
Abstract
Necroptosis is a type of programmed necrosis regulated by receptor interacting protein kinase 1 (RIP1) and RIP3. Necroptosis is found to be accompanied by an overproduction of reactive oxygen species (ROS), but the role of ROS in regulation of necroptosis remains elusive. In this study, we investigated how shikonin, a necroptosis inducer for cancer cells, regulated the signaling leading to necroptosis in glinoma cells in vitro. Treatment with shikonin (2–10 μmol/L) dose-dependently triggered necrosis and induced overproduction of intracellular ROS in rat C6 and human SHG-44, U87 and U251 glioma cell lines. Moreover, shikonin treatment dose-dependently upregulated the levels of RIP1 and RIP3 and reinforced their interaction in the glioma cells. Pretreatment with the specific RIP1 inhibitor Nec-1 (100 μmol/L) or the specific RIP3 inhibitor GSK-872 (5 μmol/L) not only prevented shikonin-induced glioma cell necrosis but also significantly mitigated the levels of intracellular ROS and mitochondrial superoxide. Mitigation of ROS with MnTBAP (40 μmol/L), which was a cleaner of mitochondrial superoxide, attenuated shikonin-induced glioma cell necrosis, whereas increasing ROS levels with rotenone, which improved the mitochondrial generation of superoxide, significantly augmented shikonin-caused glioma cell necrosis. Furthermore, pretreatment with MnTBAP prevented the shikonin-induced upregulation of RIP1 and RIP3 expression and their interaction while pretreatment with rotenone reinforced these effects. These findings suggest that ROS is not only an executioner of shikonin-induced glioma cell necrosis but also a regulator of RIP1 and RIP3 expression and necrosome assembly.
Similar content being viewed by others


Introduction
Glioma is the most common type of malignant primary brain tumor. It is very difficult to cure malignant gliomas because they are resistant to currently used chemotherapeutic agents and radiotherapeutic regimes1. However, accumulating studies have demonstrated that induction of necroptosis is an effective strategy to eliminate various cancer cells, including colon cancer cells, breast cancer cells and lung adenocarcinoma cells2,3,4. Necroptosis induction is even effective for eliminating cancer cells that are resistant to apoptosis5. Thus, necroptosis-based therapy has shed a new light on glioma treatment.
As a type of programmed necrosis, necroptosis not only contributes to cancer cell death induced by chemotherapeutic agents6 but is also involved in cell death caused by various pathological processes, such as inflammation and ischemia/reperfusion7,8. Necroptosis is morphologically similar to necrosis and characterized by increased cell volume, enlarged organelles, ruptured plasma membrane, and subsequent loss of intracellular contents9. Biochemically, necroptosis results from a sequential activation of a series of signals. Receptor interacting protein kinase 1 (RIP1) is an initiator of necroptosis, which interacts with receptor interacting protein kinase 3 (RIP3) via their RHIM domains to form the amyloid-like necrosome complex. Then, RIP3 activated by RIP1 within the necrosome recruits and phosphorylates its downstream substrate mixed lineage kinase-like domain protein (MLKL)10,11. Ultimately, the phosphorylated MLKL forms an oligomer, which causes cellular membrane leakage and cell death10,11,12,13. Thus, necrosome formation is a key step leading to necroptosis, but the factors affecting the interaction between RIP1 and RIP3 are still unclear.
Shikonin, a naphthoquinone isolated from Lithospermum erythrorhizon, has been repeatedly reported to be an effective necroptosis inducer for cancer cells3,14,15,16. Similar to necroptosis induced by other reagents17,18, shikonin-triggered necroptosis is accompanied by an overproduction of reactive oxygen species (ROS)3,14. Moreover, ROS appear to play a crucial role in glioma cell necrosis because mitigated ROS rescued glioma cell necrosis induced by not only shikonin but also other natural chemicals, such as pristimerin and deoxypodophyllotoxin14,19,20. However, it remains elusive whether ROS participate in the regulation of signals leading to necroptosis. Herein, we report that RIP1 and RIP3 regulate the shikonin-induced overproduction of intracellular ROS by excessively generating mitochondrial superoxide. Reversely, ROS improve the shikonin-induced expressional upregulation of RIP1 and RIP3 and reinforce their interaction. This finding provides novel insight into the molecular mechanism underlying glioma cell necroptosis and suggests that shikonin is a potential therapeutic agent for glioma.
Materials and methods
Reagents
Shikonin, Nec-1 (necrostatin-1), rotenone, and MnTBAP (metalloporphyrin Mn(III)tetrakis (4-benzoic acid) porphyrin) were all purchased from Sigma (St Louis, MO, USA). GSK-872 was purchased from Calbiochem (Billerica, MA, USA). Shikonin was dissolved in PBS at a storage concentration of 50 mmol/L. The anti-RIP1 and anti-RIP3 antibodies were purchased from Abcam (Cambridge, MA, USA). The anti-β-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Other reagents were purchased from Sigma.
Cell lines and culture
The rat C6 and human SHG-44, U87 and U251 glioma cell lines were obtained from the Shanghai Institute of Cell Biology at the Chinese Academy of Sciences (Shanghai, China). These cells were cultured in DMEM containing 10% fetal bovine serum, 2 mmol/L glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were maintained in a humidified environment at 37 °C and 5% CO2, and cells in the mid-log phase were used in the experiments.
Lactate dehydrogenase release cell death assay
C6 (1×105 cells/well), SHG-44 (5×104 cells/well), U87 (5×104 cells/well) and U251 (5×104 cells/well) glioma cells were treated with target compounds after being seeded onto 96-well microplates and cultured for 24 h. The cellular death rate was examined using the lactate dehydrogenase cytotoxicity assay kit (Beyotime Biotech, Haimen, China). According to the manufacturer's instructions, the absorbance value of each sample was read at 490 nm. The cell death ratio was calculated using the following formula: cell death ratio%=(Asample–Acontrol)/(Amax–Acontrol)×100, where Asample is the sample absorbance value; Acontrol is the control group absorbance value; and Amax is the positive group absorbance value.
Assessment of necrosis by flow cytometry
After being treated with the target compounds, the SHG-44 and U251 glioma cells assayed for cell death modality using the Annexin V-FITC detection kit (Invitrogen, Grand Island, NY, USA). According to the manufacturer's instruction, the cells were collected and washed twice with PBS and then suspended in 400 μL of 1× binding buffer. The cells (100 μL) were transferred to a 5-mL culture tube containing 5 μL of Annexin V-FITC and 10 μL of propidium iodide and then incubated for 15 min at room temperature in the dark. After 1× binding buffer was added to the tube, the stained cells were analyzed by flow cytometry (FACScan, Becton Dickinson, San Jose, CA, USA). The cell death rate was analyzed using CELLquest software (Becton Dickinson). Data acquisition was conducted by collecting 20 000 cells per tube and determining the number of viable and dead cells for each experimental condition.
Measuring intracellular reactive oxygen species and mitochondrial superoxide
SHG-44, U251, U87 and C6 glioma cells were seeded onto 96-well microplates and cultured for 24 h prior to being treated with the target compounds. The redox-sensitive dye DCFH-DA (Beyotime Biotech, Haimen, China) was used to evaluate the levels of intracellular ROS. Briefly, the cells were incubated with 1% Triton X-100 after being stained with 20 μmol/L DCFH-DA in the dark for 30 min. Then, the fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence spectrometer (HTS 7000, Perkin Elmer, Boston, MA, USA). The ROS levels were expressed as a percentage of the control.
MitoSOX Red (Invitrogen company, Eugene, OR, USA) was used to measure mitochondrial superoxide according to the manufacturer's instructions. The cells were incubated for 10 min with 2.0 mL of 5 μmol/L MitoSOX reagent working solution at 37 °C in the dark and then washed with PBS. The red fluorescence density was measured at an excitation wavelength of 510 nm and an emission wavelength of 580 nm. It was expressed as a ratio of the fluorescence in the control cells.
Additionally, SHG-44 glioma cells were seeded onto a 3-cm culture dish and cultured for 24 h. After being treated with the target compounds, the cells were stained with either DCFH-DA or MitoSox Red as described above and observed under a fluorescence microscope (Olympus IX71, Tokyo, Japan).
Gel electrophoresis and Western blotting
Cell collection and homogenization were performed as previously described20. Then, the homogenates were centrifuged at 1000×g for 10 min at 4 °C to obtain the supernatants, of which their protein content was determined using the Bio-Rad protein assay kit. After SDS electrophoresis and transfer, the PVDF membranes were blocked with 3% bovine serum albumin in TBS for 30 min at room temperature and then incubated overnight at 4 °C with anti-RIP1 (1:1000), anti-RIP3(1:1000), or anti-β-actin (1:1500) antibodies. After being incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2000) antibody, the blots were washed, and immunoreactive proteins were visualized on Kodak X-omat LS film (Eastman Kodak Company, New Haven, CT, USA) with an enhanced chemiluminescence substrate (Amersham Biosciences, Piscataway NJ, USA). Densitometry was performed with Kodak ID image analyses software.
Co-immunoprecipitation
Cell collection and homogenization were performed as previously described20. Then, the homogenates were centrifuged at 15 000×g for 15 min at 4 °C to obtain the supernatant. After the protein content was determined using the Bio-Rad protein assay kit and protein concentrations were normalized, 400 μg of protein samples were pre-cleared with the isotype IgG control antibody (Abcam) and Protein A/G agarose (Millipore). First, 40 μL of Protein A/G agarose prepared by incubating with 10 μL of primary antibody in 50 μL of lysis buffer overnight at 4 °C was added to the protein samples and incubated overnight at 4 °C. Then, the mixture was precipitated by high-speed freezing centrifugation at 12 000 revolutions per minute for 10 s. To remove non-specifically bound proteins, the sediment was washed three times with lysis buffer. Agarose-bound immunocomplexes were then released by denaturing solution in loading buffer prior to Western blot analysis.
Immunocytochemical, Hoechst 33342 and PI staining
SHG-44 cells (3×105 cells/well) and U251 (4×105 cells/well) grew on coverslips in 6-well culture plates for 24 h. The cells were treated with shikonin for 2 h at 37 °C, washed twice with PBS, incubated with Hoechst 33342 dye (1 μg/mL) for 5 min, and then incubated with PI (5 μg/mL) for 15 min at room temperature. After a final wash with PBS, the samples were visualized at 60× magnification under a laser scanning confocal microscope (Olympus FV1000, Tokyo, Japan).
Transmission electron microscopy
SHG-44 glioma cells were cultured and treated with shikonin at the indicated concentration, harvested using 0.25% trypsin, and then washed with PBS. Then, the cells were collected by centrifugation for 10 min at 2000 revolutions per minute and treated as described by Huang et al14. Briefly, the cells were fixed in ice-cold 2.5% glutaraldehyde in PBS (pH 7.3), rinsed with PBS, post-fixed in 1% osmium tetroxide with 0.1% potassium ferricyanide, dehydrated through a graded series of ethanol (30%–90%) and embedded in Epon resin (Energy Beam Sciences, Agawam, MA, USA). Semithin (300 nm) sections were cut using a Reichart Ultracut ultra microtome, stained with 0.5% toluidine blue and examined under a light microscope. Ultrathin sections (65 nm) were stained with 1% uranyl acetate and 0.1% lead citrate and examined using a JEM2000EX transmission electron microscope (JEOL, Pleasanton, CA, USA).
Statistical analysis
All data represent at least 4 independent experiments and are expressed as the mean±SD. Statistical comparisons were made using one-way ANOVA. P-values of less than 0.05 were considered statistically significant.
Results
Shikonin induced necrosis and ROS overproduction in glioma cells
In our previous report, we found that shikonin induced necroptosis in U87 and C6 glioma cells after 3 h of incubation, and the IC50 values were 10.0 μmol/L and 6.0 μmol/L, respectively14. Herein, our study was extended to include SHG-44 and U251 glioma cells, and the MTT assay showed IC50 values of shikonin against SHG-44 and U251 cells were, respectively, 4.0 μmol/L and 10.0 μmol/L after 3 h of incubation (data not shown).
Then, we measured the toxicity of shikonin on glioma cells using the LDH release assay and found that glioma cell death induced by shikonin was markedly increased in SHG-44, U251, U87 and C6 cells as shikonin concentration increased (Figure 1A). Furthermore, flow cytometry combined with Annexin V/PI double staining revealed that shikonin did not cause an obvious increase in the percentage of Annexin V+/PI- (early apoptosis) cells but did induce a marked increase in PI+ or Annexin V+/PI+ cells (Figure 1B). Dual staining with Hoechst 33342 and PI observed with a confocal microscope proved that the PI positive cells due to shikonin treatment did not have apoptosis features, such as nuclear condensation or fragmentation (Figure 1C). Moreover, transmission electron microscopy revealed that the SHG-44 cells treated with shikonin exhibited electron-lucent cytoplasm, lost plasma membrane integrity, and intact nuclear membranes (Supplementary Figure S1), which were consistent with the characteristics of necrosis14. Thus, both the PI+ cells and the Annexin V+/PI+ cells exhibited primary necrosis but not secondary to late apoptosis. These results suggest that shikonin induces necroptosis in glioma cells in a concentration-dependent manner.

Shikonin-induced necrosis in glioma cells was accompanied by an overproduction of intracellular ROS and mitochondrial superoxide. (A) The LDH release assay showed that shikonin induced glioma cell death in a concentration-dependent manner. (B) Flow cytometry with Annexin V/PI double staining proved that shikonin did not cause obvious increases in the percentage of Annexin V+/PI- (early apoptosis) cells but significantly increased the PI+ or Annexin V+/PI+ cells at 3 h of incubation. (C) Confocal microscopy with Hoechst 33342 and PI dual staining showed that shikonin did not induce chromatin condensation or nuclear fragmentation. (D) Representative phase-contrast and fluorescence microscopic images of SHG-44 glioma cells incubated with the ROS probe DCFH-DA at 2 h of incubation with shikonin (20×). (E) Statistical analysis of the fluorescence density detected by DCFH-DA demonstrates that shikonin induces an overproduction of intracellular ROS in a dosage-dependent manner. (F) Representative fluorescence microscopic images of the SHG-44 cells that were stained with the mitochondrial superoxide probe Mitosox Red at 2 h of incubation with shikonin (20×). (G) Statistical analysis of the red fluorescence density detected by Mitosox Red proves that the excessive generation of intracellular mitochondrial superoxide caused by shikonin was dependent on the shikonin concentration. The values are expressed as the mean±SD (n=5 per group). *P<0.01.
Given that shikonin-induced necroptosis is often accompanied by an overproduction of ROS3,14, we examined the relationship between ROS production and shikonin dosage. Fluorescence microscopy revealed that the intracellular ROS levels detected by DCFH-DA after 2 h of incubation with shikonin were significantly higher in the 6 μmol/L group than in the 3 μmol/L group (Figure 1D). Statistical analysis of the fluorescence intensity proved that the production of ROS was also dependent on shikonin concentration (Figure 1E).
Considering that ROS is primarily generated in mitochondria21, we used Mitosox Red to examine shikonin-induced changes in the generation of mitochondrial superoxide. Fluorescence microscopy proved that the levels of mitochondrial superoxide detected by Mitosox Red were markedly increased in the C6 cells treated with shikonin, and the level of mitochondrial superoxide in the 6 μmol/L group was higher than that in the 3 μmol/L group (Figure 1F). Moreover, statistical analysis of the red fluorescence intensity revealed that higher concentrations of shikonin resulted in higher levels of mitochondrial superoxide (Figure 1G). This indicates that shikonin induces abnormal levels of intracellular ROS in glioma cells via targeting mitochondria.
RIP1 and RIP3 regulated shikonin-induced necrosis and ROS overproduction
Given that RIP1 and RIP3 are major signals of necroptosis, we examined the shikonin-induced changes in RIP1 and RIP3. As revealed by Western blotting, shikonin induced a concentration-dependent upregulation of RIP1 and RIP3 in SHG-44, U251, U87 and C6 glioma cells (Figure 2A and Supplementary Figure S2A). Moreover, immunoprecipitation of RIP1 with an RIP1 antibody showed that the co-precipitated RIP3 increased as shikonin concentration increased. This indicates that the glioma cell necrosis induced by shikonin is associated with RIP1 and RIP3 (Figure 2B and Supplementary Figure S2B).

RIP1 and RIP3 regulated shikonin-induced necrosis and ROS overproduction. (A) Western blotting analysis showed that shikonin induced the upregulation of RIP1 and RIP3 expression in a concentration-dependent manner. (B) Immunoprecipitation of RIP1 with the RIP1 antibody showed that co-precipitated RIP3 increased as the shikonin concentration increased. (C) The LDH release assay showed that 100 μmol/L Nec-1 and 5 μmol/L GSK-872 inhibited shikonin-induced glioma cell death. (D) Flow cytometry analysis revealed that 5 μmol/L GSK-872 blocked shikonin-induced necrosis in glioma cells. (F) Flow cytometry analysis proved that 100 μmol/L Nec-1 prevented glioma cell necrosis triggered by shikonin. *P<0.01 versus the cells treated with shikonin alone. (F) Western blot analysis revealed that Nec-1 inhibited the shikonin-induced expressional upregulation of RIP1, and GSK-872 mitigated the increased expression of RIP3 caused by shikonin. (G) Statistical analysis of the fluorescence density detected by DCFH-DA showed that pretreatment with 100 μmol/L Nec-1 and 5 μmol/L GSK-872 inhibited the shikonin-induced overproduction of intracellular ROS. (H) Statistical analysis of the red fluorescence density detected by Mitosox Red demonstrated that 100 μmol/L Nec-1 and 5 μmol/L GSK-872 inhibited the shikonin-induced excessive generation of mitochondrial superoxide. The values are expressed as the mean±SD (n=5 per group). *P<0.01.
Then, we blocked RIP1 and RIP3 with their respective specific inhibitors Nec-1 and GSK-872, which could directly bind to the active domains of RIP1 and RIP3, and examined glioma cell death. As the LDH release assay showed, 1 h of pretreatment with either 100 μmol/L Nec-1 or 5 μmol/L GSK-872 could effectively inhibit glioma cell death caused by shikonin after 3 h of incubation (Figure 2C). Furthermore, flow cytometry analysis showed that Nec-1 and GSK-872 prevented shikonin-induced glioma cell necrosis in SHG-44 and U251 cells, respectively (Figure 2D and 2E). At the protein level, Nec-1 and GSK-872 mitigated the shikonin-induced expressional upregulation of RIP1 and RIP3, respectively (Figure 2F and Supplementary Figure S2C). This verified that shikonin-induced glioma cell necroptosis is regulated by RIP1 and RIP3.
Particularly, we found that the both the shikonin-induced increase of intracellular ROS and the excessive generation of mitochondrial superoxide were inhibited in the presence of 100 μmol/L Nec-1 or 5 μmol/L GSK872 (Figure 2G and Supplementary Figure S2D, Figure 2H and Supplementary Figure S2E), indicating that RIP1 and RIP3 account for the shikonin-induced overproduction of ROS and mitochondrial superoxide.
ROS was an executioner of shikonin-induced glioma cell necrosis
To clarify the role of ROS in shikonin-induced necroptosis, MnTBAP (a synthetic, nontoxic and cell-permeable superoxide dismutase mimetic) was used to inhibit intracellular ROS levels. Pretreatment with MnTBAP for 1 h significantly inhibited shikonin-induced overproduction of mitochondrial superoxide and increased intracellular ROS levels at 2 h of incubation (Figure 3A and Supplementary Figure S3A, Figure 3B and Supplementary Figure S3B). Correspondingly, the glioma cell death induced by shikonin was markedly inhibited in the presence of MnTBAP (Figure 3C). Furthermore, flow cytometry with Annexin V/PI double staining proved that prior administration of MnTBAP obviously attenuated shikonin-induced necroptosis in SHG-44 and U251 cells (Figure 3D). Therefore, these data indicate that intracellular ROS is responsible for shikonin-induced necroptosis in glioma cells.

ROS was an executioner of shikonin-induced glioma cell necrosis. (A) Pretreatment with 40 μmol/L MnTBAP for 1 h significantly attenuated the shikonin-induced generation of mitochondrial superoxide. (B) The increased levels of intracellular ROS resulting from shikonin treatment were markedly prevented in the presence of MnTBAP. (C) The LDH release assay showed that 40 μmol/L MnTBAP significantly inhibited shikonin-induced cell death in SHG-44, U251, U87 and C6 glioma cells. (D) Flow cytometry analysis proved that shikonin-induced glioma cell necrosis was markedly prevented by MnTBAP in SHG-44 and U251 glioma cells. The values are expressed as the mean±SD (n=5 per group). *P<0.01 versus the control group.
ROS was a regulator of shikonin-induced glioma cell necrosis
To further verify the role of ROS in shikonin-induced glioma cell necrosis, we used rotenone to improve intracellular ROS levels because rotenone generates superoxide via inhibiting the mitochondrial electron transport chain. We found that rotenone induced a marked increase of superoxide in SHG-44 and U251 cells at 4 h of incubation and triggered concomitant glioma cell death. However, both the increased mitochondrial superoxide and glioma cell death were prevented by pretreatment with 40 μmol/L MnTBAP for 1 h (Figure 4A and Supplementary Figure S4A, and Figure 4B). This indicates that rotenone induces glioma cell death via the overproduction of mitochondrial superoxide. Thus, the glioma cells were treated for 2 h with rotenone and then incubated for 2 h with shikonin at the indicated concentrations. We found that rotenone pretreatment significantly improved the shikonin-induced production of mitochondrial superoxide and intracellular ROS (Figure 4C and Supplementary Figure S4B, Figure 4D and Supplementary Figure S4C). Moreover, the LDH release assay showed that rotenone markedly augmented the shikonin-induced glioma cell death. Furthermore, flow cytometry analysis demonstrated that rotenone enhanced the shikonin-induced glioma cell necrosis (Figure 4E and 4F).

ROS regulated shikonin-induced necrosis in glioma cells. (A) Statistical analysis of the fluorescence density detected by Mitosox Red proved that 4 h of incubation with rotenone resulted in an excessive generation of mitochondrial superoxide, which was significantly prevented by pretreatment with 40 μmol/L MnTBAP for 1 h. (B) The LDH release assay showed that MnTBAP inhibited rotenone-induced cell death in SHG-44 and U251 glioma cells. (C) Statistical analysis of the fluorescence density detected by Mitosox Red demonstrated that pretreatment with rotenone for 2 h enhanced the generation of mitochondrial superoxide caused by 2 h of incubation with shikonin. (D) Statistical analysis of the fluorescence density detected by DCFH-DA proved that pretreatment with rotenone for 2 h improved the increased intracellular ROS levels induced by 2 h of treatment with shikonin. (E) The LDH release assay showed that rotenone augmented shikonin-induced glioma cell death, which was prevented by Nec-1. However, Nec-1 did not prevent glioma cell death induced by rotenone alone. (F) Flow cytometry with Annexin V/PI double staining showed that rotenone enhanced shikonin-induced necrosis in glioma cells, but this enhancement effect was blocked by Nec-1. The values are expressed as the mean±SD (n=5 per group). *P<0.01 versus control group.
To address whether the enhancement effects of rotenone on shikonin-induced glioma cell necrosis was associated with RIP1, we treated SHG-44 and U251 cells with 100 μmol/L Nec-1 for 1 h prior to sequential incubation with rotenone and shikonin. As revealed by the LDH release assay and flow cytometry analysis, Nec-1 blocked the enhancement effects of rotenone on shikonin-induced glioma cell death (Figure 4E and 4F). This indicates that rotenone augmented augments shikonin-induced glioma cell death via the necroptosis pathway.
ROS regulated the increased RIP1 and RIP3 expression and their shikonin-induced interaction
To test the effect of ROS on the necroptosis signals RIP1 and RIP3, we performed Western blotting analysis and found that prior administration of rotenone enhanced the shikonin-induced upregulation of RIP1 and RIP3 (Figure 5A and Supplementary Figure S5A), but MnTBAP prevented the expressional upregulation of RIP1 and RIP3 caused by shikonin (Figure 5B and Supplementary Figure S5B). Particularly, co-immunoprecipitation showed that the RIP3 co-precipitated with RIP1 resulting from shikonin treatment was improved by rotenone (Figure 5C and Supplementary Figure S5C) but was mitigated by MnTBAP (Figure 5D and Supplementary Figure S5D). Thus, these data indicate that the shikonin-induced upregulation of RIP1 and RIP3 and necrosome formation are both regulated by ROS.

ROS regulated increased RIP1 and RIP3 expression and their interaction caused by shikonin. (A) Western blot analysis revealed that rotenone upregulated the expression of RIP1 and RIP3 and enhanced the shikonin-induced increase of RIP1 and RIP3. However, this enhancement effect was blocked by Nec-1. (B) Western blot analysis revealed that MnTBAP prevented the shikonin-induced upregulation of RIP1 and RIP3. (C) Co-immunoprecipitation demonstrated that considerably higher amounts of RIP3 co-immunoprecipitated with RIP1 in the glioma cells treated with rotenone prior to incubation with shikonin. (D) Co-immunoprecipitation proved that less RIP3 was pulled down with RIP1 in the cells pretreated with MnTBAP compared with that of treatment with shikonin alone.
Discussion
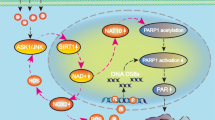
We found that shikonin triggered glioma cell necroptosis and induced the overproduction of intracellular ROS and mitochondrial superoxide in a concentration-dependent manner. Moreover, shikonin also upregulated the levels of RIP1 and RIP3 and reinforced their interaction. Inhibition of either RIP1 or RIP3 with their specific inhibitors not only prevented shikonin-induced glioma cell necroptosis but also mitigated the intracellular levels of ROS and mitochondrial superoxide, indicating that RIP1 and RIP3 regulate the shikonin-induced overproduction of ROS via targeting mitochondria. Mitigation of ROS with MnTBAP, which is a cleaner of mitochondrial superoxide, attenuated shikonin-induced glioma cell necroptosis, whereas increasing ROS with rotenone, which improves the mitochondrial generation of superoxide, augmented glioma cell necroptosis. Furthermore, we found that MnTBAP prevented the shikonin-induced expression of RIP1 and RIP3 and their interaction while rotenone reinforced these effects. Taken together, these findings suggest that ROS plays a crucial role in the regulation of shikonin-induced glioma cell necroptosis via the formation of positive feedback with RIP1 and RIP3 (Figure 6).

Schematic model for the role of ROS in shikonin-induced glioma cell necroptosis.
Necrosis was originally thought to be an accidental event, but accumulating evidence has demonstrated that it is a type of programmed cell death regulated by specific signals22. Two methods are often used to evaluate cell death by measuring changes in plasma membrane permeability. The first is to examine the uptake of DNA binding dyes that do not traverse the plasma membrane of living cells, such as propidium iodide. The other method is to assay the leakage of intracellular molecules, such as lactate dehydrogenase (LDH)23. Thus, we examined glioma cell death in this study using the LDH release assay and flow cytometry analysis combined with Annexin V/PI double staining. We found that shikonin induced dosage-dependent cell death in all four glioma cell lines (SHG-44, U251, U87 and C6 cells). Furthermore, as revealed by fluorescence microscopy with Hoechst and PI double staining, the cells treated with shikonin had no morphological features of apoptosis, such as condensed chromatin or fragmented nuclei14. Moreover, shikonin did not increase the cells that were stained with only Annexin V, thus, we excluded that necrosis induced by shikonin was secondary to late apoptosis. Our previous report also showed that the pan-caspase inhibitor z-Vad-fmk did not prevent shikonin-induced cell death in glioma cells14. Therefore, the results in this study further verify that shikonin induces necroptosis in glioma cells.
RIP1 has been reported to initiate necroptosis via binding with RIP3 to form the necrosome, in which RIP3 is activated by RIP122. Previous studies have shown that RIP1 and RIP3 contribute to glioma cell necrosis induced by either photodynamic therapy or radiation therapy24,25. Moreover, some chemical reagents, such as edelfosine and 5-benzylglycinyl-amiloride, trigger glioma cell necroptosis via the RIP1/RIP3 pathway26,27. In the current study, we found that the RIP1 inhibitor Nec-1 and the RIP3 inhibitor GSK-872 not only attenuated shikonin-induced glioma cell necroptosis but also prevented the shikonin-induced dosage-dependent upregulation of RIP1 and RIP3. Thus, these results suggest that shikonin induces RIP1/RIP3-dependent necroptosis in glioma cells.
ROS is found to be an executioner of necroptosis6,28. It can attack intracellular macromolecules, including proteins, lipids and nucleic acids29. Accumulating evidence has suggested that either RIP1 or RIP3 could regulate intracellular ROS levels via different pathways. RIP1 targets the NADPH oxidase 1 NOX1 and the small GTPase RAC1 to increase ROS production at the plasma membrane30. RIP3 interacts with and activates the mitochondrial protein GLUD1 to elevate the levels of mitochondrial superoxide and intracellular ROS31. Consistently, shikonin has been observed to increase intracellular ROS levels by targeting both NOX1 and the complex in the mitochondrial respiratory chain32. In the current study, shikonin-induced glioma cell necrosis was attenuated in the presence of MnTBAP, which inhibits the levels of mitochondrial superoxide and intracellular ROS, but was enhanced by pretreatment with rotenone, which promotes the generation of mitochondrial superoxide and intracellular ROS. Previously, we also reported that the inhibition of ROS with the antioxidant NAC attenuated shikonin-induced necrosis in glioma cells14. Thus, ROS is a downstream signal of RIP1 and RIP3 and an executioner of necroptosis induced by shikonin in glioma cells.
Notably, we found that the inhibition of ROS with MnTBAP attenuated the protein levels of RIP1 and RIP3 and mitigated their interaction in the glioma cells treated with shikonin. In contrast, increasing ROS with rotenone not only upregulated the expression of RIP1 and RIP3 but also facilitated RIP1/RIP3 necrosome formation. Consistently, recent studies have suggested that ROS plays a role in the regulation of RIP1 and RIP3 expression and their mutual binding. Using human FADD-deficient Jurkat cells challenged with BV6/TNFα, Schenk et al found that the RIP1/RIP3 necrosome was stabilized by ROS33. Similarly, ROS has been reported to promote the interaction between RIP1 and RIP3 in glioma cells stressed by photodynamic therapy34. Moreover, the interaction between RIP1 and RIP3 induced by hypoxia in colorectal cancer cells was attenuated when ROS was mitigated by BHA35. However, the protein levels of the necroptosis signals also affect necrosome assembly, as knockdown of RIP3 with an siRNA inhibited necrosome assembly in cortical neurons induced by oxygen glucose deprivation and treatment with the caspase inhibitor z-VAD36. Therefore, ROS regulates shikonin-induced necrosome assembly mainly via two pathways: by upregulating the RIP1 and RIP3 protein levels and enhancing their interaction. This also suggests that shikonin induces a positive feedback loop between ROS and necroptosis signals.
Although we did not investigate why ROS enhances the interaction between RIP1 and RIP3 in this study, Zhang et al reported that ROS activated RIP1 autophosphorylation on serine residue 161 (S161), and Cho et al found that the interaction between RIP1 and RIP3 was stabilized when they were phosphorylated37,38. In the current study, we found that rotenone treatment upregulated the expression of RIP1 and RIP3 (Figure 5A), but the RIP1 inhibitor Nec-1 did not prevent glioma cell death induced by rotenone (Figure 4E). Similarly, Xu et al reported that Nec-1 had no protective effect on the free radical-induced cell death caused by hydrogen peroxide or menadione in HT-22 cells39. Thus, we believe that ROS cannot activate RIP1 by itself in glioma cells but rather it is involved in other events that regulate RIP1 activation during the process of shikonin-induced necroptosis.
In conclusion, we have demonstrated in this study that RIP1 and RIP3 regulate shikonin-induced glioma cell necrosis by increasing intracellular ROS and mitochondrial superoxide, which improves the shikonin-induced upregulation of RIP1 and RIP3 expression and RIP1/RIP3 necrosome formation.
References
Stavrovskaya AA, Shushanov SS, Rybalkina EY . Problems of glioblastoma multiforme drug resistance. Biochemistry (Mosc) 2016; 81: 91–100.
Kang JI, Hong JY, Choi JS, Lee SK . Columbianadin inhibits cell proliferation by inducing apoptosis and necroptosis in HCT116 colon cancer cells. Biomol Ther (Seoul) 2016; 24: 320–7.
Shahsavari Z, Karami-Tehrani F, Salami S . Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac J Cancer Prev 2015; 16: 7261–6.
Diao Y, Ma X, Min W, Lin S, Kang H, Dai Z, et al. Dasatinib promotes paclitaxel-induced necroptosis in lung adenocarcinoma with phosphorylated caspase-8 by c-Src. Cancer Lett 2016; 379: 12–23.
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 2007; 6: 1641–9.
McComb S . Aguadé-Gorgorió J, Harder L, Marovca B, Cario G, Eckert C, et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med 2016; 8: 339ra70.
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 2013; 88: 31268–79.
Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, et al. MicroRNA-223-5p and -3p cooperatively suppress necroptosis in ischemic/reperfused hearts. J Biol Chem 2016; 291: 20247–59.
Cai Z, Liu ZG . Execution of RIPK3-regulated necrosis. Mol Cell Oncol 2014; 1: e960759.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–27.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 2012; 109: 5322–7.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–46.
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16: 55–65.
Huang C, Luo Y, Zhao J, Yang F, Zhao H, Fan W, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One 2013; 8: e66326.
Wada N, Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Tasaki M, et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol 2015; 46: 963–72.
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013; 13: 580.
Rohde K, Kleinesudeik L, Roesler S, Löwe O, Heidler J, Schröder K, et al. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ 2017; 24: 83–97.
Ma YM, Peng YM, Zhu QH, Gao AH, Chao B, He QJ, et al. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-κB inhibition. Acta Pharmacol Sin 2016; 37: 1381–90.
Zhao H, Wang C, Lu B, Zhou Z, Jin Y, Wang Z, et al. Pristimerin triggers AIF-dependent programmed necrosis in glioma cells via activation of JNK. Cancer Lett 2016; 374: 136–48.
Ma D, Lu B, Feng C, Wang C, Wang Y, Luo T, et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett 2016; 371: 194–204.
Pokrzywinski KL, Biel TG, Kryndushkin D, Rao VA . Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS One 2016; 11: e0168283.
Hanson B . Necroptosis: A new way of dying? Cancer Biol Ther 2016; 17: 899–910.
Chan FK, Moriwaki K, De Rosa MJ . Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol 2013; 979: 65–70.
Miki Y, Akimoto J, Moritake K, Hironaka C, Fujiwara Y . Photodynamic therapy using talaporfin sodium induces concentration-dependent programmed necroptosis in human glioblastoma T98G cells. Lasers Med Sci 2015; 30: 1739–45.
Das A, McDonald DG, Dixon-Mah YN, Jacqmin DJ, Samant VN . Vandergrift WA 3rd et al. RIP1 and RIP3 complex regulates radiation-induced programmed necrosis in glioblastoma. Tumour Biol 2016; 37: 7525–34.
Melo-Lima S, Celeste Lopes M, Mollinedo F . Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014; 1: 649–64.
Pasupuleti N, Leon L . Carraway KL 3rd, Gorin F . 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther 2013; 344: 600–15.
Dasgupta A, Nomura M, Shuck R, Yustein J . Cancer's achilles' heel: apoptosis and necroptosis to the rescue. Int J Mol Sci 2017; 18: 23.
Voss P, Grune T . The nuclear proteasome and the degradation of oxidatively damaged proteins. Amino Acids 2007; 32: 527–34.
Kim YS, Morgan MJ, Choksi S, Liu ZG . TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 2007; 26: 675–87.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–6.
Yang JT, Li ZL, Wu JY, Lu FJ, Chen CH . An oxidative stress mechanism of shikonin in human glioma cells. PLoS One 2014; 9: e94180.
Su Z, Yang Z, Xie L, DeWitt JP, Chen Y . Cancer therapy in the necroptosis era. Cell Death Differ 2016; 23: 748–56.
Belizário J, Vieira-Cordeiro L, Enns S . Necroptotic cell death signaling and execution pathway: lessons from knockout mice. Mediators Inflamm 2015; 2015: 128076.
Hannes S, Abhari BA, Fulda S . Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett 2016; 380: 31–8.
Piao JL, Cui ZG, Furusawa Y, Ahmed K, Rehman MU, Tabuchi Y, et al. The molecular mechanisms and gene expression profiling for shikonin-induced apoptotic and necroptotic cell death in U937 cells. Chem Biol Interact 2013; 205: 119–27.
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 2017; 8: 14329.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–23.
Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, et al. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem 2007; 103: 2004–14.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81372697), the Program for New Century Excellent Talents in University (NCET-12-0233), the Changbaishan Scholar Project of Jilin Province (2013026), the Scientific Research Foundation of Jilin province (20150414013GH and 20160101127JC), and the Bethune Project B of Jilin University (No 2012203).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information is available at the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Material Figure S1
Representative images of shikonin-induced morphological changes captured with a transmission electron microscope. (JPG 298 kb)
Supplementary Material Figure S2
(A) Statistical analysis of shikonin-induced changes in the RIP1 and RIP3 protein levels at 2 h of incubation. (JPG 1039 kb)
Supplementary Material Figure S3
(A and B) Representative fluorescence microscopic images of SHG-44 cells detected by the mitochondrial superoxide probe Mitosox Red and the ROS probe DCFH-DA, respectively (20×). (JPG 282 kb)
Supplementary Material Figure S4
(A) Representative images of SHG-44 cells under a fluorescence microscope showed that MnTBAP pretreatment mitigated the rotenone-induced production of mitochondrial superoxide (20×). (JPG 333 kb)
Supplementary Material Figure S5
(A) Statistical analysis of the shikonin-induced upregulation of RIP1 and RIP3 in cells pretreated with rotenone. (JPG 238 kb)
Rights and permissions
About this article

Cite this article
LU, B., GONG, X., WANG, Zq. et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin 38, 1543–1553 (2017). https://doi.org/10.1038/aps.2017.112
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.112
Keywords
This article is cited by
-
Acute exposure to polystyrene nanoparticles promotes liver injury by inducing mitochondrial ROS-dependent necroptosis and augmenting macrophage-hepatocyte crosstalk
Particle and Fibre Toxicology (2024)
-
Leveraging a disulfidptosis‑related lncRNAs signature for predicting the prognosis and immunotherapy of glioma
Cancer Cell International (2023)
-
Effect of doramectin on programmed cell death pathway in glioma cells
Clinical and Translational Oncology (2023)
-
Nanoencapsulation of Docetaxel Induces Concurrent Apoptosis and Necroptosis in Human Oral Cancer Cells (SCC-9) via TNF-α/RIP1/RIP3 Pathway
Indian Journal of Clinical Biochemistry (2023)
-
An in vitro evaluation of the antioxidant activities of necroptosis and apoptosis inhibitors: the potential of necrostatin-1 and necrostatin-1i to have radical scavenging activities
Pharmacological Reports (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}