
Abstract
(5R)-5-hydroxytriptolide (LLDT-8) is a novel triptolide analog that has been identified as a promising candidate for treating autoimmune diseases and has been shown to be effective in treating murine collagen-induced arthritis and lupus nephritis. In the present study, we investigated the therapeutic effect and possible mechanism of action of LLDT-8 in a murine anti-glomerular basement membrane (GBM) glomerulonephritis model. NZW mice were injected with rabbit anti-GBM serum (500 μL, ip). The mice were orally treated with LLDT-8 (0.125 mg/kg, every other day) or a positive control prednisolone (2 mg/kg every day) for 14 d. Blood and urine samples as well as spleen and kidney tissues were collected for analyses. LLDT-8 treatment did not affect the generation of mouse anti-rabbit antibodies. LLDT-8 significantly reversed established proteinuria, improved renal histopathology and attenuated renal dysfunction in glomerulonephritis mice. Furthermore, LLDT-8 inhibited inflammation in the kidney evidenced by significantly decreasing C3 and IgG deposition, reducing the levels of the pathogenic cytokines TNF-α, IL-6, IL-17, and IFN-γ, and reducing related chemokine expression and leukocyte infiltration in kidneys. Moreover, LLDT-8 treatment significantly increased the expression of FcγRIIB in the kidney and spleen. In addition, the treatment restored the reduced expression of FcγRIIB on the surface of kidney effector cells, CD11b+ cells, and interfered with FcγR-dependent signaling, especially FcγRIIB-mediated downstream kinases, such as BTK. These results demonstrate that LLDT-8 ameliorates anti-GBM glomerulonephritis by regulating the Fcγ receptor signaling.
Similar content being viewed by others

Introduction
Glomerular diseases cause high morbidity and disability, resulting in substantial economic losses annually. Immune mechanisms, such as antibodies, immune complex (IC) deposition, Fc receptors (FcRs), inflammatory cytokines, chemokines, and immune cells contribute to immune dysfunction and glomerular damage in glomerulonephritis (GN). Such mechanisms are largely responsible for the pathology associated with the majority of glomerular diseases1,2,3. FcRs are well-defined receptors that are expressed on the surface of effector cells, and FcR-mediated pathways contribute to GN pathogenesis4.
Glomerular damage occurs when ICs form in situ when antibodies bind to antigens distributed within the GBM, and linear binding of IgG is universally observed by immunofluorescence (IF)5. IC accumulation in the kidney subsequently triggers complement activation, the generation of inflammatory regulators, and the recruitment of leukocytes to sites of inflammation6,7. In response to these signals, mesangial cells can produce inflammatory cytokines and chemokines8. Previous studies have indicated that inflammatory cytokines (eg, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-17 (IL-17), and interferon-γ (IFN-γ)9,10,11) and chemokines (eg, CCL2, CCL5, CCL7, and CX3CL112,13) are involved in GN. Furthermore, IC may be related to inducing the expression of matrix metalloproteinases (MMPs), which are the major regulators of extracellular matrix (ECM) degradation in the glomerulus14,15.
A key feature of GN is the infiltration of inflammatory cells, especially a massive accumulation of T cells, monocytes, and macrophages, which are closely associated with poor outcomes16,17. ICs bind to FcRs that are expressed by effector cells, including B cells, monocytes and macrophages. Mouse Fc receptors for IgG (FcγRs) are composed of several activating receptors (FcγRI or CD64, FcγRIII or CD16, and FcγRIV) and one inhibitory receptor (FcγRIIB or CD32B)18, which are distinguished by their cytosolic domains. The activating FcγRs have an immunoreceptor tyrosine-based activation motif (ITAM), while the inhibitory FcγR contains an immunoreceptor tyrosine-based inhibitory motif (ITIM)18. In FcR-expressing immune cells, the activation of FcγRs is required for IgG-dependent effector functions, including immune cell activation, degranulation, phagocytosis, antibody-dependent cellular cytotoxicity (ADCC), and balanced activating and inhibitory signaling19,20. FcγRIIB is well defined as a negative regulator of IC-triggered activation, which contributes to the development and progression of autoimmune diseases. Notably, FcγRIIB-deficient NZB/NZW F1 mice suffer accelerated glomerular injuries21. Once the ITIM domain of FcγRIIB is phosphorylated, SH2-domain-containing inositol polyphosphate 5′ phosphatase (SHIP) can be recruited and activated, resulting in the hydrolysis of phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) to PtdIns(3,4)P2. This event results in an interruption of the recruitment of BTK to membrane PtdIns(3,4,5)P3, thus blocking downstream signaling pathways9.
(5R)-5-hydroxytriptolide (LLDT-8) is a novel triptolide analog extracted from the Chinese traditional herb Tripterygium wilfordii Hook. f. (TWHF). LLDT-8 possesses better immunosuppressive activities and lower toxicities than does triptolide22,23, which has shown beneficial effects on both collagen-induced arthritis in DBA/1 mice by suppressing IFN-γ signaling24 and lupus nephritis in MRL/lpr mice by preventing immune cell infiltration25. Here, we investigated the therapeutic effects of LLDT-8 on anti-GBM GN in New Zealand White (NZW) mice. Anti-GBM GN is a well-established antibody-induced GN model in which mice are immunized with heterologous anti-GBM antibodies to mimic human nephritis via the initiation of an autoantibody-mediated inflammatory response. This model shares a number of pathogenic mechanisms with human GN, including IC deposition, complement activation, immune cell infiltration, FcR expression and other mediators26,27. In addition, NZW mice are thought to be more susceptible to anti-GBM-induced diseases compared with C57BL/6 and BALB/c controls28. We show that LLDT-8 treatment suppresses proteinuria and kidney injury in this model. Furthermore, the present study highlights another underlying mechanism of LLDT-8, namely, its regulation of the expression of FcγRs and related signaling molecules. These results link FcγRIIB pathway activation to anti-GBM glomerulonephritis.
Materials and methods
Mice
NZW parental mice were purchased from Jackson Laboratory (JAX, Bar Harbor, ME, USA) and inbred in specific pathogen-free conditions (12-h light/12-h dark photoperiod, 22±1°C, 55%±5% relative humidity). Male mice aged 9 weeks were used in the experiment. All the experiments were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Bioethics Committee of the Shanghai Institute of Materia Medica.
Animal experiments
Anti-GBM serum was prepared as previously described29. Rabbit IgG, emulsified in Complete Freund' Adjuvant (CFA, Sigma, St Louis, MO, USA) at a final concentration of 2 mg/mL, was injected subcutaneously on d 0. Heat-inactivated anti-GBM serum was then intraperitoneally injected (500 μL) on d 4. The mice were followed and sacrificed to collect tissues for analysis on d 14.
LLDT-8, (purity >98.5%) was synthesized at Shanghai Pharmaceuticals Holding Co Ltd (Shanghai, China) and dissolved in 0.2% (hydroxypropyl)methyl cellulose (HPMC, Sigma). The GN mice were divided into 3 groups (n=8 per group): a vehicle group (0.2% HPMC), a prednisolone (PNS, Shanghai Sine Pharmaceutical Corp Ltd, Shanghai, China) group (2 mg·kg-1·d-1), and a LLDT-8-treated group (0.125 mg·kg-1·2d-1). The drugs were orally administered from d 0 to d 14. Eight male NZW mice were chosen as normal controls.
Assessment of urine samples and blood biochemical indexes
Spot urine was collected at d 0, 4, 7, 11 and 14 according to the 'bladder massage' method30. Urine protein was measured by performing a Coomassie brilliant blue dye-binding assay (BIO-RAD, Hercules, CA, USA), and urine creatinine was measured using Jaffe's reaction (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Serum albumin (ALB) and creatinine (CRE) levels were measured using a HITACHI-7080 automatic biochemical analyzer (Hitachi High Technologies Corporation, Tokyo, Japan), following the manufacturer's protocol.
Morphological examinations
The kidneys were fixed in 4% paraformaldehyde. The tissue sections were stained with hematoxylin and eosin (H&E) and visualized using a Leica DM 68 microscope (Leica Microsystems, Wetzlar, Germany). The histological scores were evaluated as previously described31.
ELISA
Cytokines in the serum or kidney homogenates were assayed using a mouse TNF-α ELISA kit (R&D systems, Minneapolis, MN, USA), as well as mouse IL-6, IL-17, and IFN-γ ELISA kits (BD Pharmingen, San Diego, CA, USA), according to the manufacturer's instructions. The serum anti-rabbit IgG antibodies were evaluated as previously described29.
IF
The kidney tissues were dehydrated in sucrose and mounted in OCT. Six-micron frozen sections were cut using a Leica CM950 cryostat (Leica Microsystems), air dried, and fixed in acetone. After blocking and washing, the sections were incubated with FITC-conjugated rabbit anti-mouse IgG (1:50, Beyotime, Shanghai, China), a FITC-conjugated anti-CD11b antibody (1:100, Abcam, Cambridge, MA, USA) and a rat anti-mouse C3 antibody (1:50, Santa Cruz Biotechnology, Santa Cruz, CA, USA). These steps were followed by incubation with a FITC-conjugated goat anti-rat IgG antibody (1:100, Santa Cruz Biotechnology, USA). The slides were mounted with Vectashield (Beyotime) or DAPI (Abcam). Fluorescence images were captured using a Nikon Eclipse Ti microscope (Nikon Instruments Inc, Tokyo, Japan) or a Leica LAS TCS-SP8 STED microscope (Leica Microsystems) and assembled into figures using Adobe Photoshop (Adobe Systems Incorporated, San Jose, USA). IgG and C3 deposition were semi-quantitatively analyzed using Image-Pro plus 6.0 (Media Cybernetics, MD, USA).
RNA isolation, cDNA synthesis, and quantitative reverse transcription PCR
Total RNA was extracted using a Total RNA kit (DP419, Tiangen, Shanghai, China), and reverse transcription was performed using an All-in-One cDNA Synthesis SuperMix (Biotool, Houston, Texas, USA) for TaqMan analysis. qRT-PCR was performed using a 2×SYBR Green qPCR Master Mix (High ROX) (Biotool) on an Applied Biosystems 7900 Fast Real-Time PCR System (Applied Biosystems, Stadt, CA, USA). Relative quantitation of mRNA expression was performed using the ΔΔCt method. Ct values were first normalized within the sample to the housekeeping gene β-actin before comparison across the normal group samples. The mouse primers were synthesized by Sangon Biotech Co, Ltd (Shanghai, China).
Renal leukocyte isolation
The mice were sacrificed, and the kidneys were removed aseptically. Renal cells were isolated from murine kidneys as previously described32. Briefly, kidneys were finely minced and digested for 20 min at 37 °C with 0.5 mg/mL collagenase from Clostridium histolyticum (Sigma). The cell suspensions were sequentially filtered through 70- and 40-μm nylon mesh filters (Shanghai Baolan Shiyan Yiqi Co Ltd, Shanghai, China) and washed with phosphate-buffered saline (PBS, Sigma). Single-cell suspensions were separated using Percoll (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) density gradient (70% and 35%) centrifugation. The leukocyte-enriched cell suspension was aspirated from the Percoll interface. Cell viability was assessed using trypan blue (Life Technologies, USA) staining before flow cytometry. Single-cell suspensions were washed and resuspended in RPMI-1640 media (HyClone Laboratories, Logan, UT, USA) containing 10% fetal bovine serum (FBS, HyClone Laboratories) supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin (North China Pharmaceutical Group Corporation, Shijiazhuang, China).
Flow cytometry analysis
The cells were incubated with various combinations of mAbs, including FITC-conjugated and PerCP-Cy5.5-conjugated anti-CD19, PerCP-Cy5.5-conjugated CD11b, Alexa Fluor® 647-conjugated CD64, BV421-conjugated anti-CD3 (BD Pharmingen, BD Biosciences, San Jose, CA, USA), PE-conjugated anti-CD32b (eBioscience) and FITC-conjugated anti-CD16 (BIO-RAD). Flow cytometric analysis was performed on a BD LSRFortessa (BD Biosciences) flow cytometer, and the data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Western blotting
Samples were directly lysed in sodium dodecyl sulfate (Beyotime Institute of Biotechnology) containing a protease inhibitor cocktail (Roche Life Science, Mannheim, Germany) and then boiled for 10 min at 100 °C. The aliquots were electrophoresed in a 10% polyacrylamide gel and transferred to nitrocellulose membranes (Merck Millipore Ltd, MA, USA). After blocking, the membranes were incubated with a panel of primary antibodies overnight at 4°C, including those against GAPDH (KangChen Biotechnology, Shanghai, China), SHIP1, phospho-SHIP1, BTK, and phospho-BTK (Cell Signaling Technology, Danvers, MA, USA). After washing, the membranes were incubated with HRP-conjugated anti-rabbit IgG (BIO-RAD) for 1 h. The signals were detected with an ECL system (Amersham Biosciences) and exposed on Kodak X-ray film (Kodak). The densities of the bands were quantified using a computerized densitometer (ImageJ Launcher, Broken Symmetry Software, Bristol, UK).
Statistical analysis
The data were analyzed using one-way analysis of variance (one-way ANOVA) for repeated measures, corrected with Dunnett's post-test. Student's t-test was used to assess significant differences between two groups. Statistical analyses were conducted using GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). The data are presented as the mean±SEM, where indicated.
Results
LLDT-8 ameliorated the clinical and histological symptoms of anti-GBM GN in NZW mice
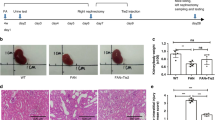
To investigate the effect of LLDT-8 on anti-GBM GN, 9-week-old male NZW mice were sensitized with rabbit anti-mouse IgG and subsequently challenged with anti-GBM serum. The mice were sacrificed 14 d after the initiation of the challenge (Figure 1A). GN mice were orally administered HPMC (0.2%, solvent control) every other day, LLDT-8 (0.125 mg/kg) every other day, or PNS (2 mg/kg, positive control) every day. In the LLDT-8-treated group, both proteinuria and the urine protein-creatinine ratio were significantly reduced compared with the vehicle group (Figure 1B). In addition, LLDT-8 prevented renal dysfunction (Figure 1C). Mildly elevated ALB was observed in the LLDT-8-treated group (Figure 1C). A kidney histopathology assay was performed to further confirm the effects of LLDT-8. The mice in the vehicle group exhibited renal injury, which was characterized by increased mesangial matrix, tubular cast deposition, and interstitial cell infiltration. By contrast, these pathological features were significantly ameliorated in LLDT-8-treated mice (Figure 1D and 1E).

LLDT-8 treatment ameliorated anti-GBM nephritis in NZW mice. (A) NZW mice were sensitized subcutaneously with rabbit IgG emulsified in CFA at a final concentration of 2 mg/mL on d 0. The mice were subsequently challenged intraperitoneally with 500 μL of anti-GBM serum on d 4 and sacrificed on d 14. LLDT-8, PNS and HPMC were administered from d 0 to d 14. (B) Left, Proteinuria levels were measured at various time points in Normal, Vehicle, PNS and LLDT-8 mice (n=8). Right, Urine albumin levels were normalized to urinary creatinine levels to adjust for urinary output. (C) The serum levels of CRE and ALB were detected at the end of the study. (D) Representative images of hematoxylin and eosin (H&E) stained kidney tissue sections are shown. H&E stain, ×200 (small) and ×400 (large). Black scale bars: 10 μm (small) and 50 μm (large). (E) Quantitative analysis of H&E scores in glomeruli and the interstitium. *P<0.05, **P<0.01 vs vehicle group.
LLDT-8 reduced the glomerular deposition of IgG and C3 without affecting serum antibodies
NZW mice immunized with heterologous anti-GBM serum generate antibodies to neutralize the foreign antigens. In our study, serum anti-rabbit antibodies were not obviously reduced by LLDT-8 (Figure 2A). We next investigated whether LLDT-8 interfered with the deposition of IC and C3. Previous work has reported that IgG binds to the glomeruli in a linear pattern and that C3 binds along the capillary wall in a fine granular pattern after induction33,34. High intensities of antibody labeling for IgG (Figure 2B) and C3 (Figure 2C), indicating significant glomerular deposition of IC, were observed in the vehicle group but not in the normal group. Compared to vehicle treatment, both IgG and C3 deposition in glomeruli were reduced in LLDT-8 treatment mice (Figure 2D).

Impacts of LLDT-8 on serum antibodies and IgG and C3 deposition in glomeruli. (A) ELISA for anti-rabbit total mouse serum IgG level (dilution 1:1000). (B and C) Frozen sections stained for glomerular deposition of IgG (B) and C3 (C). Representative IF staining images are shown. Magnification×100 (small) and ×200 (large). (D) Quantitative analysis of IgG and C3 deposition in glomeruli, presented as the IOD value. *P<0.05 vs vehicle group.
LLDT-8 inhibited the inflammatory responses in GN mice
Inflammatory cytokines play an important role in the pathogenesis of acute renal injury10. To investigate whether LLDT-8 affects inflammatory responses, we analyzed the mRNA expression of inflammatory cytokines in kidney, including TNF-α, IL-6, IL-17, and IFN-γ. LLDT-8 lowered the upregulated expression profile of TNF-α, IL-6, IL-17, and IFN-γ in vehicle mice, although the differences in IL-17 and IFN-γ mRNA expression were not significant. This lack of significance was due to clear individual differences within the vehicle group (Figure 3A). Furthermore, we detected the protein levels of these cytokines in kidney and serum by ELISA. LLDT-8 significantly decreased these cytokines in kidney cell homogenates (Figure 3B) as well as the serum levels of TNF-α and IFN-γ, but not IL-6 and IL-17 (Figure 3C).

Effects of LLDT-8 on inflammatory responses in anti-GBM GN mice. (A) Kidney tissues from normal mice and from vehicle- and LLDT-8-treated mice were used to assess the relative mRNA levels of TNF-α, IL-6, IL-17, and IFN-γ. (B) Kidney homogenates and (C) serum were collected to measure the levels of these cytokines via ELISA. *P<0.05, **P<0.01 vs vehicle group.
LLDT-8 reduced renal leukocyte recruitment
The development of disease in GN mice requires not only the presence of glomerular immune complexes35 but also the recruitment of leukocytes, including lymphocytes and monocytes, to the kidney36. CD11b+-expressing mononuclear phagocytes represent the major infiltrating monocytes/macrophages at the peak of lupus nephritis37. Thus, the proportion of renal leukocytes was analyzed by flow cytometry. LLDT-8 significantly decreased the percentage of CD11b+ cells compared with vehicle mice (Figure 4C). CD3+ T cells and CD19+ B cells (Figure 4A and 4B) were reduced after LLDT-8 treatment compared to vehicle mice, but this effect was not significant. The infiltration of CD11b+ cells in nephritis is associated with poor disease outcome37,38. The influx of CD11b+ cells was observed in vehicle mice, but little infiltration was detected in LLDT-8-treated mice. No influx of these cells was observed in normal mice (Figure 4D). The infiltration of inflammatory cells occurs due to the differential expression patterns of chemokine receptors on leukocyte subsets, the in situ production of which leads to the recruitment of circulating leukocytes to inflammatory sites39. In line with the flow cytometric data, renal chemokine expression exhibited a statistically significant decrease after LLDT-8 treatment, especially in terms of the expression of cytokine-related genes40, including CCL2, CCL5, CCL7, and CX3CL1. In addition, MMPs, including MMP2 and MMP9, which are the major regulators of ECM in the glomerulus and have been implicated in the development of glomerular injury, were significantly downregulated in LLDT-8-treated mice (Figure 4E).

Impacts of LLDT-8 on renal leukocyte infiltration and gene expression. (A-C) Representative flow cytometry analysis of CD3+ cells and the proportion in renal leukocytes (B) CD19+ cells and (C) CD11b+ cells in each group at the end of the experiments. (D) Representative confocal images of kidney cryosections for CD11b+ cells from each group are shown. The sections were stained with an anti-CD11b antibody (green) and DAPI (blue) for IF analysis. IF staining, ×200 (small) and ×630 (large). White scale bars: 10 μm (small) and 25 μm (large). (E) qRT-PCR analysis of renal chemokine and MMP mRNA expression levels. *P<0.05, **P<0.01 vs vehicle group.
LLDT-8 regulated the gene expression of FcγRs and its related signaling pathway
Self-reactive antibodies represent a significant force in autoimmune disease induction. In antibody-dependent autoimmune syndromes, such as anti-GBM GN, autoantibodies exert their inflammatory effect through FcγRs, a well-established class of cell surface receptors that interact with the Fc domain of IgG. Thus, the gene expression profiles of FcγRs in the kidney and spleen were tested. Inhibitory FcγRIIB was downregulated, while the activating genes FcγRI and FcγRIII were upregulated in kidneys from vehicle mice (Figure 5A). Interestingly, LLDT-8 restored the reduced expression of FcγRIIB in both kidney and spleen tissues (Figure 5A and 5B, left), but only in the kidney were reductions in FcγRI and FcγRIII levels observed (Figure 5A, middle and right). No effect was observed on the expression of these receptors in the spleen (Figure 5B, middle and right). We then assessed the phosphorylation levels of SHIP1, which is recruited by the cytosolic ITIM domain of FcγRIIB. SHIP1 hydrolyzes PtdIns(3,4,5)P3 to PtdIns(3,4)P2 and thereby interrupts the recruitment of BTK to membrane PtdIns(3,4,5)P3. LLDT-8 promoted the activation of SHIP1 and suppressed the phosphorylation of BTK in mouse anti-GBM GN (Figure 5C).

Effects of LLDT-8 on FcγR gene expression and related signaling pathway activation. (A) qRT-PCR analysis of renal FcγR mRNA expression. (B) qRT-PCR analysis of splenic FcγR mRNA expression. (C) Splenocytes from normal mice and from vehicle- and LLDT-8-treated mice were analyzed by Western blotting for SHIP1, p-SHIP1, BTK, and p-BTK. The blots were incubated with anti-SHIP1 Ab, anti-phospho-SHIP1 Ab, anti-BTK Ab, and anti-phospho-BTK Ab. **P<0.01 vs vehicle group.
LLDT-8 significantly upregulated FcγRIIB expression on CD11b+ cells
FcγRs have been demonstrated to be involved in the development of nephritis, the direct activation of FcR-bearing monocytes/macrophages is sufficient to induce immune responses after glomerular IC deposition38. Moreover, FcγRIIB−/− mice suffer accelerated glomerular injuries after injection with nephrotoxic serum21. Thus, the expression of FcγRIIB on CD11b+ cells was detected by flow cytometry. The expression of FcγRIIB on CD11b+ cells was upregulated in LLDT-8-treated mice when compared with vehicle-treated mice (Figure 6A). Similarly, LLDT-8 led to a slight upregulation of the expression of FcγRIIB on CD19+ B cells (Figure 6B).

LLDT-8 upregulated FcγRIIB expression on immune cells. (A) The expression of FcγRs was detected by flow cytometry analysis. The gating strategy for CD11b+ cells. (B) The gating strategy for CD19+ cells. *P<0.05 vs vehicle group.
Discussion
In the present study, we demonstrated that LLDT-8 treatment ameliorated proteinuria and renal pathology associated with anti-GBM GN in NZW mice. More importantly, we elucidated another pharmacological mechanism of LLDT-8, namely, the suppression of renal injury in murine GN by regulating the FcγR expression and related pathways, especially those linked to FcγRIIB activation.
The deposition of antibodies in kidney triggers pathogenic consequences in the anti-GBM GN model. In our research, both vehicle- and LLDT-8-treated mice were exposed to similar levels of serum antibodies. However, active disease does not always depend on high serum levels of antibodies41, and immune GN that is mediated by pathogenic antibodies can be ameliorated without affecting circulating nephrotoxic antibodies42. Glomerular damage occurs after IC deposition, which initiates complement activation3, and a potent inhibitor of C3 convertases was previously reported to dramatically protect MRL/lpr lupus mice against renal disease43. Here, despite the continued presence of serum antibodies, we confirmed a reduced glomerular deposition of IgG and C3 by IF in LLDT-8-treated mice when compared with vehicle-treated mice. This result indicates that LLDT-8 does not affect the generation of antibodies but rather interferes with IC deposition and downstream C3 activation. In addition, triptolide, the parent compound of LLDT-8, was shown to remarkably improve lupus nephritis, with little effect on serum antibodies44.
The immune system recognizes ICs as foreign, resulting in the generation of inflammatory cytokines, chemokines, and MMPs as well as the recruitment of inflammatory cells6,7,14,15. We have demonstrated that LLDT-8 treatment decreased the levels of several cytokines, such as TNF-α and IFN-γ, in serum and kidney, thereby protecting the kidney from inflammatory injury. In addition, LLDT-8 treatment reversed the increased expressions of MMP2 and MMP9, which can damage the structural integrity of the GBM in human and murine nephritis45,46. Previous studies have documented that T cells47 and monocytes/macrophages37 infiltrate the glomeruli and are attracted by chemokines to inflammatory sites48. The transfer of adoptive macrophages through tail vein injection causes disease re-acceleration in rat anti-GBM GN49, while macrophage depletion ameliorates pathogenic antibody-induced nephritis42. The present results show that LLDT-8 treatment can successfully reduce the expression of kidney chemokines, including CCL2, CCL5, CCL7, and CX3CL1. The therapeutic blockade of inflammatory chemokines and cytokines reduces renal leukocyte infiltration and ameliorates glomerular and secondary tubulointerstitial tissue injury10. In our study, both flow cytometry and IF confirmed a marked decrease of monocyte/macrophage influx in the kidney after LLDT-8 treatment.
Several studies have addressed the role of FcγRs in GN, which can occur independently of FcR in the absence of antibodies21,50. When deficient in the FcR γ chain or FcγRIIB, NZB/NZW F1 mice were protected from severe nephritis induced via exogenous ICs21,35. We further demonstrated that LLDT-8 treatment successfully upregulates FcγRIIB expression in the kidney and spleen. Treatment also results in lower gene expression levels of FcγRI and FcγRIII in the kidney but not the spleen, possibly due to reduced levels of FcγR-bearing cells in the kidney. Furthermore, we observed upregulated expression of FcγRIIB both on monocytes/macrophages and B cells using flow cytometry. FcγRIIB acts physiologically as a negative regulator. Some studies have shown that natural polymorphisms in the mouse FcγRIIB gene promoter lead to reduced receptor expression51, and the 2B.4 promoter haplotype induces high FcγRIIB expression on monocytes and primary B lymphocytes52. Thus, we speculated that LLDT-8 may play a role in FcγRIIB production. Once the FcγRIIB cytoplasmic region (ITIM) is activated, it recruits SHIP1. The latter hydrolyzes PtdIns(3,4,5)P3, interrupting BTK recruitment and thereby inhibiting downstream pathways and the release of calcium. In this way, a series of responses, including peripheral tolerance and the modulation of cell activation, are avoided18,20. Western blotting indicated that LLDT-8 treatment interfered with the FcγRIIB-dependent signaling pathway.
In summary, we have demonstrated the efficacy and mechanisms of LLDT-8 in murine anti-GBM GN. LLDT-8 can reduce IgG deposition, regulate the expression of FcγRs and activate the FcγRIIB-mediated signaling pathway. LLDT-8 can decrease C3 deposition through these effects, hampering both inflammation and leukocyte infiltration in the kidney.
Author contribution
Jian-ping ZUO, Wei TANG, Shi-jun HE, and Qing QI designed the experiments. Qing QI, Xiao-qian YANG, Feng-hua ZHU, Heng LI, Ze-min LIN, Yu-ting LIU, Mei-juan SHAO, Yan-sheng XU, Yu-xi YAN, and Lan-lan SUN performed the experiments. Jian-ping ZUO, Wei TANG, Shi-jun HE, and Qing QI analyzed the data. Jian-ping ZUO and Wei TANG contributed reagents/materials/analysis tools. Jian-ping ZUO, Wei TANG, and Qing QI wrote the manuscript. Lu-yao ZHANG edited the manuscript.
References
Collins AJ, Foley RN, Gilbertson DT, Chen SC . United States Renal Data System public health surveillance of chronic kidney disease and end-stage renal disease. Kidney Int Suppl 2015; 5: 2–7.
Breyer MD, Susztak K . The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov 2016; 15: 568–88.
Dickinson BL . Unraveling the immunopathogenesis of glomerular disease. Clin Immunol 2016; 169: 89–97.
Ravetch JV . Fc receptors: rubor redux. Cell 1994; 78: 553–60.
Kluth DC, Rees AJ . Anti-glomerular basement membrane disease. J Am Soc Nephrol 1999; 10: 2446–53.
Zipfel PF, Skerka C . Complement regulators and inhibitory proteins. Nat Rev Immunol 2009; 9: 729–40.
Harboe M, Thorgersen EB, Mollnes TE . Advances in assay of complement function and activation. Adv Drug Deliv Rev 2011; 63: 976–87.
Mayadas TN, Tsokos GC, Tsuboi N . Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation 2009; 120: 2012–24.
Kurts C, Panzer U, Anders HJ, Rees AJ . The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol 2013; 13: 738–53.
Ramesh G, Reeves WB . Inflammatory cytokines in acute renal failure. Kidney Int 2004; 66: S56–61.
Kitching AR, Holdsworth SR, Tipping PG . IFN-γ mediates crescent formation and cell-mediated immune injury in murine glomerulonephritis. J Am Soc Nephrol 1999; 10: 752–9.
Turner J, Paust H, Steinmetz OM, Peters A, Meyerschwesinger C, Heymann F, et al. CCR5 deficiency aggravates crescentic glomerulonephritis in mice. J Immunol 2008; 181: 6546–56.
Liao X, Pirapakaran T, Luo XM . Chemokines and chemokine receptors in the development of lupus nephritis. Mediators Inflamm 2016; 2016: 6012715. doi: 10.1155/2016/6012715.
Gibbs DF, Warner RL, Weiss SJ, Johnson KJ, Varani J . Characterization of matrix metalloproteinases produced by rat alveolar macrophages. Am J Respir Cell Mol Biol 1999; 20: 1136–44.
Blom AB, Radstake TR, Holthuysen AEM, Sloetjes A, Pesman GJ, Sweep FGJ, et al. Increased expression of Fcγ receptors II and III on macrophages of rheumatoid arthritis patients results in higher production of tumor necrosis factor α and matrix metalloproteinase. Arthritis Rheum 2003; 48: 1002–14.
Hu SY, Jia XY, Li JN, Zheng X, Ao J, Liu G, et al. T cell infiltration is associated with kidney injury in patients with anti-glomerular basement membrane disease. Sci China Life Sci 2016; 59: 1282–9.
Lan HY, Nikolicpaterson DJ, Mu W, Atkins RC . Local macrophage proliferation in the pathogenesis of glomerular crescent formation in rat anti-glomerular basement membrane (GBM) glomerulonephritis. Clin Exp Immunol 1997; 110: 233–40.
Nimmerjahn F, Ravetch JV . Fcγ receptors as regulators of immune responses. Nat Rev Immunol 2008; 8: 34–47.
Bruhns P . Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012; 119: 5640–9.
Takai T . Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2002; 2: 580–92.
Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, et al. Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int 1998; 54: 1166–74.
Zhou R, Zhang F, He PL, Zhou WL, Wu QL . Xu JY, et al. (5R)-5-hydroxytriptolide (LLDT-8), a novel triptolide analog mediates immunosuppressive effects in vitro and in vivo. Int Immunopharmacol 2005; 5: 1895–903.
Tang W, Zuo JP . Immunosuppressant discovery from Tripterygium wilfordii Hook f: the novel triptolide analog (5R)-5-hydroxytriptolide (LLDT-8). Acta Pharmacol Sin 2012; 33: 1112–8.
Zhou R, Tang W, Ren YX, He PL, Zhang F, Shi LP, et al. (5R)-5-hydroxytriptolide attenuated collagen-induced arthritis in DBA/1 mice via suppressing interferon-gamma production and its related signaling. J Pharmacol Exp Ther 2006; 318: 35–44.
Zhang LY, Li H, Wu YW, Cheng L, Yan YX, Yang XQ, et al. (5R)-5-Hydroxytriptolide ameliorates lupus nephritis in MRL/lpr mice by preventing infiltration of immune cells. Am J Physiol 2017; 312: F769–F777.
Assmann KJM, Tangelder MM, Lange WP, Schrijver G, Koene RA . Anti-GBM nephritis in the mouse: severe proteinuria in the heterologous phase. Virchows Arch 1985; 406: 285–99.
Fu Y, Du Y, Mohan C . Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin Immunol 2007; 124: 109–18.
Xie C, Sharma R, Wang H, Zhou XJ, Mohan C . Strain distribution pattern of susceptibility to immune-mediated nephritis. J Immunol 2004; 172: 5047–55.
Li TT, Zhang XH, Jing JF, Li X, Yang XQ, Zhu FH, et al. Artemisinin analogue SM934 ameliorates the proteinuria and renal fibrosis in rat experimental membranous nephropathy. Acta Pharmacol Sin 2015; 36: 188–99.
Wu Y, He S, Bai B, Zhang L, Xue L, Lin Z, et al. Therapeutic effects of the artemisinin analog SM934 on lupus-prone MRL/lpr mice via inhibition of TLR-triggered B-cell activation and plasma cell formation. Cell Mol Immunol 2016; 13: 379–90.
Tao X, Fan F, Hoffmann V, Longo NS, Lipsky PE . Therapeutic impact of the ethyl acetate extract of Tripterygium wilfordii Hook F on nephritis in NZB/W F1 mice. Arthritis Res Ther 2006; 8: R24.
Ascon DB, Lopezbriones S, Liu M, Ascon M, Savransky V, Colvin RB, et al. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J Immunol 2006; 177: 3380–7.
Schrijver G, Bogman MJJT, Assmann KJM, De Waal RMW, Robben HCM, Van Gasteren H, et al. Anti-GBM nephritis in the mouse: role of granulocytes in the heterologous phase. Kidney Int 1990; 38: 86–95.
Dempsey PW, Fearon DT . C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 1996; 271: 348–50.
Clynes R, Dumitru CD, Ravetch JV . Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 1998; 279: 1052–4.
Kitching AR, Holdsworth SR, Hickey MJ . Targeting leukocytes in immune glomerular diseases. Curr Med Chem 2008; 15: 448–58.
Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, et al. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus Nephritis. J Immunol 2011; 186: 4994–5003.
Bergtold A, Gavhane A, Agati VDD, Madaio MP, Clynes R . FcR-bearing myeloid cells are responsible for triggering murine lupus nephritis. J Immunol 2006; 177: 7287–95.
Cockwell P, Howie AJ, Adu D, Savage CO . In situ analysis of C-C chemokine mRNA in human glomerulonephritis. Kidney Int 1998; 54: 827–36.
Mina-Osorio P, LaStant J, Keirstead N, Whittard T, Ayala J, Stefanova S, et al. Suppression of glomerulonephritis in lupus-prone NZB×NZW mice by RN486, a selective inhibitor of Bruton's tyrosine kinase. Arthritis Rheum 2013; 65: 2380–91.
Gladman DD, Urowitz MB, Keystone EC . Serologically active clinically quiescent systemic lupus erythematosus: a discordance between clinical and serologic features. Am J Med 1979; 66: 210–5.
Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C . Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun 2014; 57: 42–52.
Bao L, Haas M, Kraus DM, Hack BK, Rakstang JK, Holers VM, et al. Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice. J Am Soc Nephrol 2003; 14: 670–9.
Tao X, Fan F, Hoffmann V, Gao CY, Longo NS, Zerfas P, et al. Effective therapy for nephritis in (NZB×NZW)F1 mice with triptolide and tripdiolide, the principal active components of the Chinese herbal remedy Tripterygium wilfordii Hook F. Arthritis Rheumatol 2008; 58: 1774–83.
Urushihara M, Kagami S, Kuhara T, Tamaki T, Kuroda Y . Glomerular distribution and gelatinolytic activity of matrix metalloproteinases in human glomerulonephritis. Nephrol Dialysis Transplant 2002; 17: 1189–96.
Tveita AA, Rekvig OP, Zykova SN . Increased glomerular matrix metalloproteinase activity in murine lupus nephritis. Kidney Int 2008; 74: 1150–8.
Tipping PG, Holdsworth SR . T cells in crescentic glomerulonephritis. J Am Soc Nephrol 2006; 17: 1253–63.
Zlotnik A, Yoshie O . Chemokines: a new classification system and their role in immunity. Immunity 2000; 12: 121–7.
Ikezumi Y, Hurst LA, Masaki T, Atkins RC, Nikolic-Paterson DJ . Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int 2003; 63: 83–95.
Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN . The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol 2014; 14: 94–108.
Jiang Y, Hirose S, Abe M, Sanokawaakakura R, Ohtsuji M, Mi X, et al. Polymorphisms in IgG Fc receptor IIB regulatory regions associated with autoimmune susceptibility. Immunogenetics 2000; 51: 429–35.
Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP . A Promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcγRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. J Immunol 2004; 172: 7192–9.
Acknowledgements
This work was supported by a grant from the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (No 2014ZX09101002-001). The authors declare no other commercial or financial conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Qi, Q., Li, H., Lin, Zm. et al. (5R)-5-hydroxytriptolide ameliorates anti-glomerular basement membrane glomerulonephritis in NZW mice by regulating Fcγ receptor signaling. Acta Pharmacol Sin 39, 107–116 (2018). https://doi.org/10.1038/aps.2017.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.88
Keywords
This article is cited by
-
Application of nanotechnology in the treatment of glomerulonephritis: current status and future perspectives
Journal of Nanobiotechnology (2024)
-
Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages
Acta Pharmacologica Sinica (2018)
