
Abstract
Recent studies have revealed extensive genetic and non-genetic variation across different geographical regions of a tumor or throughout different stages of tumor progression, which is referred to as intra-tumor heterogeneity. Several causes contribute to this phenomenon, including genomic instability, epigenetic alteration, plastic gene expression, signal transduction, and microenvironmental differences. These variables may affect key signaling pathways that regulate cancer cell growth, drive phenotypic diversity, and pose challenges to cancer treatment. Understanding the mechanisms underlying this heterogeneity will support the development of effective therapeutic strategies.
Similar content being viewed by others
Introduction
Solid tumors are heterogeneous neoplasms composed of different types of cells, including cancer cells as well as mesenchymal cells, endovascular cells, and immune cells1. In addition to the complex tissue architecture of a tumor, cancer cells within a single tumor are also heterogeneous in their molecular signatures; this is referred to as intra-tumor heterogeneity2,3,4. These cell-to-cell variations in genetic signature, gene expression, and post-translational modifications are recognized in almost every type of cancer (Table 1). However, current therapies treat cancer as a homogenous disease. Targeted drugs have been developed against single or multiple aberrant molecular signatures based on the diagnosis of the mixed populations of cancer cells, in most cases based on a single biopsy5,6. Thus, it is not surprising that drug resistance, both intrinsic and adaptive, occur extensively in all types of cancer. Understanding the specific driving forces behind different subtypes of intra-tumor heterogeneity will facilitate a better understanding of the nature of cancer, and will provide insight into the development of more effective cancer therapies.
In this review, we summarize the evidence for both genetic and non-genetic heterogeneity within a tumor, and focus on the possible origins and their impact on drug resistance. Finally, we discuss the strategies that could be adopted to improve the understanding and management of this heterogeneity in cancer treatment.
Heterogeneity in genetic mutation
Genomic aberrations in cancers have been recognized for a long time; these include base-pair substitutions, focal deletions/amplifications, tandem duplications, chromosomal rearrangements and whole-genome duplications7. Recent studies further revealed extensive variations in genomic aberrations within tumors. For example, in glioblastomas (GBMs), the copy number variation of two commonly amplified receptor tyrosine kinases (RTKs), EGFR and PDGFRA, was identified among cells from the same tumor specimen using multicolor fluorescent in situ hybridization (FISH) mapping8,9. Moreover, the majority of the cells had a mutual exclusive amplification of these two genes, whereas co-amplification was only observed in a minority of the cancer cells. There also seemed to be a spatial distribution of the differential gene amplifications, with one or the other type of gene predominating in certain areas of the same specimen. In particular, the PDGFRA-amplified cells tended to be present close to the endothelial cells. Similar intra-tumor heterogeneous genetic alterations were also observed in other types of cancers. Gerlinger et al performed whole-exome sequencing on DNA extracted from multiple regions of tumor samples obtained from one clear-cell renal cell carcinoma (ccRCC) patient10. Nine primary-tumor regions of the nephrectomy specimen and two regions of the excised chest-wall metastasis were analyzed. Among all of the nonsynonymous point mutations, insertions, and deletions that changed the protein amino acid sequences, approximately 31% of the mutations were ubiquitous across every tumor region, 46% were shared by several but not all regions, and 22% were present only in a single region. In breast cancers, spatial and temporal intra-tumor heterogeneity have been observed using comparative genomic hybridization and FISH mapping11,12. A recent genome-wide sequencing study of 21 breast cancers has shown that although every tumor has a dominant subclonal lineage of tumor cells, subclonal diversification was prominent in every tumor, and most of the mutations were found in just a fraction of the cells13.
Integrative analysis of all these genetic changes in a single cancer and in-depth evaluation of the topographical information, especially the function of this variation will greatly enhance our understanding on cancer heterogeneity and inform its use in cancer treatment.
Heterogeneity in gene expression
In addition to genetic alterations, intra-tumor heterogeneity is also common in gene expression. The clusters of differentiation (CD) antigens were among the first and are the most frequently identified proteins that are heterogeneously expressed within a tumor. For example, differential expression of CD34/CD38 in acute myeloid leukemia (AML) cells14,15, CD24/CD44 in breast cancer cells16,17, CD133 in colon cancers and brain tumors18,19,20, and CD271 in melanoma cells21,22 have been frequently found in patient samples. The so-called cancer hallmark genes, such as c-KIT23, cyclins24,25, Ki6725, Bcl-226,27,28, c-Myc29, RAS30 and EGFR31,32 have also been frequently found to be differentially expressed within a single tumor.
With the advances in deep-sequencing technology, the examination of single-cell gene expression profiles has become popular. In a large-scale study, full-length transcriptomes of 672 cells from five freshly resected human GBMs were generated using SMART-seq (switching mechanism at the 5′ end of the RNA template sequencing) technology (96–192 cells per tumor)33. After depleting the infiltrating immune cells as well as genes and cells with low sequencing coverage, the RNA-seq profiles of approximately 6000 genes in 430 cells were quantitatively analyzed. Although the gene expression profiles of individual cells from the same tumor were more similar to each other than those from different tumors, variable gene expression between individual cells from the same tumor was also common. According to this study, cell-to-cell variability was evident in genes related to multiple cellular functions, including oncogenic signaling, proliferation, and the immune and hypoxia responses. In particular, RTKs such as EGFR, PDGFRA, FGFR1 and their ligands, which are important therapeutic targets, were expressed in a mosaic fashion. Moreover, the splicing patterns of the mRNAs of these genes were also variable. For example, in one GBM sample, 7% of cells expressed wild-type EGFR, 19% expressed EGFRvIII, which is an oncogenic mutant form with a deletion of exon 2–7, and 25% expressed EGFR del4, which is another oncogenic variant with a deletion of exon 4. In addition, 1% to 2% of cells co-expressed both wild-type EGFR and the EGFRvIII variant.
Heterogeneity in protein modification
Differential posttranslational modifications of proteins have also been found in different cancer cells from the same tumor. A pioneer study of three well-characterized subpopulations of colorectal carcinoma (CRC) cells that were isolated from a single primary colon cancer has revealed a great degree of heterogeneity in phosphorylation modifications of cytosolic and membrane proteins, and a mechanism beyond differential gene expression has been demonstrated34. Since then, heterogeneous modifications of multiple signaling proteins have been identified, including STAT, ERK, p38, GSK3β, β-catenin, NF-κB, AKT, PTEN, S6 and Src family kinases28,35,36,37,38. In addition, intra-tumor heterogeneity in O-glycosylation has also been reported39.
The origins of intra-tumor heterogeneity
Intrinsic factors
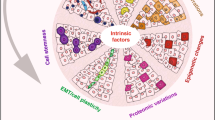
The various types of intra-tumor heterogeneity are generated by different mechanisms, which can be categorized as cell-intrinsic and cell-extrinsic (Figure 1). Intrinsic mechanisms are cell-inherent or cell-autonomous. The most well-known and extensively studied intrinsic driving force of cancer cell heterogeneity is genomic instability. Genomic alterations, which occur in the pathways of nucleotide excision repair, base excision repair, DNA mismatch repair, telomere maintenance, double-strand break repair, DNA replication, and chromosome segregation7, lead to extensive and stochastic changes across the entire genome. Recently, a large-scale mutational landscape study by The Cancer Genome Atlas (TCGA) demonstrated that cancer cells have much higher somatic mutation rates, ranging from 0.28 to 8.15 mutations per megabase in 12 major cancer types, compared to normal cells, which have approximately 10-9mutations per base pair per division48. Given the size of the human diploid genome (∼6 billion base pairs), the potential for the development of stochastic genomic mutations over the course of tumor development is vast, even without an elevated mutation rate49. Indeed, the average frequency of non-synonymous gene mutations per tumor has been reported to be in the range of 150–17050, which distribute across individual cancer cells within a tumor.

The origins of intra-tumor heterogeneity. Both cell-intrinsic and cell-extrinsic factors generate intra-tumor heterogeneity. The intrinsic mechanisms include cell-to-cell variability in genotypic alterations (as shown by different colors of cell nucleus), and non-genetic or phenotypic variations (as shown by different colors of cytoplasm), which are due to epigenetic modification, plastic gene expression, and signal transduction. The extrinsic mechanisms originate from unequal microenvironments. Multiple sources of heterogeneity within a tumor may co-exist and interact with each other over time to shape up the heterogeneous cancer.
Recent evidence has shown that genetic mutations are not the only intrinsic factors of intra-tumor heterogeneity, and epigenetic changes also play an important role. The epigenetic changes are defined as stable or heritable changes in genetic information without any changes in DNA sequences. A hierarchical composition of tumorigenic and non-tumorigenic cells in multiple human cancers, including AML, chronic myeloid leukemia (CML), GBM, CRC, breast cancer, and ovarian cancer51,52,53 has been demonstrated. The subpopulations of cancer cells within a tumor with increased tumor-initiating capacities and self-renewal potential are often termed “cancer stem cells (CSCs)”. Studies on CSCs suggest that a small number of cells in a tumor undergo epigenetic changes, similar to the differentiation of normal stem cells, to form phenotypically diverse non-tumorigenic cells that compose the bulk of the cells in a tumor. Although there is still no consensus regarding whether cancers arise from normal stem cells or whether there is a strict hierarchy or bidirectional plasticity between the differentiation and dedifferentiation states, most cancer biologists accept that at any given time in any type of cancer, there are populations of cells with self-renewal and tumor-initiating properties, which are simplified as stem-like cancer cells. Certain cancers, such as some germ cell cancers and some leukemias, have been recognized to contain a strict hierarchy of tumorigenic and non-tumorigenic cells54,55,56, suggesting possible epigenetic contributions to the differential cancer phenotypes. Direct evidence of differences in epigenetic changes between tumor subpopulations have also emerged. In AML, stem-like and non-stem-like cancer cells differ in their histone modification patterns (H3K4me3 and H3K27me3)57. Similar observations have been reported in GBMs, in which the tumorigenic cell populations exhibit aberrant activation of multiple transcription factors because of a loss of the polycomb mark H3K27me3 from their promoters58. The activated expression of one of these transcription factors, referred to as ASCL1, causes the activation of the Wnt pathway, which is required for the maintenance of the stem-like state of the cells. The epigenetic aberrations, at least in DNA methylation, occur in a chaotic shifting fashion from the very early stages of tumorigenesis59. The error rate for stochastic gain or loss of methylation has been estimated at 2×10-5 per CpG site per division in cancer cells60. The widespread loss of DNA methylation seem to be quite random and nonclonal, suggesting that at least some of the epigenome changes occur stochastically and are maintained during tumor progression4.
Differential gene expression is also a cause of intra-tumor heterogeneity. Plastic switching of the cellular state has been documented in several recent studies3,61. Certain cell surface antigens in fractions of cancer cells undergo changes in a reversible fashion based on tumorigenic assays and during drug treatment, indicating a mechanism of transient changes in gene expression other than the stable changes of epigenetic alteration. Indeed, experiments at single-cell resolution have elegantly demonstrated the stochastic gene expression as a fundamental nature of cells to cope with environmental changes, which is a phenomenon that is conserved from phage and bacteria to mammalian cells, including cancer cells62,63,64,65. An extreme example is that two genetically identical sister cells that divided from the same ancestor cell express differential amounts of cell death pathway proteins, leading to distinct cell fates in response to the same death stimuli66.
In addition to gene expression, the differential signal transduction may also contribute to the intra-tumor heterogeneity, although much fewer studies have been focused on this topic. The heterogeneous activation of proliferation-related proteins has also been reported38,67. There are two possible underlying mechanisms. One is the de novo stochasticity of enzymatic reactions in signal transduction, similar to other biochemical processes such as transcription. At the microscopic level, every protein modification chemical reaction is discrete, ie, not continuous. Although the copy numbers of the signaling proteins in a cell could be in the thousands, which greatly reduce the fluctuation of signal transduction at the macroscopic level, the number of proteins that are functional for signaling might still be small due to their sub-cellular locations. Thus, cell-to-cell signaling variation may occur due to the intrinsic stochasticity of biochemical reactions between signaling proteins. It has been shown that the biological noise can be amplified in the signal transduction cascades, and cells evolve signaling motifs, such as negative feedback loops, to attenuate the signaling noise68,69. Aberrations in the negative feedback modules, eg, mutations in protein phosphatases and protein degradation systems, are frequently observed in cancers. It would be interesting to investigate the influence of these aberrations on the cell-to-cell variation in the corresponding signaling pathways.
The other mechanism that may contribute to the heterogeneous protein modification is that some signals, eg, ERK activation, can be transduced in a digital oscillation or pulse-like manner, and individual cells are at different paces of the pulse70,71. Theoretically, the heterogeneity of signal transductions could be functional, considering that evolutionary selection operates on phenotypes, both stable and transient, but not on genotypes. To understand the detailed mechanisms and possible functions of the heterogeneous signal transduction processes, careful measurements of signaling dynamics in live cells should be performed in single cell-based experiments by using sensitive probes70,71,72,73,74,75,76. However, direct test of signaling dynamics in primary tumors is still technically infeasible.
Taken together, the intra-tumor heterogeneity may originate from four mechanisms: genetic variation, epigenetic alteration, plastic gene expression, and signal transduction. It is important to note that even in cancers that clearly contain a hierarchy of tumorigenic and non-tumorigenic cells or in cancers that are highly plastic in their phenotypic transitions, these four sources of intra-tumor heterogeneity coexist. Furthermore, they could contribute either independently, due to stochastic variations, or interactively to form the differential phenotypes of cancer cells within a tumor. For example, the gene mutations implicated in chromatin remodeling and DNA methylation dynamics could cause aberrant epigenome reprogramming in cancer cells77. The point mutations in the EGFR, KRAS, BRAF and PI3K genes could transform the gene products into constitutively active forms, and thus lead to the alterations in signal transduction6. Vice versa, the variations in signal transduction and gene expression could also lead to variations in genetic and epigenetic changes. For example, hypoxia-inducible factor 1α (HIF1α) signaling could induce genetic instability by transcriptionally down-regulating the base-mismatch protein MutSa78 or could induce genomic DNA demethylation in CpG islands by transcriptionally increasing the methionine adenosyltransferase 2A (MAT2A)79. Therefore, it is important to integrate all experimental approaches to fully understand the alterations at all levels and their distinctive roles in tumor progression.
Extrinsic factors
The extrinsic causes of intra-tumor heterogeneity include the components of the surrounding microenvironment that act on the cancer cells to influence their genotypes and phenotypes. In other words, the extrinsic factor-mediated heterogeneity originates from the spatial differences of the cancer cells (Figure 1). Multiple factors contribute to the unequal microenvironment. The most obvious example is the tumor's blood supply80. Through blood vessels, nutrients, growth factors, and oxygen are delivered, and metabolic waste is removed. The normal vascular network has a well-organized architecture, which provides nutrients by diffusion to all normal cells. In contrast, tumor blood vessels are dilated, saccular, tortuous and heterogeneously distributed81,82. Variation in the distances from individual cancer cells to the vasculature leads to differential trophic supply and metabolic status of the cancer cells. Hypovascular regions, which are located far from the blood vessels, become chronically hypoxic and acidic. Moreover, high-resolution intravital microscopy has revealed that some of the blood vessels in solid tumors carry almost no oxygen, indicating that the functioning of tumor blood vessels is also abnormal83. Both the structural and functional abnormalities of tumor blood vessels generate different microenvironments within tumors. This inequality may lead to heterogeneous signal transduction, gene expression, and genomic instability in cancer cells either directly through systemically supplied growth factors or hormones, or indirectly through oxidative stress, hypoxia, or acidosis84. In particular, it has been reported that the vasculature can provide a specialized growth niche for stem-like cancer cells in brain tumors. It has been reported that CD133+/Nestin+ cells of GBMs, medulloblastomas, ependymomas and oligodendrogliomas were located in close proximity to tumor capillaries. The self-renewal and proliferation properties of these cancer stem-like cells were maintained in culture using factors secreted by endothelial cells85. In addition to endothelial cells, other stromal cells including fibroblasts, inflammatory cells, and pluripotent mesenchymal cells, also contribute to the diversified genotypes and phenotypes of cancer cells through the secretion of cytokines, growth factors, and extracellular matrix (ECM) components, which in turn play a role in chemoattracting stromal cells including endothelial cells and inflammatory cells, further influencing the microenvironment and diversifying the cancer cells86.
Interactions between intrinsic and extrinsic factors during tumor evolution
The generation of intra-tumor heterogeneity by the interactions between intrinsic and extrinsic factors can be illustrated using the clonal evolution model, in which stochastic genetic changes during cancer development create a pool of cells with different genetic alterations and different growth potentials, but only the cancer cells that acquired the growth potentials to fit the surrounding environmental niches demonstrate growth87. In many types of cancers, such as non-small-cell lung cancer (NSCLC) and ccRCC, genetically distinct subclones in different tumor regions have been identified88,89. The spatial and temporal mapping of tumor samples also provided evidence of branched evolution of subclones across multiple cancer types. It is clear that some early somatic mutational events occur on the trunk of a tumor's phylogenetic tree, whereas the later subclonal events occur on its branches. For instance, TP53 mutations have been generally found to be early events in breast cancer, NSCLC, esophageal adenocarcinoma, ovarian cancer, and pancreatic cancer, indicating a common environmental selection for tumor-initiating TP53 mutation, whereas subclonal diversification was found to be occur in a tissue-specific manner90. For example, the BRCA2 mutation in breast cancer, the HGF and MLL3 mutations in NSCLC, and the PIK3CA, CTNNB1 and NF1 mutations in ovarian cancer were found to be late events to support the tumor maintenance and progression under specific tissue environments. Interestingly, some subclonal cancer cells could cooperate with each other to promote tumor progression. In GBMs, a low-frequency EGFRvIII-containing subpopulation of cancer cells was found to enhance the activation of the wild-type EGFR in adjacent cancer cells to facilitate the growth of the major clones through an interleukin 6 and leukemia inhibitory factor paracrine mechanism91. These studies demonstrated the interactions of the intrinsic stochastic gene mutations and extrinsic growth selection factors in the evolution of the intra-tumor heterogeneity and their functions in tumor development.
Our knowledge on the clonal evolution of epigenetic alterations is very limited despite the fact that epigenetic changes are prevalent through every step of tumor progression92. Theoretically, epigenetic changes, especially DNA methylation, are heritable and can directly influence gene expression, thereby contributing to clonal selection to create intra-tumor heterogeneity. Given that DNA demethylation is quite widespread and nonclonal and the methylation in specific promoter regions is far more clonal, possible environmental selection based on random epigenetic changes during tumor evolution may occur93. In another aspect, the environmental cues may not only select but also induce the epigenetic changes. Evidence has emerged that chronic inflammation is key for initiating progressive chromatin epigenetic alterations in the very early stages of tumor progression, most likely from the premalignant stages94. Although the detailed mechanisms of how epigenetic changes and environmental factors cooperate to function in tumor progression remain elusive, a reasonable hypothesis has recently been proposed on the basis of experimental evidence92. In this hypothesis, when cells are subject to stress, protein complexes consisting of polycomb group (PcG) proteins and DNA methyltransferases (DNMTs) quickly and transiently move into the promoter and CpG islands to protect these promoter regions from errors of DNA damage repair if transcription continues. During continued environmental insults, such as chronic inflammation, genes with specific chromatin patterns, eg, PcG marks, are kept at low levels of transcription, and the retention of the above protein complexes on the associated promoters induces abnormal DNA methylation in a time-dependent manner. If the abnormal DNA methylation silences the stem-cell-related genes, then the plasticity of cell state transitions will change and will either increase or decrease depending on the specific genes that are influenced. Reduced plasticity may help the outgrowth of stem-like cells, whereas increased plasticity may promote the emergence of heterogeneous subclones. This hypothesis provides a possible mechanism to explain how chromatin alterations may help to fix tumor phenotypes and contribute to intra-tumor heterogeneity during cancer development.
Implications of intra-tumor heterogeneity for cancer treatment
One of the clinical implications of intra-tumor heterogeneity is drug resistance. Functionally, cell-to-cell variability, either genetically or non-genetically, can compromise cellular responses to cancer therapies by increasing the repertoire of possible cellular responses and hence increasing the adaptive nature of cellular behaviors. Substantial evidence has demonstrated a correlation between intra-tumor heterogeneity and therapeutic outcomes. In a study of early-stage NSCLCs, primary tumors from patients with relapsed disease exhibited significantly larger subclonally diversified genotypes than tumors from patients with relapse-free tumors95. A single-cell exploration of GBMs revealed that proneural tumors with more diverse transcriptionally defined subclones were associated with poorer outcomes96.
Several mechanisms may contribute to the drug resistance in heterogeneous cancers. First, genotypic heterogeneity clearly confer drug resistance under certain circumstances. Targeted anti-cancer drugs, such as kinase inhibitors, specifically act on the subpopulation of cells with mutated oncogenes that they target, leaving other subpopulations without corresponding mutations unaffected. The pre-existing untargeted subclones may expand and maintain tumor progression after the drug treatment8,10,97. A very recent study on breast cancers demonstrated that resistance to chemotherapy and the acquisition of metastases could be caused by detectable subclones with different somatic DNA variations in the primary tumor42. These observations suggest that clonally dominant events are not necessarily more important in function than the minor subclonal lesions. A similar scenario has been observed in myelodysplastic syndrome (MDS), in which the presence of the driver mutations was the key determinant of the disease outcome, regardless of their clonal status98. The Deciphering Anti-tumor Response With Intratumor Heterogeneity (DARWIN, NCT02183883) trial has been launched, and it is aimed at assessing whether targeting a clonally dominant driver mutation results in improved progression free survival compared to targeting the same driver mutation when it is present in a minor subclone99. The hypothetical implication of the heterogeneous genotype on therapeutics includes either concurrent combination or sequential treatment with multiple driver mutation-targeting agents. For example, in NSCLC cells, MET amplification in certain cells is considered one of the mechanisms for the resistance to EGFR inhibitors100. The combination of a MET inhibitor and a EGFR inhibitor exhibited better efficacy than either of the individual compounds101.
Second, the population of tumorigenic cells within a tumor is intrinsically resistant to certain therapies, especially cytotoxic chemoradiotherapies. For example, the enrichment of tumorigenic cells in CRC, breast cancer, and GBMs has been observed after irradiation or cyclophosphamide treatment2. Lower levels of reactive oxygen species and the activation of ATM-dependent DNA-damage repair in these tumorigenic cells may explain the therapeutic resistance. However, whether the genetic or epigenetic changes contribute to the resistance of tumorigenic cells requires further investigation. Epigenetic therapies, such as small molecules that regulate DNA methylation and histone deacetylation or target other chromatin regulatory proteins have the potential to prevent or reverse treatment resistance102.
Third, stochastic gene expression or signal transduction-mediated cell plasticity may lead to differential cell fate determination and thus contribute to drug resistance. The fate of daughter cells derived from the same mother cell seemed to be independent of each other following the exposure to antimitotic drugs103. Strategies to overcome this resistance will not be as straightforward as targeting genetic or epigenetic sources of resistance. Studies in this field have just recently been performed. Investigating whether and how environmental conditions eliminate or control heterogeneous cancer cells hold the potential to inform novel anti-cancer strategies.
Finally, continued drug exposure imposes intense Darwinian forces on the surviving cells and thus increases the evolutionary rate, which may further facilitate selection and/or induction of more aggressive subpopulations of cancer cells with new properties84. For example, in melanomas, resistance to the BRAF inhibitor is mainly mediated by reactivation of the ERK signaling pathway through mutated RAS, and prolonged BRAF inhibitor treatment in RAS and BRAF mutant melanoma cells results in the release of proteases and tumor metastatic progression104. A recent study in breast cancers demonstrated the presence of cytotoxic chemotherapy enriched preexisted PIK3CA-mutant cells, which were resistant to HER2-targeted therapy41. In-depth investigations of the impact of intra-tumor heterogeneity on therapeutic outcome is needed, and a longitudinal study, called Tracking Non-small Cell Lung Cancer Evolution Through Therapy (TRACERx, NCT01888601) has been launched in the United Kingdom to address this issue105.
Conclusions and future directions
Genetic and non-genetic heterogeneity prevail within each individual cancer. The clonal evolution model explains how genetic variation arises. A careful examination of the heterogeneous driver mutations among various subclones would be more therapeutically relevant than simply considering a somatic event to be present or absent based on the analysis of only one biopsy. The very recent study using multiregion sequencing revealed substantial subclonal diversifications in breast cancer samples and a correlation between the degree of heterogeneity and clinical prognoses, suggesting that quantitative measurement of intra-tumor heterogeneity might have implications in understanding tumor evolution and should be included in the diagnosis and treatment of cancers42. However, targeting feasibility is another challenge for future drug development. For example, EGFR and KRAS mutations in lung cancers, KRAS and BRAF mutations in CRCs, and VHL loss-of-function mutations in ccRCCs are all considered to be functionally important events90,106, whereas only EGFR and BRAF mutations are currently targetable.
Moreover, increasing evidence of plastic phenotype switch-ing of cancer cells have been recently reported2,3,92. A revised CSC model, or the so-called plastic CSC model, has been recently proposed and explains the non-genetic origin of intra-tumor heterogeneity4. In pursuit of a better treatment, it will be crucial to determine the fraction of cancer cells that can transit reversibly between the different states and the fraction of cancer cells that follow a strict hierarchy. Experiments should be very carefully designed to distinguish cell potential, which is what cells can do, as well as cell fate, which is what cells actually do in a real scenario2. In addition, identifying the molecular mechanisms of cancer cells underlying the dynamic phenotypic switching will be of fundamental importance. Current studies focus on the reversible epigenetic changes and stochastic gene expression. It will be interesting to investigate the contribution of heterogeneous signal transduction to cell plasticity and subclonal evolution because environmental information is written, transferred, and read through signal transductions, and diverse signaling may lead to differential downstream responses, such as gene expression and protein degradation, to environmental changes.
Considering the fact that the complex bulky neoplasm evolved over a long period from several single cells, the intra-tumor heterogeneity might be regarded as a result of cell plasticity or variation from a dynamic point of view. The complexities of cancer cell plasticity emphasize the need for in-depth biological understanding of single cell behaviors in the context of cell populations. Fundamental questions include how and why heterogeneous responses arise even within an isogenic cell population. Beyond this question, how individual cells behave under tumorigenic conditions (chronic inflammation, hypoxia, or acidosis) and therapeutic conditions is of great importance to cancer biology. In fact, cellular heterogeneity is a well-recognized attribute of both normal and neoplastic tissues. The difference is that in normal tissues, an ordered developmental program underlying the heterogeneity usually leads to an early stochastic and late hierarchy pattern107. In contrast, disorder characterizes cancer cell populations. Thus, the quantification of the heterogeneity and investigation of the aberrant regulation of cell diversity in cancers hold the potential to provide hitherto unknown and important information about cancer from different angles. Until we develop a deeper understanding of the biological mechanisms underlying intra-tumor heterogeneity, a more effective strategy may be proposed to treat cancer.
Last but not least, although the ever-increasing complexities of cancers seem to be discouraging, alternative strategies, such as immunotherapy, may bypass the challenges of intra-tumor heterogeneity and attack all cancer cells because each mutation has the potential to produce a new antigen that may be recognized by corresponding immune cells108. Recently, a meta-analysis revealed that tumors expressing new antigens were associated with an improved prognosis compared to tumors without new antigens109. Removing the immunological brakes that block the induction of the anti-tumor responses, such as inhibition of CTLA-4 and PDL-1110, has become a hot topic in clinical oncology and holds great promise for treating heterologous cancer cells within a tumor.
References
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74.
Meacham CE, Morrison SJ . Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501: 328–37.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011; 146: 633–44.
Marjanovic ND, Weinberg RA, Chaffer CL . Cell plasticity and heterogeneity in cancer. Clin Chem 2013; 59: 168–79.
Cohen P, Alessi DR . Kinase drug discovery--what's next in the field? ACS Chem Biol 2013; 8: 96–104.
Gonzalez de Castro D, Clarke PA, Al-Lazikani B, Workman P . Personalized cancer medicine: molecular diagnostics, predictive biomarkers, and drug resistance. Clin Pharmacol Ther 2013; 93: 252–9.
Burrell RA, McGranahan N, Bartek J, Swanton C . The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013; 501: 338–45.
Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 2012; 109: 3041–6.
Little SE, Popov S, Jury A, Bax DA, Doey L, Al-Sarraj S, et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res 2012; 72: 1614–20.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–92.
Park SY, Gonen M, Kim HJ, Michor F, Polyak K . Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 2010; 120: 636–44.
Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR . Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat 2007; 102: 143–55.
Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell 2012; 149: 994–1007.
Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34+CD38- cells in acute myeloid leukemia. Blood 2012; 119: 3571–7.
Horton SJ, Huntly BJ . Recent advances in acute myeloid leukemia stem cell biology. Haematologica 2012; 97: 966–74.
Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res 2010; 12: R87.
Mannello F . Understanding breast cancer stem cell heterogeneity: time to move on to a new research paradigm. BMC Med 2013. doi: 10.1186/1741-7015-11-169.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401.
O' Brien CA, Pollett A, Gallinger S, Dick JE . A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445: 106–10.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007; 445: 111–5.
Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010; 466: 133–7.
Civenni G, Walter A, Kobert N, Mihic-Probst D, Zipser M, Belloni B, et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res 2011; 71: 3098–109.
Wozniak J, Kopec-Szlezak J . c-Kit receptor (CD117) expression on myeloblasts and white blood cell counts in acute myeloid leukemia. Cytometry B Clin Cytom 2004; 58: 9–16.
Erlanson M, Landberg G . Flow cytometric quantification of cyclin E in human cell lines and hematopoietic malignancies. Cytometry 1998; 32: 214–22.
Holyoake T, Jiang X, Eaves C, Eaves A . Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999; 94: 2056–64.
Laane E, Tani E, Bjorklund E, Elmberger G, Everaus H, Skoog L, et al. Flow cytometric immunophenotyping including Bcl-2 detection on fine needle aspirates in the diagnosis of reactive lymphadenopathy and non-Hodgkin's lymphoma. Cytometry B Clin Cytom 2005; 64: 34–42.
Robillard N, Pellat-Deceunynck C, Bataille R . Phenotypic characterization of the human myeloma cell growth fraction. Blood 2005; 105: 4845–8.
Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, et al. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc Natl Acad Sci U S A 2010; 107: 12747–54.
Morkve O, Halvorsen OJ, Stangeland L, Gulsvik A, Laerum OD . Quantitation of biological tumor markers (p53, c-myc, Ki-67 and DNA ploidy) by multiparameter flow cytometry in non-small-cell lung cancer. Int J Cancer 1992; 52: 851–5.
Andreeff M, Slater DE, Bressler J, Furth ME . Cellular ras onco-gene expression and cell cycle measured by flow cytometry in hematopoietic cell lines. Blood 1986; 67: 676–81.
Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov 2014; 4: 956–71.
Bai H, Wang Z, Wang Y, Zhuo M, Zhou Q, Duan J, et al. Detection and clinical significance of intratumoral EGFR mutational heterogeneity in Chinese patients with advanced non-small cell lung cancer. PLoS One 2013; 8: e54170.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014; 344: 1396–401.
Chakrabarty S, Jan Y, Miller CA, Brattain MG . Selective protein phosphorylation in heterogeneous subpopulations of human colon carcinoma cells. Cancer Res 1985; 45: 743–50.
Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 2004; 118: 217–28.
Van Meter ME, Diaz-Flores E, Archard JA, Passegue E, Irish JM, Kotecha N, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood 2007; 109: 3945–52.
Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell 2008; 14: 335–43.
Singh DK, Ku CJ, Wichaidit C, Steininger RJ 3rd, Wu LF, Altschuler SJ . Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Mol Syst Biol 2010; 6: 369.
Shuo T, Koshikawa N, Hoshino D, Minegishi T, Ao-Kondo H, Oyama M, et al. Detection of the heterogeneous O-glycosylation profile of MT1-MMP expressed in cancer cells by a simple MALDI-MS method. PLoS One 2012; 7: e43751.
Misra A, Chattopadhyay P, Dinda AK, Sarkar C, Mahapatra AK, Hasnain SE, et al. Extensive intra-tumor heterogeneity in primary human glial tumors as a result of locus non-specific genomic alterations. J Neurooncol 2000; 48: 1–12.
Janiszewska M, Liu L, Almendro V, Kuang Y, Paweletz C, Sakr RA, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet 2015 Aug 24. doi: 10.1038/ng.3391.
Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 2015; 21: 751–9.
Kuwai T, Nakamura T, Kim SJ, Sasaki T, Kitadai Y, Langley RR, et al. Intratumoral heterogeneity for expression of tyrosine kinase growth factor receptors in human colon cancer surgical specimens and orthotopic tumors. Am J Pathol 2008; 172: 358–66.
Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 2010; 18: 510–23.
Ennen M, Keime C, Kobi D, Mengus G, Lipsker D, Thibault-Carpentier C, et al. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene 2014; 34: 3251–63.
Tatematsu T, Sasaki H, Shimizu S, Hikosaka YU, Okuda K, Haneda H, et al. Intra-tumor heterogeneity of BRAF V600E mutation in lung adenocarcinomas. Exp Ther Med 2015; 9: 1719–22.
Mroz EA, Tward AD, Hammon RJ, Ren Y, Rocco JW . Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med 2015; 12: e1001786.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–9.
Lynch M . Evolution of the mutation rate. Trends Genet 2010; 26: 345–52.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW . Cancer genome landscapes. Science 2013; 339: 1546–58.
Bonnet D, Dick JE . Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine 1997; 3: 730–7.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Cacerescortes J, et al. A cell initiating human acute myeloid-leukemia after transplantation into Scid mice. Nature 1994; 367: 645–8.
Visvader JE, Lindeman GJ . Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8: 755–68.
Fearon ER, Burke PJ, Schiffer CA, Zehnbauer BA, Vogelstein B . Differentiation of leukemia cells to polymorphonuclear leukocytes in patients with acute nonlymphocytic leukemia. N Engl J Med 1986; 315: 15–24.
Illmensee K, Mintz B . Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A 1976; 73: 549–53.
Barabe F, Kennedy JA, Hope KJ, Dick JE . Modeling the initiation and progression of human acute leukemia in mice. Science 2007; 316: 600–4.
Yamazaki J, Estecio MR, Lu Y, Long H, Malouf GG, Graber D, et al. The epigenome of AML stem and progenitor cells. Epigenetics 2013; 8: 92–104.
Rheinbay E, Suvà ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Reports 2013; 3: 1567–79.
Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43: 768–75.
Yatabe Y, Tavare S, Shibata D . Investigating stem cells in human colon by using methylation patterns. Proc Natl Acad Sci U S A 2001; 98: 10839–44.
Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Reports 2014; 6: 514–27.
Elowitz MB, Levine AJ, Siggia ED, Swain PS . Stochastic gene expression in a single cell. Science 2002; 297: 1183–6.
Levine JH, Lin Y, Elowitz MB . Functional roles of pulsing in genetic circuits. Science 2013; 342: 1193–200.
Singer Zakary S, Yong J, Tischler J, Hackett Jamie A, Altinok A, Surani MA, et al. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 2014; 55: 319–31.
Raj A, van Oudenaarden A . Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 2008; 135: 216–26.
Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK . Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009; 459: 428–32.
Loo LH, Wu LF, Altschuler SJ . Image-based multivariate profiling of drug responses from single cells. Nat Methods 2007; 4: 445–53.
Balázsi G, van Oudenaarden A, Collins JJ . Cellular decision-making and biological noise: from microbes to mammals. Cell 2011; 144: 910–25.
Thattai M, van Oudenaarden A . Attenuation of noise in ultrasensitive signaling cascades. Biophys J 2002; 82: 2943–50.
Albeck JG, Mills GB, Brugge JS . Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 2013; 49: 249–61.
Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, et al. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol Cell 2013; 52: 529–40.
Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW . High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014; 157: 1724–34.
Tsai FC, Seki A, Yang HW, Hayer A, Carrasco S, Malmersjo S, et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat Cell Biol 2014; 16: 133–44.
Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T . The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013; 155: 369–83.
Lochte S, Waichman S, Beutel O, You C, Piehler J . Live cell micropatterning reveals the dynamics of signaling complexes at the plasma membrane. J Cell Biol 2014; 207: 407–18.
Cimica V, Reich NC . Nuclear trafficking of STAT proteins visualized by live cell imaging. Methods Mol Biol 2013; 967: 189–202.
Shen H, Laird PW . Interplay between the cancer genome and epigenome. Cell 2013; 153: 38–55.
Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, et al. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell 2005; 17: 793–803.
Liu Q, Liu L, Zhao Y, Zhang J, Wang D, Chen J, et al. Hypoxia induces genomic DNA demethylation through the activation of HIF-1alpha and transcriptional upregulation of MAT2A in hepatoma cells. Mol Cancer Ther 2011; 10: 1113–23.
Fukumura D, Duda DG, Munn LL, Jain RK . Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation 2010; 17: 206–25.
RK J . Determinants of tumor blood flow: a review. Cancer Res 1988; 48: 2641–58.
Brown EB, Campbell RB, Tsuzuki Y, Xu L, Carmeliet P, Fukumura D, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med 2001; 7: 864–8.
Helmlinger G, Yuan F, Dellian M, Jain RK . Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat Med 1997; 3: 177–82.
Gillies RJ, Verduzco D, Gatenby RA . Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012; 12: 487–93.
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11: 69–82.
Junttila MR, de Sauvage FJ . Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013; 501: 346–54.
Shackleton M, Quintana E, Fearon ER, Morrison SJ . Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138: 822–9.
de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014; 346: 251–6.
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46: 225–33.
McGranahan N, Swanton C . Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015; 27: 15–26.
Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DWY, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 2010; 24: 1731–45.
Easwaran H, Tsai HC, Baylin SB . Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014; 54: 716–27.
Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA methylation alterations exhibit intra-individual stability and inter-individual heterogeneity in prostate cancer metastases. Sci Transl Med 2013; 5: 169ra10–ra10.
O'Hagan HM . Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environ Mol Mutagen 2014; 55: 278–91.
Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014; 346: 256–9.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014; 344: 1396–401.
Snuderl M, Fazlollahi L, Le Long P, Nitta M, Zhelyazkova Boryana H, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell; 20: 810–7.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–27.
Hiley C, de Bruin EC, McGranahan N, Swanton C . Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol 2014; 15: 453.
Suda K, Murakami I, Katayama T, Tomizawa K, Osada H, Sekido Y, et al. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res 2010; 16: 5489–98.
Nakagawa T, Takeuchi S, Yamada T, Nanjo S, Ishikawa D, Sano T, et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming Erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther 2012; 11: 2149–57.
Azad N, Zahnow CA, Rudin CM, Baylin SB . The future of epigenetic therapy in solid tumours—lessons from the past. Nat Rev Clin Oncol 2013; 10: 256–66.
Gascoigne KE, Taylor SS . Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14: 111–22.
Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signaling 2014; 7: ra30–ra.
Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking genomic cancer evolution for precision medicine: the Lung TRACERx Study. PLoS Biol 2014; 12: e1001906.
Bedard PL, Hansen AR, Ratain MJ, Siu LL . Tumour heterogeneity in the clinic. Nature 2013; 501: 355–64.
Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012; 150: 1209–22.
Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014; 124: 453–62.
Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014; 24: 743–50.
Quezada SA, Peggs KS . Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer 2013; 108: 1560–5.
Acknowledgements
This work was supported by the China Ministry of Science and Technology Key New Drug Creation and Manufacturing Program grant No 2013ZX09102015, 2014ZX09102001002, the China Ministry of Science and Technology Research grant 2013ZX10002010-009, the National Natural Science Foundation of China grants No 81302792, 81373447, 91413121, 91213304, and the China National Key Basic Research Program grants No 2012CB910704 and 2013CB910900. We would like to thank Shensi SHEN, Chiraj K DALAL, Peter S SWAIN and Steven J ALTSCHULER for stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Sun, Xx., Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin 36, 1219–1227 (2015). https://doi.org/10.1038/aps.2015.92
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.92
Keywords
This article is cited by
-
Multimodal epigenetic sequencing analysis (MESA) of cell-free DNA for non-invasive colorectal cancer detection
Genome Medicine (2024)
-
Clustering on hierarchical heterogeneous data with prior pairwise relationships
BMC Bioinformatics (2024)
-
Deciphering cellular plasticity in pancreatic cancer for effective treatments
Cancer and Metastasis Reviews (2024)
-
A robust optimal control framework for controlling aberrant RTK signaling pathways in esophageal cancer
Journal of Mathematical Biology (2024)
-
USP13 drives lung squamous cell carcinoma by switching lung club cell lineage plasticity
Molecular Cancer (2023)
