
Abstract
Aim:
To investigate the effects Astragalus polysaccharides (APS) on tumor necrosis factor (TNF)-α-induced inflammatory reactions in human umbilical vein endothelial cells (HUVECs) and to elucidate the underlying mechanisms.
Methods:
HUVECs were treated with TNF-α for 24 h. The amounts of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) were determined with Western blotting. HUVEC viability and apoptosis were detected using cell viability assay and Hoechst staining, respectively. Reactive oxygen species (ROS) production was measured by DHE staining. Monocyte and HUVEC adhesion assay was used to detect endothelial cell adhesive function. NF-κB activation was detected with immunofluorescence.
Results:
TNF-α (1-80 ng/mL) caused dose- and time-dependent increases of ICAM-1 and VCAM-1 expression in HUVECs, accompanied by significant augmentation of IκB phosphorylation and NF-κB translocation into the nuclei. Pretreatment with APS (10 and 50 μg/mL) significantly attenuated TNFα-induced upregulation of ICAM-1 VCAM-1 and NF-κB translocation. Moreover, APS significantly reduced apoptosis, ROS generation and adhesion function damage in TNF-α-treated HUVECs.
Conclusion:
APS suppresses TNFα-induced adhesion molecule expression by blocking NF-κB signaling and inhibiting ROS generation in HUVECs. The results suggest that APS may be used to treat and prevent endothelial cell injury-related diseases.
Similar content being viewed by others


Introduction
Endothelial cell injury is an important cause of serious cardiovascular diseases, such as atherosclerosis, which can cause serious clinical consequences such as myocardial infarction, heart failure and stroke. Accumulated evidence demonstrates that atherosclerosis is closely related to the inflammatory and proliferative responses of the endothelium after injury1. During the early stages of atherosclerosis, adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), are secreted by activated endothelial cells in atherosclerotic lesions, stimulating immune cell and monocyte recruitment and migration into the intimal area of the vascular wall2. Tumor necrosis factor (TNF)-α, one of the most potent pro-inflammatory cytokines3, is critically implicated not only in the induction of endothelial apoptosis4 but also in the development and progression of atherosclerotic lesions in humans5.
Nuclear factor-κB (NF-κB) plays an important role in the transcriptional regulation of inflammatory proteins such as cyclooxygenase-2 (COX-2), ICAM-1, VCAM-1, and E-selectin6. NF-κB exists in the cytoplasm of unstimulated cells and is bound to its inhibitory protein, IκBα. IκBα phosphorylation leads to its degradation and the subsequent translocation of NF-κB to the nucleus where it activates target gene transcription. During atherosclerosis, NF-κB functions as a regulator of pro-inflammatory and anti-inflammatory gene transcription and as a regulator of cell survival and proliferation.
Many herbs are used in traditional Chinese medicine. The roots of Astragalus membranaceus (Huangqi) belong to the Fabaceae family, which contains some of the most popular health-promoting herbs in China7,8. Astragalus is a crude drug that has been widely used in traditional Chinese medicine for thousands of years to treat various renal diseases9. Polysaccharides from Radix Astragali are a class of macromolecules that have shown strong anti-tumor and anti-glomerulonephritis activities10,11. Studies have also shown that Astragalus polysaccharides (APS) have a beneficial effect on nephrotic syndrome, possess immunopotentiative functions and improve early diabetic nephropathy8,12. However, the effects and molecular mechanisms of APS on endothelial cell protection are far from clear. To address this issue, this study was designed to investigate whether APS can inhibit TNF-α-induced inflammatory reactions in human umbilical vein endothelial cells (HUVECs) and illustrate the intercellular signaling mechanisms.
Materials and methods
Preparation of APS
APS were purchased from the Gracia Chemical Technology Co, Chengdu, China. APS were diluted to 10 mg/mL in M199 culture medium containing 10% FBS.
Reagents
Anti-ICAM-1, VCAM-1, NF-κB, phosphorylated-NF-κB, IκBα, phosphorylated-IκBα, Bax, Bcl-2, and MCP-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Recombinant human TNF-α was obtained from Peprotech (Rocky Hill, NJ, USA). Secondary antibody against rabbit or goat was purchased from Cell Signaling Technology (Danvers, MA, USA). Unless otherwise indicated, all chemicals were purchased from Sigma (St Louis, CA, USA) or Amresco (Solon, OH, USA).
Cell culture
Human umbilical cords were obtained from healthy donors from whom we received informed consent. HUVECs were isolated from fresh umbilical veins using the procedure described by Jaffe et al13 and cultured in M199 containing 20% fetal bovine serum, 2 mmol/L glutamine, antibiomycins (10 μmol/L penicillin G and 10 μmol/L streptomycin) at 37 °C in a humidified 5%-CO2 atmosphere. HUVECs at passages 3–5 were used in the current study.
TNF-α treatment
HUVECs were pretreated with APS for 4 h and then treated with the indicated concentrations of TNF-α for 24 h in 2% FBS medium. The cells were lysed in lysis buffer (Santa Cruz Biotechnology, CA, US), and the samples were then sonicated and centrifuged at 15 000×g for 20 min. The lysates were subjected to Western blotting with specific antibodies.
Western blotting analysis
Cell lysates were analyzed with SDS-PAGE and electrotransfered to PVDF membranes. Membranes were then blocked with 1% bovine serum albumin for 1 h and incubated with specific antibodies for 2 h. After five washes in TBST (containing 0.1% Tween 20 in TBS), the membranes were incubated with horseradish peroxidase–conjugated secondary antibodies in TBST for 1 h. The bands were detected by chemiluminescence detection agents. Blot densitometry was performed, and the bands were analyzed with ImageJ software.
Cell viability assay
Cell viability was examined by the MTT assay according to the instructions of the manufacturer. HUVECs (5000 cells/well) were plated onto 96-well plates. HUVECs were pretreated with APS for 4 h and then treated with the indicated concentrations of TNF-α for 24 h. All assays were performed in triplicate. The cells were incubated with 0.5 mg/mL 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenylterazolium bromide for 4 h, and the absorbance at 490 nm was measured, as described previously14. The MTT kit was purchased from Roche Applied Science (Indianapolis, Indiana).
Hoechst 33342 staining
Nuclear fragmentation was detected by incubating fixed cells (70% alcohol and 30% acetone) in 10 mmol/L Hoechst 33342, as previously described15. A total of 500–700 cells in 10 randomly chosen fields from each dish were counted to determine the percentage of apoptotic nuclei. Each data point is the result of 5000–7000 cells of 4-8 independent experiments.
In situ detection of reactive oxygen species (ROS) production
To evaluate cell ROS production in situ, living HUVECs were stained with 10 μmol/L DHE (Sigma) for 30 min in a dark humidified chamber at 37 °C. ROS generation was labeled with red fluorescence and visualized by fluorescence microscopy, as previously described16.
Adhesion assay for monocytes and HUVECs
HUVECs were cultured in 6-well plates in M199 with 20% FBS for 48 h to obtain 90%–95% confluence and were then stimulated with 40 ng/mL TNF-α for 12 h. The human monocyte cell line U937 was incubated in the RPMI-1640 medium with 10% FBS and 50 μg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide at 37 °C for 2 h. The treated HUVECs were washed and co-cultured with a suspension of prepared U937 cells (1×106 cells/mL) for 30 min and then washed 3 times with fresh medium 1640. Non-adherent U937 cells were removed by washing. Adherent cells were incubated in 400 μL DMSO at 37 °C for 10 min, and the absorbance at 490 nm was measured.
Immunofluorescence of NF-κB
NF-κB expression in HUVECs was detected by immunofluorescence as described previously17. Cells were seeded onto sterilized cover slips placed in a 6-well tissue culture plate. After being treated with TNF-α for 8 h, the cells were fixed for 15 min in 4% (w/v) paraformaldehyde/PBS and made permeable by the addition of 0.2% Triton X-100/PBS for 15 min. Blocking solution was added and incubated overnight at 4 °C, and anti-NF-κB p65 antibody was then added to each well for 2 h at 37 °C. After washing, Alexa 488-conjugated anti-rabbit IgG antibody was added and incubated for 0.5 h at 37°C. Cells were then incubated with Hoechst 33342 for 20 min to stain the nuclei. NF-κB p65 was imaged by a fluorescence microscope (BX60; Olympus, Ina, Japan). NF-κB p65 was observed as green fluorescence and the nucleus as blue fluorescence.
Statistical analyses
Data were expressed as the mean±SEM. A one-way ANOVA followed by the Bonferroni procedure was used for multiple-group statistical comparisons. P<0.05 was considered statistically significant.
Results
APS inhibit TNF-α-induced ICAM-1 and VCAM-1 expression
Activation of endothelial cells by TNF-α has been known to upregulate the expression of adhesion molecules, such as ICAM-1 and VCAM-1. We first determined the concentration of TNF-α that induced HUVEC injury and adhesive molecule upregulation. As shown in Figure 1A, 40 ng/mL TNF-α enhanced the expression of several adhesion molecules, including ICAM-1 and VCAM-1. Next, we evaluated adhesion molecule expression at multiple time points (Figure 1B). To be consistent with our previous finding, 40 ng/mL TNF-α was used in this experiment. Thus, we examined whether APS could suppress the upregulation of ICAM-1 and VCAM-1 induced by TNF-α in HUVECs. As shown in Figure 1C, 40 ng/mL TNF-α increased ICAM-1 and VCAM-1 expression at 12 h, while pretreatment with APS abrogated TNF-α-induced ICAM-1 and VCAM-1 expression in a concentration-dependent manner. In particular, HUVEC pretreatment with 10 μg/mL APS significantly decreased ICAM-1 and VCAM-1 expression by 58.37% and 43.54%, respectively, relative to the control (P<0.01). The results indicate that APS suppress the expression of adhesion molecules induced by TNF-α.

APS 0.1-50 μg/mL inhibit TNF-α-induced ICAM-1 and VCAM-1 expression. (A) TNF-α 40 ng/mL up-regulated ICAM-1 and VCAM-1 in HUVECs, as assayed by Western blotting. (B) ICAM-1 and VCAM-1 expression in HUVECs treated by 40 ng/mL of TNF-α for different time, as assayed by Western blotting. (C) The average data of ICAM-1 and VCAM-1 expression in HUVECs treated by different concentrations of APS and 40 μg/mL TNF-α. n=3. Mean±SD. bP<0.05, cP<0.01 vs control.
APS attenuate the reduced HUVEC viability and increased apoptosis induced by TNF-α
Mounting evidence has demonstrated that TNF-α can reduce cell viability and induce apoptosis, which contribute to the development of atherosclerosis. Cultured HUVECs treated with TNF-α showed a significant reduction in cell viability compared to the control (Figure 2A). HUVEC pretreatment with 10 μg/mL APS strongly enhanced the cell viability compared to treatment with TNF-α alone (P<0.05). Moreover, the apoptotic effect of TNF-α was further manifested by TNF-α-induced increases in chromatin condensation and fragmentation, as revealed in Hoechst 33342 nuclear staining (Figure 2B). Accordingly, chromatin condensation and fragmentation in HUVECs pre-treated with APS drastically decreased with increased cell density. The results suggest that pre-treatment with APS can relieve the reduced HUVEC viability and increased apoptosis induced by TNF-α.

APS attenuate TNFα-induced HUVEC viability injury and apoptosis. (A) The HUVEC viability as shown by MTT assay. (B) The apoptosis in HUVECs, as analyzed by Hoechst 33342 staining. n=3. Mean±SD. bP<0.05, cP<0.01 vs control.
APS block TNF-α-induced oxidative stress and adhesion function damage in HUVECs
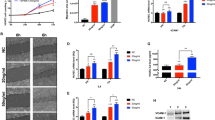
Atherosclerosis is associated with increased intracellular oxidative stress18. Oxidative stress can lead to adhesion function damage of HUVECs. TNF-α is believed to play a role in ROS production in injured endothelium. Therefore, we examined the inhibitory effects of APS on ROS production. As shown in Figure 3A, treatment of HUVECs with 40 ng/mL TNF-α for 12 h significantly increased intracellular ROS production as measured by dihydroethidium. Remarkably, TNF-α–induced ROS generation was significantly inhibited when HUVECs were pretreated with APS at concentrations of 10 μg/mL for 4 h. As shown in Figure 3B, the adhesion function of HUVECs treated by TNF-α for 12 h was significantly damaged; however, the effect was inhibited by pretreatment with APS at concentrations of 10 μg/mL for 4 h. These results indicate that APS can markedly inhibit the oxidative stress and adhesion function damage induced by TNF-α in HUVECs.

APS block TNF-α-induced oxidative stress and adhesion function damage in HUVECs. (A) DHE staining in control (Con), TNF-α-treated HUVECs (TNF-α), and APS-pretreated and TNF-α-treated HUVECs (APS+TNF-α). (B) Adhesion assay for monocytes (human U937 cell) and HUVECs. n=3. Mean±SD. bP<0.05, cP<0.01.
APS downregulate the expression of adhesion molecules by inhibiting NF-κB activation
NF-κB is known to regulate the expression of inflammatory proteins such as ICAM-1 and VCAM-1 in many types of cells. IκBα is the main regulator of NF-κB activation. Inactive NF-κB, bound to its inhibitor IκBα as a complex, is restricted to the cytoplasm. IκBα phosphorylation results in its ubiquitination and degradation; NF-κB is then released from the NF-κB-IκBα complex and translocated to the nucleus. Because we found that APS inhibited the ICAM-1 and VCAM-1 expression induced by TNF-α, we speculated that APS might affect NF-κB activation. Therefore, intracellular activation of NF-κB and IκBα was examined by Western blotting. As shown in Figure 4, ICAM-1 and VCAM-1 were upregulated after stimulation by 40 ng/mL TNF-α, which was accompanied by NF-κB and IκBα activation. Pretreatment with 10 μg/mL APS prevented the upregulation of ICAM-1 and VCAM-1 and decreased the levels of phosphorylated NF-κB and IκBα in TNF-α-treated HUVECs. In addition, APS attenuated TNF-α-induced Bax expression in HUVECs. Immunofluorescence was used to analyze NF-κB translocation. As shown in Figure 5, in unstimulated cells, NF-κB was predominantly localized in the cytoplasm, whereas in cells stimulated with TNF-α, NF-κB was almost completely translocated to the nucleus. However, in TNF-α-stimulated cells, pretreatment with 10 μg/mL APS led to the localization of NF-κB in the cytoplasm. The results reveal that APS suppress the activation of NF-κB and IκBα, leading to reduced adhesion molecule expression.

APS down-regulate the expression of adhesion molecules by inhibition of NF-κB activation. (A) Western blotting analysis of ICAM-1, VCAM-1, pIκBα, IκBα, pNF-κB, NF-κB, Bax, and Bcl-2 abundance in HUVECs. (B) The average data of ICAM-1, VCAM-1, IκBα phosphorylation, and NF-κB phosphorylation in HUVECs. n=3. Mean±SD. bP<0.05, cP<0.01.

APS attenuate the nuclear translocation of NF-κB. The NF-κB translocation in control, TNFα-treated and APS+TNFα-treated HUVECs was analyzed by immunofluorescence using NF-κB p65 subunit antibody. Images are representatives of three independent experiments.
Discussion
Although the beneficial effects of APS have been known for years, this is the first study to demonstrate the protective effects of APS on TNF-α-impaired endothelial adhesion functions and to evaluate the associated mechanism. The major finding of the present study was that pretreatment with APS significantly suppressed ICAM-1 and VCAM-I expression in HUVECs stimulated by TNF-α. The activation of NF-κB and IκBα was decreased and oxidative stress and apoptosis in HUVECs were inhibited by the administration of APS.
Inflammation is involved in the initiation, rupture, and thrombosis of atherosclerotic plaques. TNF-α is involved in nearly every step of inflammation19. During the early stages of atherosclerosis, inflammatory cell recruitment plays a central role20. TNF-α is a key cytokine in the recruitment and activation of inflammatory cells. ICAM-1 and VCAM-1 play a major role in the initiation of early atherosclerosis21, preferentially contributing to monocyte adhesion22. Inhibition of the inflammatory response is widely known to be beneficial in the early stages of atherosclerosis23,24. Our results showed ICAM-1 and VCAM-1 expression increased in TNF-α-treated HUVECs, and this upregulation of ICAM-1 and VCAM-1 was significantly suppressed by pretreatment with APS in a concentration-dependent manner.
The literature demonstrates that the intracellular signaling initiated by TNF-α is mediated through reactive oxygen intermediates25. Furthermore, oxidative stress can result in adhesion function damage, cell survival and apoptosis, which is mediated through apoptosis-associated proteins such as Bax and Bcl-2. In the present study, we found that TNF-α could increase intracellular ROS production, Bax and cell adhesion molecule expression and decrease Bcl-2 expression in HUVECs. Normal cell adhesion function and cell viability were drastically impaired. However, APS significantly attenuated TNF-α-induced oxidative stress and adhesion function damage in HUVECs by inhibiting NF-κB activation and regulating Bax and Bcl-2 expression. Although the signal transduction mechanism was not completely investigated in the present study, a significant decrease in ROS production and a significant protective effect on adhesion function and cell viability in HUVECs pretreated with APS were observed.
There are ample data suggesting that TNF-α increases the binding of NF-κB to its recognition site in the VCAM-1 promoter, promoting its expression and, subsequently, monocyte adhesion to vascular endothelial cells. Recently, reports have shown that both VCAM-1 and ICAM-1 expression are regulated by the TNF-α-mediated NF-κB signaling pathway26. In the present study, we observed the degradation of IκBα in the cytoplasm and activation of NF-κB in HUVECs by Western blotting. The TNF-α-induced increase in IκBα degradation in concert with NF-κB p65 activation was inhibited by pretreatment with APS. These findings indicate that APS strongly inhibits NF-κB activation and p65 translocation into the nucleus. In conclusion, we demonstrated that APS suppressed TNF-α-induced phosphorylation of IκBα and NF-κB activation, leading to the downregulation of adhesion molecules and a protective effect on the adhesion function of HUVECs.
In summary, the results of this study demonstrate that the endothelial dysfunction induced by TNF-α, which is associated with the upregulation of adhesion molecules, reduced viability and apoptosis, and elevated ROS generation and adhesion function damage in HUVECs, can be reversed by pretreatment with APS. APS have a preventive effect against endothelial dysfunction by inhibiting oxidative stress, improving endothelial survival and preventing adhesion function damage. Moreover, APS also regulated TNF-α production in endothelial cells (see the Supplementary Data). Therefore, our present findings indicate some novel pharmacological activity of APS, which might be used as a target in the prevention and treatment of endothelial cell injury-related diseases.
Author contribution
Jian LI, Tao SHEN, and Yu-ping ZHU designed the study; Yu-ping ZHU, Tao SHEN, Ya-jun LIN, Bei-dong CHEN, Yang RUAN, Yuan CAO, Yue QIAO, Yong MAN, and Shu WANG performed the research; Yu-ping ZHU and Tao SHEN analyzed the data; and Jian LI, Tao SHEN, and Yu-ping ZHU wrote the paper.
References
Ross R . Atherosclerosis: current understanding of mechanisms and future strategies in therapy. Transplant Proc 1993; 25: 2041–3.
Reape TJ, Groot PH . Chemokines and atherosclerosis. Atherosclerosis 1999; 147: 213–25.
Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, et al. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 2009; 35: 341–6.
Dimmeler S, Hermann C, Zeiher AM . Apoptosis of endothelial cells. Contribution to the pathophysiology of atherosclerosis? Eur Cytokine Netw 1998; 9: 697–8.
Meyer JW, Holland JA, Ziegler LM, Chang MM, Beebe G, Schmitt ME . Identification of a functional leukocyte-type NADPH oxidase in human endothelial cells: a potential atherogenic source of reactive oxygen species 2. Endothelium 1999; 7: 11–22.
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta 2008; 1783: 886–95.
Zhang Y, Shi P, Yao H, Shao Q, Fan X . Metabolite profiling and pharmacokinetics of herbal compounds following oral administration of a cardiovascular multi-herb medicine (Qishen yiqi pills) in rats. Curr Drug Metab 2012; 13: 510–23.
Liu J, Hu X, Yang Q, Yu Z, Zhao Z, Yi T, et al. Comparison of the immunoregulatory function of different constituents in radix astragali and radix hedysari 2. J Biomed Biotechnol 2010; 2010: 479426.
Yesilada E, Bedir E, Calis I, Takaishi Y, Ohmoto Y . Effects of triterpene saponins from Astragalus species on in vitro cytokine release. J Ethnopharmacol 2005; 96: 71–7.
Liu QY, Yao YM, Zhang SW, Sheng ZY . Astragalus polysaccharides regulate T cell-mediated immunity via CD11c(high)CD45RB(low) DCs in vitro. J Ethnopharmacol 2011; 136: 457–64.
Yin X, Chen L, Liu Y, Yang J, Ma C, Yao Z, et al. Enhancement of the innate immune response of bladder epithelial cells by Astragalus polysaccharides through upregulation of TLR4 expression. Biochem Biophys Res Commun 2010; 397: 232–8.
Chen W, Yu MH, Li YM, Chen WJ, Xia YP . Beneficial effects of astragalus polysaccharides treatment on cardiac chymase activities and cardiomyopathy in diabetic hamsters. Acta Diabetol 2010; 47: 35–46.
Jaffe EA, Nachman RL, Becker CG, Minick CR . Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest 1973; 52: 2745–56.
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis 1. J Biol Chem 2007; 282: 23354–61.
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis 1. J Biol Chem 2007; 282: 23354–61.
Lu Y, Qin W, Shen T, Dou L, Man Y, Wang S, et al. The antioxidant N-acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-kappaB in ApoE-deficient mice. J Atheroscler Thromb 2011; 18: 998–1008.
Shen T, Aneas I, Sakabe N, Dirschinger RJ, Wang G, Smemo S, et al. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J Clin Invest 2011; 121: 4640–54.
Lee EN, Choi YW, Kim HK, Park JK, Kim HJ, Kim MJ, et al. Chloroform extract of aged black garlic attenuates TNF-alpha-induced ROS generation, VCAM-1 expression, NF-kappaB activation and adhesiveness for monocytes in human umbilical vein endothelial cells. Phytother Res 2011; 25: 92–100.
Landsberger M, Wolff B, Jantzen F, Rosenstengel C, Vogelgesang D, Staudt A, et al. Cerivastatin reduces cytokine-induced surface expression of ICAM-1 via increased shedding in human endothelial cells. Atherosclerosis 2007; 190: 43–52.
Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG . An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med 2006; 203: 2763–77.
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 2001; 107: 1255–62.
Zhang H, Issekutz AC . Down-modulation of monocyte transendothelial migration and endothelial adhesion molecule expression by fibroblast growth factor: reversal by the anti-angiogenic agent SU6668. Am J Pathol 2002; 160: 2219–30.
Ross R . Atherosclerosis: current understanding of mechanisms and future strategies in therapy. Transplant Proc 1993; 25: 2041–3.
Hernandez–Presa MA, Martin-Ventura JL, Ortego M, Gomez-Hernandez A, Tunon J, Hernandez-Vargas P, et al. Atorvastatin reduces the expression of cyclooxygenase-2 in a rabbit model of atherosclerosis and in cultured vascular smooth muscle cells. Atherosclerosis 2002; 160: 49–58.
Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res 1999; 85: 199–207.
Collins T, Cybulsky MI . NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 2001; 107: 255–64.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (No 2012CB517502), the National Natural Science Foundation of China (No 81200221, 81070634, and 30900627) and the Scientific Research Foundation for Returned Chinese Scholars of the Ministry of Human Resource and Social Security of China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhu, Yp., Shen, T., Lin, Yj. et al. Astragalus polysaccharides suppress ICAM-1 and VCAM-1 expression in TNF-α-treated human vascular endothelial cells by blocking NF-κB activation. Acta Pharmacol Sin 34, 1036–1042 (2013). https://doi.org/10.1038/aps.2013.46
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.46
Keywords
This article is cited by
-
Myricetin ameliorates ox-LDL-induced HUVECs apoptosis and inflammation via lncRNA GAS5 upregulating the expression of miR-29a-3p
Scientific Reports (2021)
-
The role of the adipocytokines vaspin and visfatin in vascular endothelial function and insulin resistance in obese children
BMC Endocrine Disorders (2019)
-
The influence of ICAM1 rs5498 on diabetes mellitus risk: evidence from a meta-analysis
Inflammation Research (2019)
-
Effects of fermented Sorghum bicolor L. Moench extract on inflammation and thickness in a vascular cell and atherosclerotic mice model
Journal of Natural Medicines (2019)
-
Upregulation of ICAM-1 in diabetic rats after transient forebrain ischemia and reperfusion injury
Journal of Inflammation (2014)
