
Abstract
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by the loss of tolerance to self-nuclear antigens. Accumulating evidence shows that Toll-like receptors (TLRs), previously proven to be critical for host defense, are implicated in the pathogenesis of autoimmune diseases by recognition of self-molecules. Genome-wide association studies, experimental mouse models and clinical sample studies have provided evidence for the involvement of TLRs, including TLR2/4, TLR5, TLR3 and TLR7/8/9, in SLE pathogenesis. A number of downstream proteins in the TLR signaling cascade (such as MyD88, IRAKs and IFN-α) are identified as potential therapeutic targets for SLE treatment. Numerous antagonists targeting TLR signaling, including oligonucleotides, small molecular inhibitors and antibodies, are currently under preclinical studies or clinical trials for SLE treatment. Moreover, the emerging new manipulation of TLR signaling by microRNA (miRNA) regulation shows promise for the future treatment of SLE.
Similar content being viewed by others

Introduction
Toll-like receptors (TLRs) are a family of evolutionarily conserved, innate immune receptors that play a crucial role in the first-line defense against foreign molecules called pathogen-associated molecular patterns (PAMP)1,2. These receptors are expressed on/in various immunocompetent cells (including macrophages, dendritic cells and B lymphocytes) and some cells with non-immune functions (such as epithelial and mesangial cells of the kidney)3. The identified TLRs include cell surface receptors that recognize a variety of bacterial or fungal molecules, including lipopeptide (TLR2), lipopolysaccharide (TLR4) and flagellin (TLR5)4. There are also intracellular receptors, which are expressed in the endosomal compartments of cells. The receptors sense nucleic acids, such as double-stranded RNA (TLR3), single-stranded RNA (TLR7 and TLR8) or unmethylated single-stranded DNA containing cytosine–phosphate–guanine (CpG) motifs (TLR9), which are common in viral and bacterial genomes4.
However, recent findings have revealed that TLRs also recognize and respond to endogenous ligands produced during inflammation or tissue damage5,6,7,8,9,10,11,12. Systemic autoimmune diseases are commonly associated with the production of autoantibodies specific for self-determinants13. Many of the autoantigens are derived from structures such as chromatin and small nuclear ribonucleoproteins (snRNPs), which are normally sequestered from the immune system by virtue of their intracellular location14. However, these nuclear antigens become more accessible for TLRs as a result of cell death or apoptosis. Once engaged, these TLRs act rapidly via adaptor proteins to induce transcription factors for type I IFNs and other pro-inflammatory mediators, which further contribute to the development and progression of autoimmune diseases15. Therefore, the identification and characterization of endogenous ligands of TLRs provide a novel perspective for exploring the etiology of autoimmune diseases.
Systemic lupus erythematosus (SLE) is a typical autoimmune disease characterized by the loss of tolerance to self-nuclear antigens13. Approximately 40% of SLE patients exhibit defects in the clearance of apoptotic cells, which are removed rapidly by phagocytes in healthy individuals16. A lupus murine model showed a defect in the clearance of cellular debris17. The inefficient clearance of cellular debris leads to increased release of host DNA and RNA, which can be detected by TLRs and induces the production of autoantibodies16. Therefore, targeting TLRs and modulating TLR signaling have emerged as important strategies for the treatment of SLE.
TLRs in SLE
TLRs fulfil many of the criteria required for potential therapeutic targets in SLE. For specific TLRs, evidence includes gene polymorphisms of TLRs contributing to disease susceptibility, overexpression of TLRs in patients and mouse models, TLR knockout mice being resistant to disease and TLR ligands exacerbating and TLR inhibitors alleviating disease. However, different TLRs provide distinct or synergistic contributions to the pathogenesis of SLE.
TLR2 and TLR4
Environmental factors play a role in the onset of SLE. The recognition of these factors is mediated by TLRs, in particular, TLR2 and TLR4, which bind PAMP of Gram+ and Gram− bacteria, respectively. Although the gene polymorphism of TLR2 (Arg677Trp and Arg753Gln) and TLR4 (Asp299Gly and Thr399 Ile) has no effects on the predisposition and clinical characteristics of SLE18, the expression levels of TLR2 and TLR4 mRNAs in SLE patients' PBMCs are much higher than those in healthy subjects'19. Deficiency in TLR4 and, to a lesser extent, in TLR2 down-regulates the production of autoantibodies and attenuates the development of renal injuries in lpr mutation-induced mouse lupus20. TLR4−/− mice also have decreased pathogenic cytokines, anti-dsDNA and anti-RNP antibodies and attenuated renal injury in pristane-induced experimental lupus21. In addition, TLR4 up-regulation at the protein or gene level is a potent trigger to induce lupus-like autoimmune disease22. Furthermore, TLR2 and TLR4 may be involved in anti-DNA autoantibody-induced kidney damage in lupus nephritis by recognizing HMGB1 (high mobility group box-1 protein), which binds with DNA and pathogenic anti-DNA autoantibodies and is implicated in the pathogenesis of SLE9.
TLR5
The chromosomal region lq41-42 contains major susceptibility genes of SLE23,24. Intriguingly, the TLR5 gene maps to chromosome lq41 and contains a common stop codon polymorphism (allele C1174T). Allele 1174C, but not 1174T (with the stop codon), was preferentially transmitted to SLE-affected offspring25. Additionally, populations with this stop codon produce reduced levels of pro-inflammatory cytokines, suggesting that the TLR5 stop codon polymorphism is associated with protection from the development of SLE25. Other evidence revealed that the expression level of TLR5 mRNA correlates significantly with IFN-α mRNA in the PBMCs of SLE patients19. Furthermore, because of its central role in regulating inflammatory pathways, the biological plausibility of TLR5′s association with SLE is compelling.
TLR3
TLR3 recognizes double-stranded RNA from viruses and the synthetic mimic Poly I:C1. Although it is also intracellularly localized, TLR3 signaling is distinct from the TLR7/8/9 signaling pathways. Rather than utilizing Myeloid differentiation factor 88 (MyD88) as an adaptor protein, TLR3 uses TIR-domain-containing adaptor-inducing interferon-β (TRIF)26. In MRL/lpr mice, TLR3, TLR7 and TLR9 are expressed by intrarenal macrophages, whereas only TLR3 is expressed by mesangial cells27. Additionally, the expression of TLR3 mRNA increases with the progression of glomerulonephritis, and Poly I:C aggravates lupus nephritis through TLR3 on glomerular mesangial cells and APC in MRL/lpr mice27,28. Nevertheless, Poly I:C injection does not increase the titer of anti-dsDNA antibodies, and ablation of TLR3 does not inhibit the formation of autoantibodies, suggesting that TLR3 participates in the pathogenesis of SLE in a B cell-independent way28,29.
TLR7, TLR9 and TLR8
Production of autoantibodies is the main feature of SLE13. Disease-related autoantibodies in SLE focus on particular targets, including DNA-containing antigens, such as dsDNA, and RNA-containing antigens, such as Sm/RNP14,30. A variety of in vitro studies in mouse cells show that RNA- and DNA-containing immune complexes, respectively, activate TLR7 and TLR9 through BCR-mediated internalization in B cells or through FcγR-mediated internalization in dendritic cells (DCs)31,32,33. TLR engagement in B cells increases BCR signaling and antibody production, whereas in pDCs, TLR induces IFN-α production, which causes mDCs to release B-cell activating factor (BAFF) and further activates autoreactive B cells34. Additionally, upregulated TLR7 and TLR9 mRNA expression has been reported in PBMCs from SLE patients, and the levels correlate with the expression of IFN-α19,35.
The link between TLR7 and RNA-associated antibody production is supported by studies of lupus-prone mice harboring the Y-linked autoimmune acceleration (Yaa) cluster, which includes a duplication of the TLR7 gene. The overexpression of TLR7 is the cause of the autoimmune phenotypes associated with yaa36 and autoreactive B cell responses to RNA-related antigens due to TLR7 gene replication in male BXSB mice37. Ablated TLR7 signaling in the BXSB mouse models results in decreased autoantibody production38. IFN-α production in pristane-induced mice is also dependent on TLR739. Murine TLR7−/− pDCs stimulated with U1snRNP/anti-Sm ICs produce markedly reduced levels of IFN-α and IL-640. The overall impact of TLR7 on SLE has also been investigated. MRL/lpr mice lacking TLR7 display ameliorated disease manifestation, vanished anti-Sm antibodies, decreased serum IgG and decreased lymphocyte activation41.
Whereas the pathological role of TLR7 in human SLE and lupus nephritis in mouse models is relatively accepted, the role of TLR9 remains controversial. Multiple mouse studies have shown the indispensable role of TLR9 in B cells for the production of anti-dsDNA, anti-chromatin and anti-nucleosome autoantibodies29,41,42. However, the deletion of TLR9 in these lupus-prone models does not lead to disease amelioration as predicted but to disease exacerbation, suggesting a protective role of TLR9 in lupus in mice41,42. In addition, although TLR7 and TLR9 act in parallel on different subsets of autoantibodies, TLR9 suppresses the production of TLR7-dependent, RNA-associated autoantibodies42,43. The proportions of TLR9-expressing B cells, plasma cells and monocytes increase in SLE patients, and the increase in TLR9-expressing B cells is correlated with the production of anti-dsDNA antibodies44,45. Additionally, B cells and monocytes from patients with active disease express higher TLR9 levels compared to patients with inactive disease44,46. Paradoxically, the increased expression of TLR9 does not give rise to stronger responsiveness to TLR9 ligands. Despite increased TLR9 expression in B cells and DCs from patients with severe disease, cells are less activated and are hyporesponsive to ODN-CpG stimulation47. Overall, further studies are required to establish the role of TLR9 in mediating the progression of SLE.
TLR8, phylogenetically similar to TLR7, also recognizes single-stranded RNA (ssRNA) and synthetic ligands, such as R848. To date, few studies on the function of TLR8 in SLE have been published. TLR8 may participate in the pathogenesis of SLE by recognition of RNA autoantigens11. However, recent studies reveal that TLR8 in dendritic cells restrains TLR7-mediated lupus manifestations in C57BL/6 mice43. TLR8 deletion accelerates autoimmunity in lupus-prone mice through a TLR7-dependent mechanism48. Moreover, TLR8 contributes to the gender differences in the development of SLE49.
Other potential targets involved in the TLR signaling cascade
MyD88 is a common adaptor protein present in most TLR signaling50. Because both TLR7 and TLR9 utilize this protein, MyD88 is an excellent target to intervene in the abnormal signaling in SLE. MyD88-knockout MRL/lpr mice exhibit a prolonged lifespan with no apparent development of autoimmune nephritis in comparison with wild-type mice51. Shlomchik's group further investigated the separate pathogenetic mechanism of MyD88 in different cell types, revealing that MyD88 in B cells controls lupus nephritis, whereas MyD88 in DCs is critical for dermatitis in MRL/lpr mice52. The intracellular tyrosine kinase Lyn mediates the inhibitory receptor function in B cells and myeloid cells, and Lyn−/− mice spontaneously develop an autoimmune and inflammatory disease that closely resembles human SLE. Production of germinal center, anti-nuclear antibody in Lyn-deficient mice also requires MyD88 signaling in B cells and DCs53. Deletion of MyD88 in DCs alone completely reversed the lupus manifestation shown in Lyn-mutant mice54.
Another potential target for abolishing aberrant TLR signaling in SLE is IL-1R–associated kinases (IRAKs). IRAK1 and IRAK4 function as active kinases and as scaffolds for interacting with MyD88 and TRAF6 in TLR signaling. All TLRs, except TLR3, require IRAKs and MyD88 for signaling55. IRAK4-deficient patients and MyD88-deficient patients do not display autoreactive antibodies in their serum and do not develop autoimmune diseases, suggesting that IRAK4 and MyD88 pathway blockades may thwart autoimmunity in humans56. An IRAK1/4 kinase inhibitor abrogated the TLR7/9-induced IFN-α responses in both mouse and human pDCs57.
IFN-α, one of the end-product effectors of TLR signaling, has been an intense research focus in SLE pathogenesis. Genome-wide association studies provide strong genetic evidence that type I IFNs (with IFN-α as the dominant mediator) are important for SLE risk. Of 47 genetic variants associated with SLE, over half (27/47, 57%) can be linked to type I IFN production or signaling58. The accumulated data indicate that the levels of IFN-α in circulation are significantly elevated among lupus patients compared with control subjects, and the high levels of IFN-α are associated with worsened measures of disease activity59,60,61. Recombinant IFN-α, when administered as a therapy to patients with malignancy or hepatitis infection, can induce SLE60. In addition, deficiency of the type I IFN receptor protects mice from experimental lupus62,63.
Potential therapeutic treatment by blocking TLR signaling transduction
Currently, there are diverse agents under development for lupus treatment by targeting TLRs or TLR accessory proteins (including MyD88, IRAK-4 and IFN-α) at different stages of TLR signaling pathways (Figure 1). Here, we focus on a series of agents in the discovery phase or in clinical trials, including oligonucleotides (Table 1), small molecular inhibitors (SMIs) (Table 2), antibodies (Table 3) and new emerging modulators, such as microRNAs, which might offer further possibilities for therapeutic manipulation.

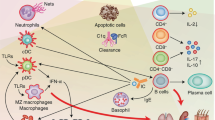
Potential targets involved in TLR signaling pathways for SLE treatment. TLR-mediated responses are controlled mainly in a MyD88-dependent way, which is used by all TLRs except TLR3. In the pathogenesis of SLE, both exogenous ligands from infection and endogenous ligands from apoptotic debris contribute to the activation of TLRs and initiate the downstream signaling cascade, resulting in the production of type I IFN and inflammatory cytokines. A series of potential therapeutics, including oligonucleotides, SMIs and antibodies, modulate TLR signaling pathways at different stages. Some oligonucleotides, SMIs and TLR-neutralized antibodies directly block TLRs; some SMIs target TLR accessory proteins, such as MyD88 and IRAKs. Anti-IFN-α antibodies are also under intensive development for the treatment of SLE. TAK1, transforming growth factor-β-activated kinase; IRF, interferon regulatory factor.
Oligonucleotides
Several TLR ligand mimics that are oligonucleotides that bind to endosomal TLR7 and/or TLR8 and/or TLR9 have been designed and synthesized to treat lupus64,65. Immunoregulatory sequence-954 (IRS-954) is an oligonucleotide-based bifunctional inhibitor of TLR7 and TLR9 and is currently under pre-clinical research66. In vitro experiments showed that IRS-954 inhibits the induction of IFN-α by human pDCs in response to DNA/RNA viruses and isolated immune complexes from lupus patients66. Preclinical data from animal model studies showed a significant reduction of the serum levels of nucleic acid-specific autoantibodies, as well as decreased proteinuria, reduced glomerulonephritis, end-organ damage and increased survival67. The glucocorticoid resistance characteristics of SLE mediated via TLR7 and TLR9 was also reversed by IRS-95468.
DV-1179, another TLR7/9 dual antagonist developed by the same company that produced IRS-954, is being tested for the treatment of SLE. A phase I study of DV-1179 was conducted to assess its safety and tolerability in healthy volunteers followed by a phase Ib/IIa study of the safety and pharmacodynamics in patients with active SLE. In the SLE study, doses up to 60 mg/week for 8 weeks were well-tolerated, and the most common adverse events were injection site reaction. However, DV-1179 did not achieve the pharmacodynamic endpoints related to reduction in IFN-α-regulated genes69.
A number of oligonucleotides-based immune modulatory oligonucleotides (IMO) are also in the pipeline for use in autoimmune disease treatment. IMO-3100, a TLR7 and TLR9 antagonist, is in phase II clinical trials for the treatment of psoriasis. The company evaluated the compound pre-clinically for SLE treatment. IMO-3100 inhibits TLR-induced increases in the gene expression of TNF-α, IFN-α and IL-17 in human PBMCs and inhibits disease development in lupus-prone NZBW/F1 mice70,71. However, no recent development has been reported for this research. IMO-8400, which antagonizes TLR7, TLR8 and TLR9, has shown efficacy in mouse models of lupus72,73. Early clinical trials are now underway for the treatment of SLE.
At the end of 2013, preclinical research on INH-ODN-24888, a guanine-modified inhibitory oligonucleotide (INH-ODN) derived from INH-ODN-2088, was initiated for the treatment of lupus based on activity as a TLR7 and TLR9 antagonist. The guanine modification of INH-ODN potentiates the suppressive function of its prototypic agent and efficiently impairs the TLR7- and TLR9-mediated immune responses of human immune cells74,75. Thus, INH-ODN-24888 represents a promising therapeutic agent for the treatment of SLE.
Small molecular inhibitors (SMIs)
Small molecule inhibitors (SMIs) can be taken orally and are promising agents to penetrate the cell membrane, effectively targeting endosomal TLRs and downstream signaling proteins. Thus, SMIs have been designed to treat SLE based on multiple targets in the TLR signaling pathway, including TLRs, MyD88 and IRAKs.
In the treatment of SLE, antimalarial drugs, such as hydroxychloroquine sulfate (HCQ), chloroquine and quinacrine, have been used clinically since 1956 and are now known to act as TLR7/8/9 antagonists. Their suppression of endosomal TLR activation was attributed to the inhibition of endosomal acidification, which is a prerequisite for the activation of these receptors76,77,78. Recently, a new mechanistic model of these antimalarial drugs has proposed that they directly interact with nucleic acids and consequently cause structural modifications of the TLR ligand to prevent the ligand from binding to TLR79,80,81.
The quinazoline derivative CpG-52364 is a specific SMI of TLR7/8/9. CpG-52364 was designed to interfere at an early stage of the immune cascade by blocking the inappropriate immune activation of all the three TLRs and to treat the underlying cause of the disease without causing general suppression of immune function. CpG-52364 has been proved to be safer and more efficacious than HCQ in preclinical animal studies82,83, and it is currently in phase I clinical trials for the oral treatment of SLE.
A novel artemisinin analogue, β-aminoarteether maleate (SM934), has recently been authorized by the Chinese SFDA to enter clinical trials for SLE treatment. In preclinical study, SM934 exhibited significant protective effects on both MRL/lpr mice and NZBW/F1 mice84,85,86. The therapeutic mechanism of SM934 involves inhibiting TLR7/9-triggered B cell activation in a MyD88-dependent way87. Since 1956, no new chemical drug for SLE treatment has reached the market in the USA, and no original class I chemical drugs have been developed in China. Therefore, the clinical study of SMI SM934 could be a milestone in the development of lupus treatment drugs in China.
In addition, E-6446 and AT-791, targeting TLR7/9 and IL-6, were designed for the treatment of SLE88. However, research on the two SMIs was suspended at the preclinical stage. The TLR8-specific antagonist VTX-763 and the TLR7-specific antagonist TMX-302 are both under active preclinical research for autoimmune disease therapy. IMO-9200, is under phase I clinical trials as a small molecule poly-inhibitor of TLR7/8/9 to treat autoimmune diseases. However, greater therapeutic effects are reported in the treatment of rheumatoid arthritis (RA)89,90.
A series of peptido-mimetic compounds was synthesized as MyD88 dimerization inhibitors, including ST-2825, ST-3324, ST-2928, ST-2797, ST-2804, ST-2807, ST-2565 and ST-3375, among which ST-2825 was the most effective. ST-2825 interferes with the recruitment of IRAK4 and IRAK1 via MyD88, causing inhibition of pro-inflammatory factor overproduction91. In human PBMCs, ST2825 suppressed B cell proliferation and differentiation into plasma cells in response to CpG-induced activation of TLR991. ST-2825 also blocks autoantibody production in B cells from SLE patients92.
Given the critical role of IRAK-4 in inflammatory processes, modulation of IRAK-4 kinase activity presents an attractive therapeutic approach for the treatment of SLE. Several IRAK-4 inhibitors have been identified, among which PF-05387252, PF-05388169 and AS-2444697 are in preclinical studies and PF-06650833 is in phase I clinical studies93,94.
Abnormal post-translational protein modifications involved in TLR signaling also play a significant role in SLE pathogenesis. We and others have reported DZ2002, a reversible SAHH (S-adenosyl-l-homocysteine hydrolase) inhibitor, as a candidate compound for lupus treatment95,96. In our studies, DZ2002 showed significant therapeutic effects on lupus-prone NZB/W F1 mice via interference with the TLR-mediated APC response95. Similarly, Lawson's group demonstrated that DZ2002 could prevent lupus-like disease from developing in both BXSB and MRL/lpr mouse models by reducing the TLR-induced activation of immune cells96. However, the exact target protein of DZ2002 still requires further exploration.
Antibodies
Several antibodies have been designed to block ligands from binding to their respective TLRs. OPN-305, under phase II clinical research, is the first humanized IgG4 monoclonal antibody against TLR2 in development and is intended for the prevention of reperfusion injury following renal transplantation, among other indications97. Recent data suggest a potential role of OPN-305 for TLR2 in SLE98.
Similarly, TLR4 antibodies are being developed to block the excessive immune responses associated with autoimmune diseases, among which NI-0101 is the most advanced under phase I clinical research. NI-0101 is capable of binding an epitope on TLR4, thereby inhibiting TLR4 dimerization and reducing pro-inflammatory cytokine production99.
IFN-α signaling may be suppressed by various strategies: direct neutralization by an anti-IFN-α antibody or suppression of the IFN-α signature using an anti-IFN-α receptor antibody. Although a number of projects aiming to develop antibody agents targeting IFN-α have been launched, only a few of the projects are still under active development. With the discontinued development of MAb 9F3, 13H5, ACO-1 and Rontalizumab100, two anti-IFN-α monoclonal antibodies (mAbs), Sifalimumab (MEDI-545) and AGS-009, have achieved a safe and dose-dependent reduction of the IFN-α signature in their clinical trials for SLE treatment101,102,103. Sifalimumab is a fully human mAb in phase II clinical trials for the treatment of SLE. Preclinical data indicate that the levels of IFN-α are elevated in many patients with active SLE61. Sifalimumab binds to IFN-α and has been shown to neutralize its activity in previous studies104. AGS-009 is a humanized IgG4 mAb for the intravenous treatment of mild to moderate SLE, and it has recently completed a phase I clinical trial. The results announced at the 2012 EULAR Congress show that patients receiving the drug trend toward normal IFN-α signatures after only a single dose, whereas none of the patients receiving a placebo show a similar shift105.
Anifrolumab (MEDI-546), a fully human mAb against huIFNAR, is now in phase II/III clinical trials for the treatment of SLE. The safety, tolerability, pharmacokinetics, and pharmacodynamics of intravenously administered Anifrolumab in patients with active SLE were assessed in a phase II, open-label, dose-escalation study conducted on 17 Japanese patients. This study exhibited promising results, indicating that Anifrolumab causes increased and more sustained suppression of the IFN gene signature (IFNGS) target compared to Sifalimumab106,107.
MicroRNAs
MicroRNAs (miRNAs) are small, non-coding regulatory RNAs of 21–25 nucleotides in length. miRNAs work as post-transcriptional regulators by binding to a target sequence located within the 3′ UTR of the mRNA, promoting degradation of the mRNA or inhibiting translation of the mRNA. Accumulating studies provide evidence for the connection between the dysregulated miRNA network and excessively activated TLR signaling in SLE, indicating the potential of miRNA as a novel SLE therapeutic108,109,110. In this review, we briefly summarize pilot miRNAs involved in SLE therapy.
MiR-146a, targeting TRAF6 and IRAK1, is a negative regulator in autoimmunity111. Mice deficient in miR-146a manifest severe autoimmune phenotypes with elevated autoantibodies, splenomegaly and lymphadenopathy112. Restoring the loss of miR-146a using an MS2 VLP-based delivery system was effective in eliminating the production of autoantibodies and ameliorating SLE progression in lupus-prone mice113. miR-146a negatively regulates abnormal production of type I IFN in human lupus by targeting key proteins in the TLR signaling pathway111,114. miR-155 can be induced by a number of immune cell stimuli, including TLR ligands. Further studies found that several TLR ligands up-regulate miR-155 expression by either MyD88 or TRIF signaling, uncovering the therapeutic potential of miR-155 in SLE115,116.
Benefiting from miRNA's fine tuning of gene expression, the potential miRNA therapeutics should be more effective and safer than traditional approaches. Several companies are developing approaches to mimic or block the miRNA regulation of its targeting mRNA, resulting in the design of so-called agomirs or antagomirs. These approaches, if used to target TLR-relating miRNAs, are likely to be successful in the treatment of SLE.
Perspectives
SLE is a polygenic disease with a high degree of heterogeneity in clinical manifestations; therefore, drug development within this area has been challenging and has a high rate of attrition. Currently, the existing therapeutics revolve around non-steroidal anti-inflammatory drugs, antimalarials, steroids, immunosuppressives and biologics. However, several critical defects of those traditional drugs, such as modest therapeutic effects, severe side effects and extremely high costs, necessitate the development of novel therapeutic agents. Further exploration of the exact role of TLRs in SLE will aid in the development of improved therapeutics. We anticipate that new modulators targeting TLR signaling could alleviate disease severity with minimal side effects and lighten the financial burden of SLE patients. Although it is unlikely that all of the therapeutic agents currently under development will progress to approval for marketing, many show promise in clinical trials and will hopefully continue to progress along the approval process. The emergence of novel regulatory molecules, such as miRNAs, presents encouraging new opportunities for drug discovery and development in SLE treatment.
References
Kawai T, Akira S . The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11: 373–84.
Takeda K, Akira S . Toll-like receptors in innate immunity. Int Immunol 2005; 17: 1–14.
Zarember KA, Godowski PJ . Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 2002; 168: 554–61.
Barton G, Medzhitov R . Toll-like receptors and their ligands. Toll-like receptor family members and their ligands: Springer; 2002. p81–92.
Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates toll-like receptor 4. J Biol Chem 2001; 276: 10229–33.
Smiley ST, King JA, Hancock WW . Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 2001; 167: 2887–94.
Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 2002; 195: 99–111.
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H . HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J Biol Chem 2002; 277: 15107–12.
Park JS, Svetkauskaite D, He Q, Kim J-Y, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279: 7370–7.
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D . RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum 2005; 52: 2656–65.
Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med 2005; 202: 1575–85.
Yasuda K, Richez C, Uccellini MB, Richards RJ, Bonegio RG, Akira S, et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J Immunol 2009; 183: 3109–17.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25: 1271–7.
Muro Y . Antinuclear antibodies. Autoimmunity 2005; 38: 3–9.
Marshak-Rothstein A . Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 2006; 6: 823–35.
Kruse K, Janko C, Urbonaviciute V, Mierke CT, Winkler TH, Voll RE, et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis 2010; 15: 1098–113.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR . Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum 1998; 41: 1241–50.
Sanchez E, Orozco G, López-Nevot M, Jimenez-Alonso J, Martin J . Polymorphisms of toll-like receptor 2 and 4 genes in rheumatoid arthritis and systemic lupus erythematosus. Tissue Antigens 2004; 63: 54–7.
Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol 2008; 152: 482–7.
Lartigue A, Colliou N, Calbo S, François A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol 2009; 183: 6207–16.
Summers S, Hoi A, Steinmetz O, O'Sullivan K, Ooi J, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun 2010; 35: 291–8.
Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, et al. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol 2006; 177: 6880–8.
Tsao BP, Cantor RM, Kalunian KC, Chen CJ, Badsha H, Singh R, et al. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. J Clin Invest 1997; 99: 725–31.
Wakeland EK, Liu K, Graham RR, Behrens TW . Delineating the genetic basis of systemic lupus erythematosus. Immunity 2001; 15: 397–408.
Hawn TR, Wu H, Grossman JM, Hahn BH, Tsao BP, Aderem A . A stop codon polymorphism of Toll-like receptor 5 is associated with resistance to systemic lupus erythematosus. Proc Natl Acad Sci U S A 2005; 102: 10593–7.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003; 301: 640–3.
Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, et al. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant 2006; 21: 3062–73.
Patole PS, Gröne HJ, Segerer S, Ciubar R, Belemezova E, Henger A, et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol 2005; 16: 1326–38.
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ . Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med 2005; 202: 321–31.
Egner W . The use of laboratory tests in the diagnosis of SLE. J Clin Pathol 2000; 53: 424–32.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A . Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002; 416: 603–7.
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A . Activation of autoreactive B cells by CpG dsDNA. Immunity 2003; 19: 837–47.
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 2005; 202: 1171–7.
Kim W-U, Sreih A, Bucala R . Toll-like receptors in systemic lupus erythematosus; prospects for therapeutic intervention. Autoimmun Rev 2009; 8: 204–8.
Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, et al. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol 2014; 61: 38–43.
Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A 2006; 103: 9970–5.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S . Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006; 312: 1669–72.
Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A 2009; 106: 12061–6.
Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J Exp Med 2008; 205: 2995–3006.
Savarese E, Chae OW, Trowitzsch S, Weber G, Kastner B, Akira S, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood 2006; 107: 3229–34.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell Richard A, Shlomchik MJ . Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006; 25: 417–28.
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7-and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010; 184: 1840–8.
Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, et al. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A 2014; 111: 1497–502.
Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: Implications for the induction and maintenance of the autoimmune process. Arthritis Rheum 2006; 54: 3601–11.
Migita K, Miyashita T, Maeda Y, Nakamura M, Yatsuhashi H, Kimura H, et al. Toll-like receptor expression in lupus peripheral blood mononuclear cells. J Rheumatol 2007; 34: 493–500.
Nakano S, Morimoto S, Suzuki J, Nozawa K, Amano H, Tokano Y, et al. Role of pathogenic auto-antibody production by Toll-like receptor 9 of B cells in active systemic lupus erythematosus. Rheumatology (Oxford) 2008; 47: 145–9.
Zorro S, Arias M, Riano F, Paris S, Ramirez L, Uribe O, et al. Response to ODN-CpG by B Cells from patients with systemic lupus erythematosus correlates with disease activity. LUPUS 2009; 18: 718–26.
Tran NL, Manzin-Lorenzi C, Santiago-Raber ML . TLR8 deletion accelerates autoimmunity in a mouse model of lupus through a TLR7-dependent mechanism. Immunology 2014; 145: 60–70.
Umiker BR, Andersson S, Fernandez L, Korgaokar P, Larbi A, Pilichowska M, et al. Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. Eur J Immunol 2014; 44: 1503–16.
Deguine J, Barton GM . MyD88: a central player in innate immune signaling. F1000Prime Rep 2014; 6: 97.
Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T, et al. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum 2007; 56: 1618–28.
Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ . Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity 2013; 38: 528–40.
Hua Z, Gross AJ, Lamagna C, Ramos-Hernández N, Scapini P, Ji M, et al. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 2014; 192: 875–85.
Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA . Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A 2013; 110: E3311–20.
Cohen P . The TLR and IL-1 signalling network at a glance. J Cell Sci 2014; 127: 2383–90.
Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, et al. IRAK-4-and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 2008; 29: 746–57.
Chiang EY, Yu X, Grogan JL . Immune complex-mediated cell activation from systemic lupus erythematosus and rheumatoid arthritis patients elaborate different requirements for IRAK1/4 kinase activity across human cell types. J Immunol 2011; 186: 1279–88.
Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR . The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol 2012; 24: 530–7.
Elkon KB, Stone VV . Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res 2011; 31: 803–12.
Crow MK, Kirou KA . Interferon-α in systemic lupus erythematosus. Curr Opin Rheumatol 2004; 16: 541–7.
Crow MK . Type I interferon in the pathogenesis of lupus. J Immunol 2014; 192: 5459–68.
Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 2003; 197: 777–88.
Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum 2007; 56: 3770–83.
Barrat FJ, Coffman RL . Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev 2008; 223: 271–83.
Kandimalla ER, Bhagat L, Wang D, Yu D, Sullivan T, La Monica N, et al. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res 2013; 41: 3947–61.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 2005; 202: 1131–9.
Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL . Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol 2007; 37: 3582–6.
Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010; 465: 937–41.
Dynavax regains full rights to investigational TLR 7/9 inhibitor DV1179 following expiration of collaboration with GSK [Nov 28, 2014]. Available from: http://investors.dynavax.com/releasedetail.cfm?releaseid=885172.
Zhu F, Yu D, Kandimalla E, La Monica N, Agrawal S . Treatment with IMO-3100, a novel TLR7 and TLR9 dual antagonist, inhibits disease development in lupus prone NZBW/F1 mice. Keystone Symposia: Dendritic Cells and the Initiation of Adaptive Immunity ed. 2011.
Jiang W, Bhagat L, Yu D, Kandimalla ER, Agrawal S . IMO-3100, an antagonist of Toll-like receptors 7 and 9, modulates gene expression and regulatory networks induced by ligands. J Immunol 2009; 182 (Meeting Abstracts 1): 48.25.
Zhu FG, Jiang W, Dong Y, Kandimalla E, La Monica N, Agrawal S . IMO-8400, a novel TLR7, TLR8 and TLR9 antagonist, inhibits disease development in lupus-prone NZBW/F1 mice. J Immunol 2012; 188 (Meeting Abstracts 1): 119.12.
Kandimalla ER, Wang D, Li Y, Yu D, Zhu F, Bhagat L, et al. Immune regulatory oligonucleotide (iro) compounds to modulate toll-like receptor based immune response. ed: Google Patents 2012.
Römmler F, Jurk M, Uhlmann E, Hammel M, Waldhuber A, Pfeiffer L, et al. Guanine modification of inhibitory oligonucleotides potentiates their suppressive function. J Immunol 2013; 191: 3240–53.
Römmler F, Hammel M, Waldhuber A, Müller T, Jurk M, Uhlmann E, et al. Guanine-modified Inhibitory oligonucleotides efficiently impair TLR7-and TLR9-mediated immune responses of human immune cells. PLoS One 2015; 10: e 0116703.
Häcker H, Mischak H, Miethke T, Liptay S, Schmid R, Sparwasser T, et al. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J 1998; 17: 6230–40.
Macfarlane DE, Manzel L . Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine, and structurally related compounds. J Immunol 1998; 160: 1122–31.
Yi AK, Tuetken R, Redford T, Waldschmidt M, Kirsch J, Krieg AM . CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J Immunol 1998; 160: 4755–61.
Kužnik A, Benčina M, Švajger U, Jeras M, Rozman B, Jerala R . Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186: 4794–804.
Venuturupalli S, Gudsoorkar V, Wallace D . Reconsidering antimalarials in systemic lupus erythematosus: developments of translational clinical interest. J Rheumatol 2012; 39: 1769–71.
Wallace DJ, Gudsoorkar VS, Weisman MH, Venuturupalli SR . New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat Rev Rheumatol 2012; 8: 522–33.
Lipford G, Forsbach A, Zepp C, Nguyen T, Weeratna R, McCluskie M, et al. Selective toll-like receptor 7/8/9 antagonists for the oral treatment of autoimmune diseases. American College of Rheumatology 2007 Annual Scientific Meeting.
Parkinson T . The future of toll-like receptor therapeutics. Curr Opin Mol Ther 2008; 10: 21–31.
Hou LF, He SJ, Wang JX, Yang Y, Zhu FH, Zhou Y, et al. SM934, a water-soluble derivative of arteminisin, exerts immunosuppressive functions in vitro and in vivo. Int Immunopharmacol 2009; 9: 1509–17.
Hou LF, He SJ, Li X, Yang Y, He PL, Zhou Y, et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum 2011; 63: 2445–55.
Hou LF, He SJ, Li X, Wan CP, Yang Y, Zhang XH, et al. SM934 treated lupus-prone NZB x NZW F1 mice by enhancing macrophage interleukin-10 production and suppressing pathogenic T cell development. PLoS One 2012; 7: e 32424.
Wu Y, He S, Bai B, Zhang L, Xue L, Lin Z, et al. Therapeutic effects of the artemisinin analog SM934 on lupus-prone MRL/lpr mice via inhibition of TLR-triggered B-cell activation and plasma cell formation. Cell Mol Immunol 2015 Mar 16. doi: 10.1038/cmi.2015.13. [Epub ahead of print].
Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, et al. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol 2014; 85: 429–40.
Mullen LM, Chamberlain G, Sacre S . Pattern recognition receptors as potential therapeutic targets in inflammatory rheumatic disease. Arthritis Res Ther 2015; 17: 122.
Thwaites R, Chamberlain G, Sacre S . Emerging role of endosomal toll-like receptors in rheumatoid arthritis. Front Immunol 2014; 5: 1.
Loiarro M, Capolunghi F, Fantò N, Gallo G, Campo S, Arseni B, et al. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc Biol 2007; 82: 801–10.
Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (Oxford) 2010: keq226.
Kondo M, Tahara A, Hayashi K, Abe M, Inami H, Ishikawa T, et al. Renoprotective effects of novel interleukin-1 receptor-associated kinase 4 inhibitor AS2444697 through anti-inflammatory action in 5/6 nephrectomized rats. Naunyn Schmiedebergs Arch Pharmacol 2014; 387: 909–19.
Pfizer. study to evaluate safety and tolerability of single ascending doses of multiple formulations of PF-06650833 in healthy subjects under fasted and fed conditions. ed. August 21, 2014.
He SJ, Lin ZM, Wu YW, Bai BX, Yang XQ, He Pl, et al. Therapeutic effects of DZ2002, a reversible SAHH inhibitor, on lupus-prone NZB× NZW F1 mice via interference with TLR-mediated APC response. Acta Pharmacol Sin 2014; 35: 219–29.
Tardif V, Manenkova Y, Berger M, Hoebe K, Zuo JP, Yuan C, et al. Critical role of transmethylation in TLR signaling and systemic lupus erythematosus. Clin Immunol 2013; 147: 133–43.
Reilly M, Miller R, Thomson M, Patris V, Ryle P, McLoughlin L, et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther 2013; 94: 593–600.
Hennessy EJ, Parker AE, O'Neill LA . Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 2010; 9: 293–307.
Monnet E, Shang L, Lapeyre G, Hatterer E, Buatois V, Elson G, et al. AB0451 NI-0101, a monoclonal antibody targeting toll like receptor 4 (TLR4) being developed for rheumatoid arthritis (RA) treatment with a potential for personalized medicine. Ann Rheum Dis 2015; 74: 1046.
Kalunian K, Merrill JT, Maciuca R, Ouyang W, McBride J, Townsend MJ, et al. Efficacy and safety of rontalizumab (anti-interferon alpha) in SLE subjects with restricted immunosuppressant use: results of a randomized, double-blind, placebo-controlled phase 2 study. [abstract]. Arthritis Rheum 2012; 64: 2622. Doi: 10.1002/art.40353.
Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis 2011; 70: 1905–13.
Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a human anti-Interferon-α monoclonal antibody, in systemic lLupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum 2013; 65: 1011–21.
Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, et al. Neutralization of interferon-α/β-inducible genes and downstream effect in a phase I trial of an anti-interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 2009; 60: 1785–96.
Wallace D, Petri M, Olsen N, Kirou K, Dennis G, Yao Y . MEDI-545, an anti-interferon alpha monoclonal antibody, shows evidence of clinical activity in systemic lupus erythematosus. Arthritis Rheum 2007; 56: S526.
Tcherepanova I, Curtis M, Sale M, Miesowicz F, Nicolette C . Results of a randomized placebo controlled phase I a study of ags-009, a humanized anti-interferon-α monoclonal antibody in subjects with systemic lupus erythematosus. Abstract SAT0193. Ann Rheum Dis 2012; 71: 536.
Mathian A, Hie M, Cohen-Aubart F, Amoura Z . Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs 2015: 1–12.
Morehouse C, Chang L, Wang L, Brohawn P, Ueda S, Illei G, et al. Target modulation of a type I interferon (IFN) gene signature with sifalimumab or anifrolumab in systemic lupus erythematosus (SLE) patients in two open label phase 2 Japanese trials. 2014 ACR/ARHP Annual Meeting. Abstract number: 719.
Yan S, Yim LY, Lu L, Lau CS, Chan VS-F . MicroRNA regulation in systemic lupus erythematosus pathogenesis. Immune Netw 2014; 14: 138–48.
Shen N, Liang D, Tang Y, de Vries N, Tak P-P . MicroRNAs-novel regulators of systemic lupus erythematosus pathogenesis. Nat Rev Rheumatol 2012; 8: 701–9.
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, et al. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 2007; 16: 939–46.
Taganov KD, Boldin MP, Chang KJ, Baltimore D . NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006; 103: 12481–6.
Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med 2011; 208: 1189–201.
Pan Y, Jia T, Zhang Y, Zhang K, Zhang R, Li J, et al. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int J Nanomedicine 2012; 7: 5957–67.
Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum 2009; 60: 1065–75.
Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 2007; 179: 5082–9.
O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D . MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 2007; 104: 1604–9.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81273524, 81273525, 81322049) and the National Key Basic Research Program (973 Program, 2014CB541906).
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
About this article

Cite this article
Wu, Yw., Tang, W. & Zuo, Jp. Toll-like receptors: potential targets for lupus treatment. Acta Pharmacol Sin 36, 1395–1407 (2015). https://doi.org/10.1038/aps.2015.91
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.91
Keywords
This article is cited by
-
The role of toll-like receptors (TLRs) and their therapeutic applications in glomerulonephritis
International Urology and Nephrology (2023)
-
Hydroxychloroquine in nephrology: current status and future directions
Journal of Nephrology (2023)
-
Toll-like receptor 5 and Toll-like receptor 9 single nucleotide polymorphisms and risk of systemic lupus erythematosus and nephritis in Egyptian patients
Egyptian Rheumatology and Rehabilitation (2021)
-
Interferon-directed therapies for the treatment of systemic lupus erythematosus: a critical update
Clinical Rheumatology (2021)
-
Mesangial Cells in Lupus Nephritis
Current Rheumatology Reports (2021)
