes. It showed a 36% ORR as a single agent in a pilot study of 14 patients with refractory/recurrent PTCL. However, excessive infectious complications prompted premature closure of this study after five patients suffered from treatment-related mortality.158 More recently, alemtuzumab was combined with CHOP as front-line therapy. This regimen had 41% 1-year EFS, with an acceptable toxicity profile.159 Other monoclonal antibodies have been explored in cutaneous T-cell lymphomas and their role in PTCL is unclear. These include the anti-CD4 antibody zanolimumab, and the anti-CD25 antibodies daclizumab and denileukin diftitox.160, 161 Despite this wide arsenal of novel therapeutics and the guardedly optimistic results achieved by reduced intensity allo-SCT in PTCL, it remains crucial to pursue the biologic underpinnings of these diseases. Given their rare occurrence, it is unlikely they will ever be amenable to large-scale clinical trials to empirically test all possible therapies. It will therefore continue to be important to identify biological markers that provide scientific rationale to help guide treatment choices that optimize therapeutic outcomes for this challenging and diverse group of patients.
Rare T- and NK-cell lymphomas in children and adolescents
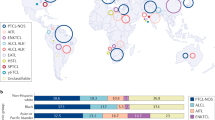
A number of rare lymphomas originating from mature T and NK cells account for an incredibly diverse spectrum of malignancies (Figure 1).162 These lymphomas vary widely by geography and ethnicity with a considerably higher prevalence in Asia.131 Although certain diseases have well-described etiologies (for example, human T-cell lymphotropic virus-1 (HTLV-1) in ATLL and EBV in EBV+ T-LPD), for most rare T- and NK-cell lymphoams, a link is not determined.3
Over 20 mature T- and NK-cell lymphomas are listed in Table 1. The 2008 WHO Classification of Lymphoid Neoplasms subdivides T- and NK-cell lymphomas into four clinical presentation-based groups: leukemic/disseminated, extranodal, extranodal-cutaneous, and nodal. Among mature T- and NK-cell lymphomas, the leukemic/disseminated group includes HTLV-1+ ATLL and systemic EBV+ T-cell LPD of childhood. Extranodal diseases include extranodal NK/T-cell lymphoma, hepatosplenic lymphoma, enteropathy-associated T-cell lymphoma, and others. Cutaneous forms include mycosis fungoides/Sezary syndrome, primary cutaneous CD30+ LPD (discussed previously with ALCL), SPTCL, and primary cutaneous gamma/delta (γ/δ) T-cell lymphoma. Nodal entities include ALCL, AITL, and PTCL-NOS. The remainder of this review will discuss several rare T- and NK-cell lymphomas in children and adolescents.
Hepatosplenic γ/δ T-cell lymphoma
Characterized by involvement of liver, spleen, and marrow, hepatosplenic γ/δ T-cell lymphoma is a rare and aggressive peripheral T-cell neoplasm. It is a disease of young adults with distinct male predominance. Marked hepatosplenomegaly with frequent marrow infiltration is typical, and patients notably present without lymphadenopathy. Up to 20% of cases arise in patients with chronic immune suppression, often after solid organ transplantation or prolonged exposure to azathioprine and infliximab. Recently, EBV-negative T-cell lymphomas with features of hepatosplenic T-cell disease were reported in inflammatory bowel disease patients receiving infliximab.163 Karyotypic studies consistently show isochromosome 7q, often with trisomy 8, and TCR rearrangements. Expression profiling showed high levels of NK-cell messages such as killer cell immunoglobulin-like and lectin-like receptors, compared with PTCL specimens with α/β phenotype.164 Ontology analysis revealed enrichment of genetic pathways for cellular defense responses, signal transduction, receptor activity, transmembrane receptor activity, and immunoglobulin G binding. Prognosis is grim, as most patients experience recurrent disease after brief responses to conventional chemotherapy.165 Long-term remissions have been reported in pediatric and adult patients receiving allo-SCT.166, 167
Subcutaneous panniculitis-like T-cell lymphoma
The characteristic presentation of subcutaneous lesions distinguishes SPTCL. Although more common in adults, there are several reports of SPTCL in children and adolescents. Patients present with atypical T-cell infiltrations confined to subcutaneous tissues, resulting in panniculitic-appearing skin nodules and/or ulcerated lesions. SPTCL is usually local, with organ or node involvement uncommon. Clinical courses can be protracted or aggressive, and patients may present with symptoms of hemophagocytic syndrome.168 T-cell phenotyping has delineated two distinct entities. The α/β type is more common and differs from the ominous primary cutaneous γ/δ T-cell lymphoma. Cutaneous γ/δ disease has similar clinical features to SPTCL, but is considered a variant of the hepatosplenic γ/δ T-cell lymphomas discussed above. α/β SPTCL and cutaneous γ/δ disease have different immunophenotypes (α/β: CD8+56−/βF1+; γ/δ: CD8−56+/βF1−), and α/β disease has less frequent features of hemophagocytosis and markedly better outcome. Patients with α/β SPTCL have been treated with multi-agent regimens with reasonable efficacy, whereas patients with cutaneous γ/δ disease have poor outcomes like cases of γ/δ hepatosplenic lymphoma.169
EBV-positive T-cell lymphoproliferative disorders
The 2008 WHO classification incorporated two types of EBV-associated T-cell LPD affecting the pediatric population: systemic EBV+ T-cell LPD of childhood and hydroa vacciniforme-like T-cell lymphoma. Both diseases occur predominantly in Asians and individuals of Native American descent from various regions of Latin America.
Systemic EBV-positive T-cell LPD of childhood
Most patients present with symptoms of a systemic hyper-inflammatory response with disease mimicking the presentation of hemophagocytic lymphohistiocytosis. Acute onset of fever and malaise often precedes the eventual development of hepatosplenomegaly and liver failure, with or without lymphadenopathy. This disease has rapid progression to multi-organ failure, hemophagocytic syndrome and death. Chronic active EBV infection has been documented in some patients before developing EBV+ T-cell LPD, but most cases are fulminant and arise with acute primary EBV infection. Fulminant cases are CD8+, whereas chronic active EBV-associated cases are CD4+. EBV-encoded RNA-1 is positive in neoplastic T cells that exhibit clonal TCR gene rearrangement and harbor EBV in a clonal episomal form. This severe illness is characterized by clonal proliferation of EBV-infected T cells with an activated cytotoxic phenotype. All cases are type A EBV, with either the wild type or a specific 30 base pair-deletion of the viral LMP1 gene. Frequent sites of involvement include liver, spleen, nodes, and marrow. The typical immunophenotype of tumor cells is CD2+CD3+CD56−. Transient chemotherapy responses are reported, but almost all cases have rapid disease progression ending in death.170
Hydroa vacciniforme-like lymphoma
Hydroa vacciniforme-like T-cell lymphoma is an EBV+ cutaneous malignancy occurring mostly in children. Linked to sun sensitivity, this condition primarily affects sun-exposed skin, and the face in particular. Papulovesicular eruptions precede ulceration and scarring. The clinical course is variable and skin lesions may be recurrent. Late in the disease, systemic symptoms develop such as fever, wasting, lymphadenopathy, and hepatosplenomegaly. Similar to EBV+ T-cell LPD, neoplastic cells have clonal TCR rearrangements and harbor EBV in a clonal episomal form. It is not clear whether the severe mosquito-bite allergy seen in chronic active EBV infection, which has similar cutaneous lesions of NK-cell derivation, is related to the hydroa vacciniforme-like lymphoma or a distinct entity in the spectrum of EBV-associated disorders. Both diseases are considered part of the spectrum of severe chronic active EBV, demonstrating a broad variation in clinical aggressiveness.171 Allo-SCT provides the only curative option for patients, but it remains to be seen whether novel EBV-specific cytotoxic T-lymphocyte (CTL) immunotherapies have a role in this unique disorder.172
Adult T-cell leukemia/lymphoma
ATLL is another rare T-cell malignancy associated with viral infection. It is associated with HTLV-1 infection, a virus endemic to regions including Japan, the Caribbean, parts of South America, West and Central Africa, and some of the Oceanic Islands.173 Infection with the HTLV-1 retrovirus carries an approximate 5% lifetime risk of developing ATLL.174 Although trophic for CD4+ T cells, HTLV-1 infection does not cause cytopathic destruction of CD4+ T cells. Rather, upon integration, it governs its own transcription using its regulatory protein Tax. Tax interacts with host proteins that can promote lymphomagenesis including the NF-κB pathway,175 trans-activating proteins that promote T-cell proliferation and apoptosis inhibition, proteins repressing cell cycle control and DNA repair, and multiple tumor suppressors.176 A second retroviral protein, HBZ, is also linked to the NF-κB177 and T-cell proliferation pathways.178 These retroviral proteins contribute to HTLV-1 pathogenesis, but the virus itself is not considered to be directly oncogenic. HTLV-1 infection induces humoral and cellular immune responses, and like EBV-induced lymphomas, impaired anti-HTLV-1 CTL responses are associated with ATLL.179, 180, 181
There are four clinical variants of ATLL: acute, lymphomatous, chronic, and smoldering. Acute is most common, with patients presenting with disseminated disease, systemic symptoms, organomegaly, and circulating malignant cells. The ATLL immunophenotype is usually CD2+/4+/5+/25+, with loss of pan-T marker CD7. CD8+ variants also exist.182 ATLL patients are typically immunocompromised with increased risk of opportunistic infection. Prognosis for acute and lymphomatous ATLL is dismal with median survival <1 year using standard chemotherapy.183 Evidence for a link between HTLV-1 and ATLL is substantiated by improved prognosis (30–40% survival) in patients treated with allo-SCT.184 Clearance of HTLV-1 proviral loads and suggestion of a graft-vs-lymphoma effect have been documented in multiple clinical trials, revealing promise for this immunotherapeutic approach.185
Extranodal NK/T-cell lymphoma
Extremely rare in the western hemisphere, extranodal NK/T-cell lymphoma occurs primarily in Far and Southeastern Asia. The majority of cases (60–90%) present with nasal or upper airway masses, but extranasal disease is also described. Virtually all cases are EBV+, with cytoplasmic CD3 and CD56 surface expression. Most cases also test positive for cytotoxic proteins such as TIA-1, granzyme B, and perforin.186 Genomic profiling studies have shown several genomic losses and gains associated with NK-cell malignancies—deletion in 6q21 being most common.187 Meanwhile, expression profiling reveals high transcript levels for genes in the p53 pathway, anti-apoptosis pathways, and cytokine receptors in NK lymphoma cell lines as compared with normal NK cells. A tandem mass spectrometry-based proteomic analysis established overexpression of potential targets implicated in disease pathogenesis, including HSP90 and proliferating cell nuclear antigen. Moreover, in vitro inhibition of HSP90 with geldanamycin demonstrates reduced cell viability and increased apoptosis.188 NK-cell lymphomas are very aggressive and have poor outcomes, with 5-year OS of 32% in recent studies. Of note, extranasal NK disease has strikingly worse prognosis (9% 5-year OS) vs the more common nasal type (42% 5-year OS).189
Angioimmunoblastic T-cell lymphoma
In contrast to the extranodal presentation of NK-cell lymphoma, AITL usually presents with lymphadenopathy. Clinical hallmarks include systemic disease features, with most patients manifesting generalized lymphadenopathy, B symptoms, elevated LDH, and hepatosplenomegaly. Many patients also present with pruritic skin rash. EBV and human herpesvirus-6 are detectable in many cases, but the implications of these findings are not clear.190 Histologically, AITL is characterized by proliferation of high endothelial venules and follicular dendritic cells. Tumors arise from CD4+ cells belonging to the follicular helper T-cell lineage.191 Determination of AITL molecular signatures has yielded a classifier schema comprised of three prominent patterns: a B-cell signature, a follicular dendritic-cell signature, and a cytokine signature, reflecting the relationship between the tumor cells and their microenvironment. Gene expression analyses in AITL show activation of the NFκB pathway and enrichment of genes denoting dendritic cell function and interleukin-6 signaling. Importantly, this study identified prognostic features predictive of poor outcome in the form of gene signatures reflecting immunosuppression: tolerogenic dendritic cells and CD31− stromal cells. Additionally, involvement of NFκB may provide a potential means to develop novel treatments. Like extranodal NK-cell lymphoma cases, AITL patients have poor survival outcomes with 5-year OS approximating 30%.192
Rare T- and NK-cell lymphomas summary
Ultimately, PTCL lymphomas remain a diagnostic and therapeutic challenge. Although biologic studies have been performed in adult series, the rarity of pediatric PTCL cases hinders large-scale investigations on both the basic science and clinical fronts. To address this problem, COG has initiated collection of rare pediatric lymphomas and LPDs via study ANHL04B1. A central institutional review board facilitates prompt patient enrollment and expedited pathologic review of specimens to confirm these scarce diagnoses by a three-expert hematopathology panel. This study also gathers information regarding therapy chosen by the treating physicians and outcome. As these cases require individualized care, no specific therapy is linked to the ANHL04B1 study. Instead, its aim is to collect diagnostic, treatment, and outcome data on these rare diseases in children and adolescents. Meanwhile, as new therapeutics are continually tested in adults, it is likely that extrapolation from these studies will be necessary to improve outcomes in rare pediatric cases. As our biologic understanding of these diseases advances and new targets emerge, therapies will become more enlightened. Until such regimens have been proven, children with all subtypes of advanced stage, refractory, and recurrent PTCL may benefit from induction therapy followed by allo-SCT with reduced intensity conditioning regimens.
Conclusion
T- and NK-cell lymphomas in pediatric patients are a complex group of neoplasms arising from precursor T lymphoblasts or other mature T- and NK-cell subsets. T-LBL is the most common, representing one-third of childhood and adolescent NHL. Although the genomic and genetic underpinnings of T-ALL have been thoroughly scrutinized, much less is known about T-LBL molecular pathogenesis. It has been generally accepted that T-ALL and T-LBL are different manifestations of the same disease, but recent studies show that T-ALL and T-LBL have distinct genetic aberrations and non-identical expression patterns. Advances in T-ALL biology have led to novel therapeutics currently under investigation, but a deeper understanding of T-LBL and its relation to T-ALL is needed to enhance treatment outcomes for these patients. Among mature T-cell neoplasms, ALK+ ALCL is most prevalent. The genetic aberration defining this disease is, logically, a target for therapeutic intervention. ALK tyrosine kinase inhibitors are currently being tested in relapsed ALCL. Pediatric PTCLs arising from mature T cells are so rare that they are challenging to study. However, our increasing knowledge of their biologic features, coupled with attention to adult treatment strategies, will undoubtedly offer insights for pediatric PTCL. As our understanding of PTCL molecular pathogenesis continues to grow, there is cause for optimism. Ultimately, there is no inherent reason why childhood and adolescent T- and NK-cell lymphomas should not be equally curable to other pediatric lymphocyte cancers like HL, B-NHL, and ALL.
References
T lyphoblastic leukaemia/lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2008, pp. 176–178.
Mature T- and NK-cell neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2008, pp. 270–319.
Rodriguez-Abreu D, Filho VB, Zucca E . Peripheral T-cell lymphomas, unspecified (or not otherwise specified): a review. Hematol Oncol 2008; 26: 8–20.
El-Mallawany NK, Van Vlierberghe P, Ferrando AA, Lim M, Cairo MS . T-cell malignancies in children and adolescents: state of the clinical and biological science. In: Foss F (ed). T-Cell Lymphoproliferative Disorders. Humana Press: New York, 2012 (in press).
Cairo MS . Non-Hodgkin's lymphoma and lymphoproliferative disorders in children. In: Carroll WL, Finlay J (eds). Cancer in Children and Adolescents. Jones and Bartlett Publishers: Sudbury, MA, 2010.
Cairo MS, Bradley MB . Lymphoma. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF (eds). Nelson Textbook of Pediatrics, 18th edn. Elsevier: Phildelphia, 2007, pp. 2123–2126.
Cairo MS, Raetz E, Perkins SL . Non-Hodgkin's lymphoma in children. In: Kufe DW, Bast RC, Hait WN, Hong WK, Pollock R, Weichselbaum RR, et al. (eds). Cancer Medicine, 7th edn. BC Decker Inc.: Hamilton, Ontario, Canada, 2005, pp 1962–1976.
Burkhardt B, Moericke A, Klapper W, Greene F, Salzburg J, Damm-Welk C et al. Pediatric precursor T lymphoblastic leukemia and lymphoblastic lymphoma: differences in the common regions with loss of heterozygosity at chromosome 6q and their prognostic impact. Leuk Lymphoma 2008; 49: 451–461.
Raetz EA, Perkins SL, Bhojwani D, Smock K, Philip M, Carroll WL et al. Gene expression profiling reveals intrinsic differences between T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Pediatr Blood Cancer 2006; 47: 130–140.
Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R et al. Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the Berlin-Frankfurt-Muenster group. J Clin Oncol 2009; 27: 3363–3369.
Reiter A, Schrappe M, Ludwig WD, Tiemann M, Parwaresch R, Zimmermann M et al. Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma: a BFM group report. Blood 2000; 95: 416–421.
Schrappe M, Reiter A, Zimmermann M, Harbott J, Ludwig WD, Henze G et al. Long-term results of four consecutive trials in childhood ALL performed by the ALL-BFM study group from 1981 to 1995. Berlin-Frankfurt-Munster. Leukemia 2000; 14: 2205–2222.
Bertsch H, Rudoler S, Needle MN, Malloy P, Sutton L, Belasco J et al. Emergent/urgent therapeutic irradiation in pediatric oncology: patterns of presentation, treatment, and outcome. Med Pediatr Oncol 1998; 30: 101–105.
Link MP, Weinstein HJ . Malignant non-hodgkin lymphomas in children. In: Pizzo PA, Poplack DG (eds). Principles and Practice of Pediatric Oncology, 5th edn. Lippincott, Williams and Wilkins: Philadelphia, 2006, pp. 722–747.
Meadows AT, Sposto R, Jenkin RD, Kersey JH, Chilcote RR, Siegel SE et al. Similar efficacy of 6 and 18 months of therapy with four drugs (COMP) for localized non-Hodgkin's lymphoma of children: a report from the Childrens Cancer Study Group. J Clin Oncol 1989; 7: 92–99.
Link MP, Shuster JJ, Donaldson SS, Berard CW, Murphy SB . Treatment of children and young adults with early-stage non-Hodgkin's lymphoma. N Engl J Med 1997; 337: 1259–1266.
Wollner N, Burchenal JH, Lieberman PH, Exelby P, D’Angio G, Murphy ML . Non-Hodgkin's lymphoma in children. A comparative study of two modalities of therapy. Cancer 1976; 37: 123–134.
Pillon M, Piglione M, Garaventa A, Conter V, Giuliano M, Arcamone G et al. Long-term results of AIEOP LNH-92 protocol for the treatment of pediatric lymphoblastic lymphoma: a report of the Italian association of pediatric hematology and oncology. Pediatr Blood Cancer 2009; 53: 953–959.
Burkhardt B, Woessmann W, Zimmermann M, Kontny U, Vormoor J, Doerffel W et al. Impact of cranial radiotherapy on central nervous system prophylaxis in children and adolescents with central nervous system-negative stage III or IV lymphoblastic lymphoma. J Clin Oncol 2006; 24: 491–499.
Abromowitch M, Sposto R, Perkins S, Zwick D, Siegel S, Finlay J et al. Shortened intensified multi-agent chemotherapy and non-cross resistant maintenance therapy for advanced lymphoblastic lymphoma in children and adolescents: report from the Children's Oncology Group. Br J Haematol 2008; 143: 261–267.
Abromowitch M, Termuhlen A, Lynch J, Chang M, Perkins SL, Gross T et al. High-dose methotrexate and early intensification of therapy do not improve 3 year EFS in children and adolescents with disseminated lymphoblastic lymphoma. Results of the randomized arms of COG A5971. Hematol Meeting Rep 2009, (abstract); 3: 33.
Uyttebroeck A, Suciu S, Laureys G, Robert A, Pacquement H, Ferster A et al. Treatment of childhood T-cell lymphoblastic lymphoma according to the strategy for acute lymphoblastic leukaemia, without radiotherapy: long term results of the EORTC CLG 58881 trial. Eur J Cancer 2008; 44: 840–846.
Sandlund JT, Pui CH, Zhou Y, Behm FG, Onciu M, Razzouk BI et al. Effective treatment of advanced-stage childhood lymphoblastic lymphoma without prophylactic cranial irradiation: results of St Jude NHL13 study. Leukemia 2009; 23: 1127–1130.
Anderson JR, Jenkin RD, Wilson JF, Kjeldsberg CR, Sposto R, Chilcote RR et al. Long-term follow-up of patients treated with COMP or LSA2L2 therapy for childhood non-Hodgkin's lymphoma: a report of CCG-551 from the Childrens Cancer Group. J Clin Oncol 1993; 11: 1024–1032.
Hartmann O, Pein F, Beaujean F, Kalifa C, Patte C, Parmentier C et al. High-dose polychemotherapy with autologous bone marrow transplantation in children with relapsed lymphomas. J Clin Oncol 1984; 2: 979–985.
Mills W, Chopra R, McMillan A, Pearce R, Linch DC, Goldstone AH . BEAM chemotherapy and autologous bone marrow transplantation for patients with relapsed or refractory non-Hodgkin's lymphoma. J Clin Oncol 1995; 13: 588–595.
Won SC, Han JW, Kwon SY, Shin HY, Ahn HS, Hwang TJ et al. Autologous peripheral blood stem cell transplantation in children with non-Hodgkin's lymphoma: a report from the Korean Society of Pediatric Hematology-Oncology. Ann Hematol 2006; 85: 787–794.
Kleiner S, Kirsch A, Schwaner I, Kingreen D, Schwella N, Huhn D et al. High-dose chemotherapy with carboplatin, etoposide and ifosfamide followed by autologous stem cell rescue in patients with relapsed or refractory malignant lymphomas: a phase I/II study. Bone Marrow Transplant 1997; 20: 953–959.
Kobrinsky NL, Sposto R, Shah NR, Anderson JR, DeLaat C, Morse M et al. Outcomes of treatment of children and adolescents with recurrent non-Hodgkin's lymphoma and Hodgkin's disease with dexamethasone, etoposide, cisplatin, cytarabine, and l-asparaginase, maintenance chemotherapy, and transplantation: Children's Cancer Group Study CCG-5912. J Clin Oncol 2001; 19: 2390–2396.
Bureo E, Ortega JJ, Munoz A, Cubells J, Madero L, Verdaguer A et al. Bone marrow transplantation in 46 pediatric patients with non-Hodgkin's lymphoma. Spanish Working Party for Bone Marrow Transplantation in Children. Bone Marrow Transplant 1995; 15: 353–359.
Levine JE, Harris RE, Loberiza Jr FR, Armitage JO, Vose JM, Van Besien K et al. A comparison of allogeneic and autologous bone marrow transplantation for lymphoblastic lymphoma. Blood 2003; 101: 2476–2482.
Gross TG, Hale GA, He W, Camitta BM, Sanders JE, Cairo MS et al. Hematopoietic stem cell transplantation for refractory or recurrent non-Hodgkin lymphoma in children and adolescents. Biol Blood Marrow Transplant 2010; 16: 223–230.
Mitsui T, Mori T, Fujita N, Inada H, Horibe K, Tsurusawa M . Retrospective analysis of relapsed or primary refractory childhood lymphoblastic lymphoma in Japan. Pediatr Blood Cancer 2009; 52: 591–595.
Berg SL, Blaney SM, Devidas M, Lampkin TA, Murgo A, Bernstein M et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 2005; 23: 3376–3382.
Balakrishnan K, Nimmanapalli R, Ravandi F, Keating MJ, Gandhi V . Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2006; 108: 2392–2398.
Haioun C, Itti E, Rahmouni A, Brice P, Rain JD, Belhadj K et al. [18F]fluoro-2-deoxy-D-glucose positron emission tomography (FDG-PET) in aggressive lymphoma: an early prognostic tool for predicting patient outcome. Blood 2005; 106: 1376–1381.
Lovisa F, Mussolin L, Corral L, Pillon M, Cazzaniga G, Biondi A et al. TCR-based RQ-PCR assay for MDD and MRD assessment in T-cell lymphoblastic lymphoma of childhood. Hematol Meeting Rep 2009, (abstract); 3: 37.
Stark B, Avigad S, Luria D, Manor S, Reshef-Ronen T, Avrahami G et al. Bone marrow minimal disseminated disease (MDD) and minimal residual disease (MRD) in childhood T-cell lymphoblastic lymphoma stage III, detected by flow cytometry (FC) and real-time quantitative polymerase chain reaction (RQ-PCR). Pediatr Blood Cancer 2009; 52: 20–25.
Coustan-Smith E, Sandlund JT, Perkins SL, Chen H, Chang M, Abromowitch M et al. Minimal disseminated disease in childhood T-cell lymphoblastic lymphoma: a report from the children's oncology group. J Clin Oncol 2009; 27: 3533–3539.
Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009; 10: 147–156.
Aifantis I, Raetz E, Buonamici S . Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008; 8: 380–390.
Gilliland DG . Hematologic malignancies. Curr Opin Hematol 2001; 8: 189–191.
De Keersmaecker K, Marynen P, Cools J . Genetic insights in the pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 2005; 90: 1116–1127.
Van Vlierberghe P, Pieters R, Beverloo HB, Meijerink JP . Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol 2008; 143: 153–168.
Burkhardt B . Paediatric lymphoblastic T-cell leukaemia and lymphoma: one or two diseases? Br J Haematol 2010; 149: 653–668.
Krieger D, Moericke A, Oschlies I, Zimmermann M, Schrappe M, Reiter A et al. Frequency and clinical relevance of DNA microsatellite alterations of the CDKN2A/B, ATM and p53 gene loci: a comparison between pediatric precursor T-cell lymphoblastic lymphoma and T-cell lymphoblastic leukemia. Haematologica 2010; 95: 158–162.
O’Neil J, Look AT . Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene 2007; 26: 6838–6849.
Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A . Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia 2006; 20: 1496–1510.
Teitell MA, Pandolfi PP . Molecular genetics of acute lymphoblastic leukemia. Annu Rev Pathol 2009; 4: 175–198.
Ferrando AA, Look AT . Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol 2003; 40: 274–280.
Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87.
Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005; 106: 274–286.
Hebert J, Cayuela JM, Berkeley J, Sigaux F . Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood 1994; 84: 4038–4044.
Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306: 269–271.
Bar-Eli M, Ahuja H, Foti A, Cline MJ . N-RAS mutations in T-cell acute lymphocytic leukaemia: analysis by direct sequencing detects a novel mutation. Br J Haematol 1989; 72: 36–39.
Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 2008; 205: 751–758.
Paietta E, Ferrando AA, Neuberg D, Bennett JM, Racevskis J, Lazarus H et al. Activating FLT3 mutations in CD117/KIT(+) T-cell acute lymphoblastic leukemias. Blood 2004; 104: 558–560.
Balgobind BV, Van Vlierberghe P, van den Ouweland AM, Beverloo HB, Terlouw-Kromosoeto JN, van Wering ER et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood 2008; 111: 4322–4328.
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 2007; 13: 1203–1210.
Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S et al. WT1 mutations in T-ALL. Blood 2009; 114: 1038–1045.
Sambandam A, Maillard I, Zediak VP, Xu L, Gerstein RM, Aster JC et al. Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol 2005; 6: 663–670.
Radtke F, Wilson A, MacDonald HR . Notch signaling in T- and B-cell development. Curr Opin Immunol 2004; 16: 174–179.
Radtke F, Wilson A, Mancini SJ, MacDonald HR . Notch regulation of lymphocyte development and function. Nat Immunol 2004; 5: 247–253.
Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991; 66: 649–661.
Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med 1996; 183: 2283–2291.
Chen J, Jette C, Kanki JP, Aster JC, Look AT, Griffin JD . NOTCH1-induced T-cell leukemia in transgenic zebrafish. Leukemia 2007; 21: 462–471.
Lewis HD, Leveridge M, Strack PR, Haldon CD, O’Neil J, Kim H et al. Apoptosis in T cell acute lymphoblastic leukemia cells after cell cycle arrest induced by pharmacological inhibition of notch signaling. Chem Biol 2007; 14: 209–219.
Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 2009; 15: 50–58.
Palomero T, Dominguez M, Ferrando AA . The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle 2008; 7: 965–970.
Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ . Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007; 110: 278–286.
Cullion K, Draheim KM, Hermance N, Tammam J, Sharma VM, Ware C et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009; 113: 6172–6181.
Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med 2007; 13: 70–77.
Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009; 459: 1000–1004.
Langenau DM, Feng H, Berghmans S, Kanki JP, Kutok JL, Look AT . Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA 2005; 102: 6068–6073.
Frazer JK, Meeker ND, Rudner L, Bradley DF, Smith AC, Demarest B et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia 2009; 23: 1825–1835.
Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985; 66: 848–858.
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G . The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008; 8: 11–23.
World Health Organization. Tumours of haematopoietic and lymphoid tissues. In: Jaffe E, Harris N, Stein H, Vardiman J (eds). World Health Organization Classification of Tumors. IARC Press: Washington, DC, 2000.
Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 2000; 96: 3681–3695.
Duyster J, Bai RY, Morris SW . Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001; 20: 5623–5637.
Lamant L, de Reynies A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood 2007; 109: 2156–2164.
Salaverria I, Bea S, Lopez-Guillermo A, Lespinet V, Pinyol M, Burkhardt B et al. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br J Haematol 2008; 140: 516–526.
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998; 91: 2076–2084.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–1392.
Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999; 93: 2697–2706.
Le Deley MC, Reiter A, Williams D, Delsol G, Oschlies I, McCarthy K et al. Prognostic factors in childhood anaplastic large cell lymphoma: results of a large European intergroup study. Blood 2008; 111: 1560–1566.
Jaffe ES . Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod Pathol 2001; 14: 219–228.
Salzburg J, Burkhardt B, Zimmermann M, Wachowski O, Woessmann W, Oschlies I et al. Prevalence, clinical pattern, and outcome of CNS involvement in childhood and adolescent non-Hodgkin's lymphoma differ by non-Hodgkin's lymphoma subtype: a Berlin-Frankfurt-Munster Group Report. J Clin Oncol 2007; 25: 3915–3922.
Seidemann K, Tiemann M, Schrappe M, Yakisan E, Simonitsch I, Janka-Schaub G et al. Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma: a report of the Berlin-Frankfurt-Munster Group Trial NHL-BFM 90. Blood 2001; 97: 3699–3706.
Sandlund JT, Pui CH, Santana VM, Mahmoud H, Roberts WM, Morris S et al. Clinical features and treatment outcome for children with CD30+ large-cell non-Hodgkin's lymphoma. J Clin Oncol 1994; 12: 895–898.
Brugieres L, Deley MC, Pacquement H, Meguerian-Bedoyan Z, Terrier-Lacombe MJ, Robert A et al. CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood 1998; 92: 3591–3598.
Laver JH, Kraveka JM, Hutchison RE, Chang M, Kepner J, Schwenn M et al. Advanced-stage large-cell lymphoma in children and adolescents: results of a randomized trial incorporating intermediate-dose methotrexate and high-dose cytarabine in the maintenance phase of the APO regimen: a Pediatric Oncology Group phase III trial. J Clin Oncol 2005; 23: 541–547.
Lowe EJ, Sposto R, Perkins SL, Gross TG, Finlay J, Zwick D et al. Intensive chemotherapy for systemic anaplastic large cell lymphoma in children and adolescents: final results of Children's Cancer Group Study 5941. Pediatr Blood Cancer 2009; 52: 335–339.
Rosolen A, Pillon M, Garaventa A, Burnelli R, d’Amore ES, Giuliano M et al. Anaplastic large cell lymphoma treated with a leukemia-like therapy: report of the Italian Association of Pediatric Hematology and Oncology (AIEOP) LNH-92 protocol. Cancer 2005; 104: 2133–2140.
Williams DM, Hobson R, Imeson J, Gerrard M, McCarthy K, Pinkerton CR . Anaplastic large cell lymphoma in childhood: analysis of 72 patients treated on The United Kingdom Children's Cancer Study Group chemotherapy regimens. Br J Haematol 2002; 117: 812–820.
Brugieres L, Quartier P, Le Deley MC, Pacquement H, Perel Y, Bergeron C et al. Relapses of childhood anaplastic large-cell lymphoma: treatment results in a series of 41 children--a report from the French Society of Pediatric Oncology. Ann Oncol 2000; 11: 53–58.
Kraveka JM, Weitzman S, Smith L, Lynch JC, Chang M, Kinney M et al. Advanced-stage anaplastic large-cell lymphoma in children and adolescents: results of ANHL0131, a randomized phase III trial with standard APO (doxorubicin, prednisone, vincristine) versus consolidation with a regimen including vinblastine: a report from the Children's Oncology Group. Hematol Meeting Rep 2009, (abstract); 3: 41.
Brugieres L, Le Deley MC, Rosolen A, Williams D, Horibe K, Wrobel G et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: results of a randomized trial of the EICNHL Group. J Clin Oncol 2009; 27: 897–903.
Massimino M, Spreafico F, Luksch R, Giardini R . Prognostic significance of p80 and visceral involvement in childhood CD30 anaplastic large cell lymphoma (ALCL). Med Pediatr Oncol 2001; 37: 97–102.
Damm-Welk C, Busch K, Burkhardt B, Schieferstein J, Viehmann S, Oschlies I et al. Prognostic significance of circulating tumor cells in bone marrow or peripheral blood as detected by qualitative and quantitative PCR in pediatric NPM-ALK-positive anaplastic large-cell lymphoma. Blood 2007; 110: 670–677.
Mussolin L, Bonvini P, Ait-Tahar K, Pillon M, Tridello G, Buffardi S et al. Kinetics of humoral response to ALK and its relationship with minimal residual disease in pediatric ALCL. Leukemia 2009; 23: 400–402.
Ait-Tahar K, Damm-Welk C, Burkhardt B, Zimmermann M, Klapper W, Reiter A et al. Correlation of the autoantibody response to the ALK oncoantigen in pediatric anaplastic lymphoma kinase-positive anaplastic large cell lymphoma with tumor dissemination and relapse risk. Blood 2010; 115: 3314–3319.
Lamant L, McCarthy K, d’Amore ESG, Klapper W, Nakagawa A, Fraga M et al. Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic large cell lymphoma (ALCL): results of the ALCL99 study. Hematol Meeting Rep 2009, (abstract); 3: 42.
Sandlund JT, Pui CH, Roberts WM, Santana VM, Morris SW, Berard CW et al. Clinicopathologic features and treatment outcome of children with large-cell lymphoma and the t(2;5)(p23;q35). Blood 1994; 84: 2467–2471.
Vecchi V, Burnelli R, Pileri S, Rosito P, Sabattini E, Civino A et al. Anaplastic large cell lymphoma (Ki-1+/CD30+) in childhood. Med Pediatr Oncol 1993; 21: 402–410.
Reiter A, Schrappe M, Tiemann M, Parwaresch R, Zimmermann M, Yakisan E et al. Successful treatment strategy for Ki-1 anaplastic large-cell lymphoma of childhood: a prospective analysis of 62 patients enrolled in three consecutive Berlin-Frankfurt-Munster group studies. J Clin Oncol 1994; 12: 899–908.
Mora J, Filippa DA, Thaler HT, Polyak T, Cranor ML, Wollner N . Large cell non-Hodgkin lymphoma of childhood: analysis of 78 consecutive patients enrolled in 2 consecutive protocols at the Memorial Sloan-Kettering Cancer Center. Cancer 2000; 88: 186–197.
Woessmann W, Zimmermann M, Lenhard M, Burkhardt B, Rossig C, Kremens B et al. Relapsed or refractory anaplastic large-cell lymphoma in children and adolescents after Berlin-Frankfurt-Muenster (BFM)-type first-line therapy: a BFM-group study. J Clin Oncol 2011; 29: 3065–3071.
Mori T, Takimoto T, Katano N, Kikuchi A, Tabuchi K, Kobayashi R et al. Recurrent childhood anaplastic large cell lymphoma: a retrospective analysis of registered cases in Japan. Br J Haematol 2006; 132: 594–597.
Le Deley MC, Rosolen A, Williams DM, Horibe K, Wrobel G, Attarbaschi A et al. Vinblastine in children and adolescents with high-risk anaplastic large-cell lymphoma: results of the randomized ALCL99-vinblastine trial. J Clin Oncol 2010; 28: 3987–3993.
Cesaro S, Pillon M, Visintin G, Putti MC, Gazzola MV, D’Amore E et al. Unrelated bone marrow transplantation for high-risk anaplastic large cell lymphoma in pediatric patients: a single center case series. Eur J Haematol 2005; 75: 22–26.
Woessmann W, Peters C, Lenhard M, Burkhardt B, Sykora KW, Dilloo D et al. Allogeneic haematopoietic stem cell transplantation in relapsed or refractory anaplastic large cell lymphoma of children and adolescents--a Berlin-Frankfurt-Munster group report. Br J Haematol 2006; 133: 176–182.
Bartlett NL, Younes A, Carabasi MH, Forero A, Rosenblatt JD, Leonard JP et al. A phase 1 multidose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30+ hematologic malignancies. Blood 2008; 111: 1848–1854.
Forero-Torres A, Leonard JP, Younes A, Rosenblatt JD, Brice P, Bartlett NL et al. A phase II study of SGN-30 (anti-CD30mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol 2009; 146: 171–179.
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 2010; 363: 1812–1821.
Bartlett N, Forero-Torres A, Rosenblatt J, Fanale M, Horning SJ, Thompson S et al. Complete remissions with SGN-35 weekly dosing: a phase I dose-escalation study in relapsed/refractory Hodgkin lymphoma or systemic ALCL patients. J Clin Oncol 2009, (abstract); 17: 8500.
Younes A, Forero-Torres A, Bartlett NL, Leonard JP, Lynch C, Kennedy DA et al. Multiple complete responses in a phase 1 dose-escalation study of the antibody-drug conjugate SGN-35 in patients with relapsed or refractory CD30-positive lymphomas. Blood 2008, (abstract); 112: 1006.
Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci USA 2007; 104: 270–275.
Gambacorti-Passerini C, Messa C, Pogliani EM . Crizotinib in anaplastic large-cell lymphoma. N Engl J Med 2011; 364: 775–776.
Palmer RH, Vernersson E, Grabbe C, Hallberg B . Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 2009; 420: 345–361.
Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G et al. Germline mutations of the perforin gene are a frequent occurrence in childhood anaplastic large cell lymphoma. Cancer 2007; 109: 2566–2571.
Clementi R, Locatelli F, Dupre L, Garaventa A, Emmi L, Bregni M et al. A proportion of patients with lymphoma may harbor mutations of the perforin gene. Blood 2005; 105: 4424–4428.
Singh RR, Cho-Vega JH, Davuluri Y, Ma S, Kasbidi F, Milito C et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res 2009; 69: 2550–2558.
Lim MS, Carlson ML, Crockett DK, Fillmore GC, Abbott DR, Elenitoba-Johnson OF et al. The proteomic signature of NPM/ALK reveals deregulation of multiple cellular pathways. Blood 2009; 114: 1585–1595.
Lim MS, Elenitoba-Johnson KS . Mass spectrometry-based proteomic studies of human anaplastic large cell lymphoma. Mol Cell Proteomics 2006; 5: 1787–1798.
Lim M, Tygeson J, Seiler C, Crockett D, Satwani P, Perkins S et al. Proteomic analysis of denileukin diftitox (Ontak) as a potential therapeutic agent for ALCL. Hematol Meeting Rep 2009, (abstract); 3: 44.
Miles RR, Cairo MS, Satwani P, Zwick DL, Lones MA, Sposto R et al. Immunophenotypic identification of possible therapeutic targets in paediatric non-Hodgkin lymphomas: a children's oncology group report. Br J Haematol 2007; 138: 506–512.
Bonvini P, Gastaldi T, Falini B, Rosolen A . Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res 2002; 62: 1559–1566.
Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res 2001; 61: 2194–2199.
Windsor R, Stiller C, Webb D . Peripheral T-cell lymphoma in childhood: population-based experience in the United Kingdom over 20 years. Pediatr Blood Cancer 2008; 50: 784–787.
A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood 1997; 89: 3909–3918.
Rudiger T, Geissinger E, Muller-Hermelink HK . ‘Normal counterparts’ of nodal peripheral T-cell lymphoma. Hematol Oncol 2006; 24: 175–180.
Nelson M, Horsman DE, Weisenburger DD, Gascoyne RD, Dave BJ, Loberiza FR et al. Cytogenetic abnormalities and clinical correlations in peripheral T-cell lymphoma. Br J Haematol 2008; 141: 461–469.
Zettl A, Rudiger T, Konrad MA, Chott A, Simonitsch-Klupp I, Sonnen R et al. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am J Pathol 2004; 164: 1837–1848.
Martinez-Delgado B, Cuadros M, Honrado E, Ruiz de la Parte A, Roncador G, Alves J et al. Differential expression of NF-kappaB pathway genes among peripheral T-cell lymphomas. Leukemia 2005; 19: 2254–2263.
Ballester B, Ramuz O, Gisselbrecht C, Doucet G, Loi L, Loriod B et al. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas. Oncogene 2006; 25: 1560–1570.
Hutchison RE, Laver JH, Chang M, Muzzafar T, Desai S, Murphy S et al. Non-anaplastic peripheral t-cell lymphoma in childhood and adolescence: a Children's Oncology Group study. Pediatr Blood Cancer 2008; 51: 29–33.
Gisselbrecht C, Gaulard P, Lepage E, Coiffier B, Briere J, Haioun C et al. Prognostic significance of T-cell phenotype in aggressive non-Hodgkin's lymphomas. Groupe d’Etudes des Lymphomes de l’Adulte (GELA). Blood 1998; 92: 76–82.
Savage KJ . Peripheral T-cell lymphomas. Blood Rev 2007; 21: 201–216.
Savage KJ, Chhanabhai M, Gascoyne RD, Connors JM . Characterization of peripheral T-cell lymphomas in a single North American institution by the WHO classification. Ann Oncol 2004; 15: 1467–1475.
Kewalramani T, Zelenetz AD, Teruya-Feldstein J, Hamlin P, Yahalom J, Horwitz S et al. Autologous transplantation for relapsed or primary refractory peripheral T-cell lymphoma. Br J Haematol 2006; 134: 202–207.
Paolo C, Lucia F, Anna D . Hematopoietic stem cell transplantation in peripheral T-cell lymphomas. Leuk Lymphoma 2007; 48: 1496–1501.
Corradini P, Tarella C, Zallio F, Dodero A, Zanni M, Valagussa P et al. Long-term follow-up of patients with peripheral T-cell lymphomas treated up-front with high-dose chemotherapy followed by autologous stem cell transplantation. Leukemia 2006; 20: 1533–1538.
Mollee P, Lazarus HM, Lipton J . Why aren’t we performing more allografts for aggressive non-Hodgkin's lymphoma? Bone Marrow Transplant 2003; 31: 953–960.
Corradini P, Dodero A, Zallio F, Caracciolo D, Casini M, Bregni M et al. Graft-versus-lymphoma effect in relapsed peripheral T-cell non-Hodgkin's lymphomas after reduced-intensity conditioning followed by allogeneic transplantation of hematopoietic cells. J Clin Oncol 2004; 22: 2172–2176.
Gallamini A, Stelitano C, Calvi R, Bellei M, Mattei D, Vitolo U et al. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood 2004; 103: 2474–2479.
Sallah S, Wan JY, Nguyen NP . Treatment of refractory T-cell malignancies using gemcitabine. Br J Haematol 2001; 113: 185–187.
Czuczman MS, Porcu P, Johnson J, Niedzwiecki D, Canellos GP, Cheson BD . CALGB 59901: results of a phase II study of 506U78 in CTCL and PTCL. Blood 2004, (abstract); 104: 2486.
Piekarz R, Wright J, Frye R, Allen SL, Craig M, Geskin L et al. Results of a phase 2 NCI multicenter study of romidepsin in patients with relapsed peripheral T-cell lymphoma (PTCL). 2008, (abstract); 112: 1567.
Aguiar Bujanda D . Complete response of relapsed angioimmunoblastic T-cell lymphoma following therapy with bevacizumab. Ann Oncol 2008; 19: 396–397.
Bruns I, Fox F, Reinecke P, Kobbe G, Kronenwett R, Jung G et al. Complete remission in a patient with relapsed angioimmunoblastic T-cell lymphoma following treatment with bevacizumab. Leukemia 2005; 19: 1993–1995.
O’Connor OA, Horwitz S, Hamlin P, Portlock C, Moskowitz CH, Sarasohn D et al. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 2009; 27: 4357–4364.
O’Connor OA, Pro B, Pinter-Brown L, Popplewell L, Barlett N, Shustov A et al. PROPEL: a multi-center phase 2 open-label study of pralatrexate (PDX) with vitamin B12 and folic acid supplementation in patients with replapsed or refractory peripheral T-cell lymphoma. 2008, (abstract); 112: 261.
Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T et al. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood 2005; 105: 3058–3065.
Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol 2007; 25: 4293–4297.
Lee J, Suh C, Kang HJ, Ryoo BY, Huh J, Ko YH et al. Phase I study of proteasome inhibitor bortezomib plus CHOP in patients with advanced, aggressive T-cell or NK/T-cell lymphoma. Ann Oncol 2008; 19: 2079–2083.
Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol 2009; 82: 440–449.
Enblad G, Hagberg H, Erlanson M, Lundin J, MacDonald AP, Repp R et al. A pilot study of alemtuzumab (anti-CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy-refractory peripheral T-cell lymphomas. Blood 2004; 103: 2920–2924.
Gallamini A, Zaja F, Patti C, Billio A, Specchia MR, Tucci A et al. Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 2007; 110: 2316–2323.
Cheson BD . Novel therapies for peripheral T-cell non-Hodgkin's lymphomas. Curr Opin Hematol 2009; 16: 299–305.
Horwitz SM . Novel therapies and role of transplant in the treatment of peripheral T-cell lymphomas. Hematol Am Soc Hematol Educ Program 2008, 289–296.
Lim MS . Commentary on the WHO 2008 classification of neoplasms arising from histiocytic and other accessory cells. J Hematopathol 2009; 2: 75–76.
Mackey AC, Green L, Liang LC, Dinndorf P, Avigan M . Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2007; 44: 265–267.
Miyazaki K, Yamaguchi M, Imai H, Kobayashi T, Tamaru S, Nishii K et al. Gene expression profiling of peripheral T-cell lymphoma including gammadelta T-cell lymphoma. Blood 2009; 113: 1071–1074.
Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood 2003; 102: 4261–4269.
Domm JA, Thompson M, Kuttesch JF, Acra S, Frangoul H . Allogeneic bone marrow transplantation for chemotherapy-refractory hepatosplenic gammadelta T-cell lymphoma: case report and review of the literature. J Pediatr Hematol Oncol 2005; 27: 607–610.
Konuma T, Ooi J, Takahashi S, Tomonari A, Tsukada N, Kobayashi T et al. Allogeneic stem cell transplantation for hepatosplenic gammadelta T-cell lymphoma. Leukemia Lymphoma 2007; 48: 630–632.
Medhi K, Kumar R, Rishi A, Kumar L, Bakhshi S . Subcutaneous panniculitislike T-cell lymphoma with hemophagocytosis: complete remission with BFM-90 protocol. J Pediatr Hematol Oncol 2008; 30: 558–561.
Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008; 111: 838–845.
Ohshima K, Kimura H, Yoshino T, Kim CW, Ko YH, Lee SS et al. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol Int 2008; 58: 209–217.
Cohen JI, Kimura H, Nakamura S, Ko YH, Jaffe ES . Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8–9 September 2008. Ann Oncol 2009; 20: 1472–1482.
Sato E, Ohga S, Kuroda H, Yoshiba F, Nishimura M, Nagasawa M et al. Allogeneic hematopoietic stem cell transplantation for Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disease in Japan. Am J Hematol 2008; 83: 721–727.
Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL . Global epidemiology of HTLV-I infection and associated diseases. Oncogene 2005; 24: 6058–6068.
Matsuoka M . Human T-cell leukemia virus type I and adult T-cell leukemia. Oncogene 2003; 22: 5131–5140.
Li XH, Gaynor RB . Mechanisms of NF-kappaB activation by the HTLV type 1 tax protein. AIDS Res Hum Retroviruses 2000; 16: 1583–1590.
Yoshida M . Multiple viral strategies of HTLV-1 for dysregulation of cell growth control. Ann Rev Immunol 2001; 19: 475–496.
Zhao T, Yasunaga J, Satou Y, Nakao M, Takahashi M, Fujii M et al. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 2009; 113: 2755–2764.
Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL . Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 2008; 112: 3788–3797.
Arnulf B, Thorel M, Poirot Y, Tamouza R, Boulanger E, Jaccard A et al. Loss of the ex vivo but not the reinducible CD8+ T-cell response to Tax in human T-cell leukemia virus type 1-infected patients with adult T-cell leukemia/lymphoma. Leukemia 2004; 18: 126–132.
Kannagi M . Immunologic control of human T-cell leukemia virus type I and adult T-cell leukemia. Int J Hematol 2007; 86: 113–117.
Kozako T, Arima N, Toji S, Masamoto I, Akimoto M, Hamada H et al. Reduced frequency, diversity, and function of human T cell leukemia virus type 1-specific CD8+ T cell in adult T cell leukemia patients. J Immunol 2006; 177: 5718–5726.
Matutes E . Adult T-cell leukaemia/lymphoma. J Clin Pathol 2007; 60: 1373–1377.
Ishitsuka K, Tamura K . Treatment of adult T-cell leukemia/lymphoma: past, present, and future. Eur J Haematol 2008; 80: 185–196.
Okamura J, Uike N, Utsunomiya A, Tanosaki R . Allogeneic stem cell transplantation for adult T-cell leukemia/lymphoma. Int J Hematol 2007; 86: 118–125.
Shiratori S, Yasumoto A, Tanaka J, Shigematsu A, Yamamoto S, Nishio M et al. A retrospective analysis of allogeneic hematopoietic stem cell transplantation for adult T cell leukemia/lymphoma (ATL): clinical impact of graft-versus-leukemia/lymphoma effect. Biol Blood Marrow Transplant 2008; 14: 817–823.
Au WY, Weisenburger DD, Intragumtornchai T, Nakamura S, Kim WS, Sng I et al. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood 2009; 113: 3931–3937.
Iqbal J, Kucuk C, Deleeuw RJ, Srivastava G, Tam W, Geng H et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia 2009; 23: 1139–1151.
Cairo M, Day N, Shereck E, Ayello J, Elenitoba-Johnson K, Lim M (eds). Genomic and proteomic characterization of natural killer cell lymphoma: identification of therapeutic targets. T-cell Lymphoma Forum; 28–30 January 2010; Maui, HI.
Vose J, Armitage J, Weisenburger D . International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26: 4124–4130.
Zhou Y, Attygalle AD, Chuang SS, Diss T, Ye H, Liu H et al. Angioimmunoblastic T-cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol 2007; 138: 44–53.
Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res 2007; 67: 10703–10710.
Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010; 115: 1026–1036.
Adam P, Katzenberger T, Seeberger H, Gattenlohner S, Wolf J, Steinlein C et al. A case of a diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol 2003; 27: 1473–1476.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994; 263: 1281–1284.
Onciu M, Behm FG, Downing JR, Shurtleff SA, Raimondi SC, Ma Z et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood 2003; 102: 2642–2644.
Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi T, Takeshita M et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood 1995; 86: 1954–1960.
Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B . A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999; 93: 3088–3095.
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000; 157: 377–384.
Siebert R, Gesk S, Harder L, Steinemann D, Grote W, Schlegelberger B et al. Complex variant translocation t(1;2) with TPM3-ALK fusion due to cryptic ALK gene rearrangement in anaplastic large-cell lymphoma. Blood 1999; 94: 3614–3617.
Hernandez L, Bea S, Bellosillo B, Pinyol M, Falini B, Carbone A et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am J Pathol 2002; 160: 1487–1494.
Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 1999; 94: 3265–3268.
Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M . ATIC-ALK: a novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol 2000; 156: 781–789.
Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood 2000; 95: 2144–2149.
Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG et al. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res 2000; 60: 793–798.
Bridge JA, Kanamori M, Ma Z, Pickering D, Hill DA, Lydiatt W et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol 2001; 159: 411–415.
Reichard KK, McKenna RW, Kroft SH . ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature. Mod Pathol 2007; 20: 310–319.
Touriol C, Greenland C, Lamant L, Pulford K, Bernard F, Rousset T et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 2000; 95: 3204–3207.
Tort F, Campo E, Pohlman B, Hsi E . Heterogeneity of genomic breakpoints in MSN-ALK translocations in anaplastic large cell lymphoma. Hum Pathol 2004; 35: 1038–1041.
Tort F, Pinyol M, Pulford K, Roncador G, Hernandez L, Nayach I et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest 2001; 81: 419–426.
Meech SJ, McGavran L, Odom LF, Liang X, Meltesen L, Gump J et al. Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4--anaplastic lymphoma kinase gene fusion. Blood 2001; 98: 1209–1216.
Cools J, Wlodarska I, Somers R, Mentens N, Pedeutour F, Maes B et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2002; 34: 354–362.
Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003; 37: 98–105.
Lamant L, Gascoyne RD, Duplantier MM, Armstrong F, Raghab A, Chhanabhai M et al. Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer 2003; 37: 427–432.
Debelenko LV, Arthur DC, Pack SD, Helman LJ, Schrump DS, Tsokos M . Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest 2003; 83: 1255–1265.
Stachurski D, Miron PM, Al-Homsi S, Hutchinson L, Harris NL, Woda B et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma with a complex karyotype and cryptic 3′ ALK gene insertion to chromosome 4 q22–24. Hum Pathol 2007; 38: 940–945.
Panagopoulos I, Nilsson T, Domanski HA, Isaksson M, Lindblom P, Mertens F et al. Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer 2006; 118: 1181–1186.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448: 561–566.
Acknowledgements
This work was supported in part by grants from the Pediatric Cancer Research Foundation, Marisa Fund, and Paul Luisi Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
AAF has sponsored research from Merck and Pfizer on the use of GSIs for the treatment of T-ALL. The remaining authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
El-Mallawany, N., Frazer, J., Van Vlierberghe, P. et al. Pediatric T- and NK-cell lymphomas: new biologic insights and treatment strategies. Blood Cancer Journal 2, e65 (2012). https://doi.org/10.1038/bcj.2012.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2012.8
Keywords
This article is cited by
-
Primary peripheral T-cell lymphoma, not otherwise specified, of the central nervous system in a child
Brain Tumor Pathology (2015)
-
Unusual presentation of primary T-cell lymphoblastic lymphoma: description of two cases
Diagnostic Pathology (2014)
