
Abstract
Aim:
To investigate whether atorvastatin treatment could prevent Aβ1-42 oligomer (AβO)-induced synaptotoxicity and memory dysfunction in rats, and to elucidate the mechanisms involved in the neuroprotective actions of atorvastatin.
Methods:
SD rats were injected with AβOs (5 nmol, icv). The rats were administrated with atorvastatin (10 mg·kg−1·d−1, po) for 2 consecutive weeks (the first dose was given 5 d before AβOs injection). The memory impairments were evaluated with Morris water maze task. The expression of inflammatory cytokines in the hippocampus was determined using ELISA assays. The levels of PSD-95 and p38MAPK proteins in rat hippocampus were evaluated using Western blot analysis. For in vitro experiments, cultured rat hippocampal neurons were treated with AβOs (50 nmol/L) for 48 h. The expression of MAP-2 and synaptophysin in the neurons was detected with immunofluorescence.
Results:
The AβO-treated rats displayed severe memory impairments in Morris water maze tests, and markedly reduced levels of synaptic proteins synaptophysin and PSD-95, increased levels of inflammatory cytokines (IL-1β, IL-6 and TNF-α) and p38MAPK activation in the hippocampus. All these effects were prevented or substantially attenuated by atorvastatin administration. Pretreatment of cultured hippocampal neurons with atorvastatin (1 and 5 μmol/L) concentration-dependently attenuated the AβO-induced synaptotoxicity, including the loss of dendritic marker MAP-2, and synaptic proteins synaptophysin and PSD-95. Pretreatment of the cultured hippocampal neurons with the p38MAPK inhibitor SB203580 (5 μmol/L) blocked the AβO-induced loss of synaptophysin and PSD-95.
Conclusion:
Atorvastatin prevents AβO-induced synaptotoxicity and memory dysfunction through a p38MAPK-dependent pathway.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease and the most common cause of dementia among the elderly. The characteristic pathological hallmarks of AD include the presence of intracellular neurofibrillary tangles and the formation of senile plaques outside the neurons and in cerebral blood vessels. These senile plaques are amyloid-β peptide (Aβ) aggregates, which are deposited in brain areas involved in cognitive functions. It is assumed that they initiate a pathological cascade that results in synaptic dysfunction, synaptic loss, and neuronal death1,2. Aβ spontaneously self-aggregates into multiple, coexisting, physical forms. One form consists of oligomers (ranging from dimers to dodecamers), which coalesce into intermediate assemblies. Accumulating evidence suggests that soluble Aβ oligomers (AβOs) and intermediate amyloid are the most neurotoxic forms, and AβOs are elevated strikingly in AD brain tissue and transgenic mouse AD models3,4.
Importantly, recent studies in animals have established links between AβOs and cognitive impairment5. AβOs have been shown to inhibit long-term potentiation (LTP), a classic experimental paradigm for synaptic plasticity, and acutely disrupt cognitive function after being infused into the central nervous system (CNS)1,2,6. Synapse loss is the most robust correlate of AD-associated cognitive deficits. In both AD patients and animal models of this disease, the greastest synapse loss is near senile plaques, indicating a link between Aβ pathology and synaptotoxicity in vivo7,8. Furthermore, AβOs have been shown to downregulate the levels of two synaptic proteins, postsynaptic density-95 (PSD-95) and synaptophysin9. PSD-95 is an abundant postsynaptic scaffolding protein that plays a critical role in synapse maturation and synaptic plasticity10. AβOs bind to synaptic sites that are immunopositive for PSD-957. Clusters of PSD-95 have been previously established as definitive markers for postsynaptic terminals11.
A number of studies have shown that Aβ can affect the function of NMDA-type glutamate receptors (NMDARs) and abolish the induction of NMDAR-dependent LTP at the neuronal plasma membrane12,13. Aβ-mediated spine loss requires the activity of NMDARs. Aβ binds to NR1 and NR2B subunits of NMDARs on the hippocampal neuron7,14. Shankar et al demonstrated that AβOs induced a marked decrease in the density of dendritic spines and the number of electrophysiologically active synapses of pyramidal neurons13. Furthermore, the NR2B subunit of NMDARs plays a role in regulating the effects of AβOs by increasing intracellular calcium in dendritic spines13. Additionally, the stimulation of NR2B by AβOs triggers the activation of mitogen-activated protein kinase (MAPK) and the subsequent down-regulation of cyclic AMP-responsive element-binding protein15. Thus, early AβO-induced synaptotoxicity and the underlying mechanisms constitute major targets in the development of novel therapeutic strategies for AD.
To date, there is no satisfactory treatment available for AD. The development of novel pharmacological strategies for treatment is of critical importance. Statins are widely prescribed drugs for the treatment of hypercholesterolemia and act to reduce plasma cholesterol levels by inhibiting the rate-limiting enzyme in the cholesterol biosynthetic pathway, 3-hydroxy-3-methy-lglutaryl-CoA reductase. In addition to the cholesterol lowering effect, statins have many pleiotropic effects, such as reducing Aβ production, suppressing inflammatory responses, protecting neurons from excitotoxins, apoptosis, and oxidative stresses, and promoting synaptogenesis16,17,18,19. In particular, statins have been linked to the reduced prevalence of AD in statin-prescribed populations20,21, the improved cognition in normo-cholesterolemic patients22, and the slowed cognitive decline in mild-to-moderate AD patients23. It has been shown that simvastatin was effective in reversing learning and memory deficits in an aged AD mouse model24. Atorvastatin is a member of the statin family.The safety of high doses of atorvastatin has been demonstrated25. Clarke et al demonstrated that rats treated with atorvastatin for 3 weeks showed increased production of the anti-inflammatory cytokine interleukin (IL)-4 in the hippocampus and that the rats were protected against a deficiency in LTP caused by the acute injection of Aβ1–4226. Notably, memory impairment resulting from AβOs involves synaptotoxicity. This observation suggests that statins prevent memory impairment by selectively controlling synaptotoxicity, which would provide a molecular basis for the neuroprotective action of statins.
The present study tested the ability of atorvastatin to prevent AβO-induced synaptotoxicity and memory impairment and investigated the underlying mechanisms. The results show that atorvastatin prevents AβO-induced synaptotoxicity and subsequent memory dysfunction by a mechanism involving the control of the p38 MAPK pathway.
Materials and methods
Atorvastatin was obtained from LKT Laboratories (St Paul, MN, USA). SB203580 was obtained from Calbiochem (Darmstadt, Germany).
Preparation and characterization of AβOs
Rat Aβ1–42 (Product number, SCP0038) was purchased from Sigma (St Louis, MO, USA). AβOs were prepared according to a previously described method27. Aβ1-42 was dissolved in sterile water at a concentration of 2 mmol/L and incubated at 37 °C for 24 h. The preparation was centrifuged at 14 000×g for 10 min at 4 °C, and the supernatant containing soluble AβOs was transferred to clean tubes and stored at 4 °C. Oligomer solutions were used within 24 h after preparation. The qualitative analysis of the oligomerization status of the Aβ peptide solution was evaluated by Western blot analysis using a rabbit polyclonal anti-Aβ1–42 antibody (ab10148, Abcam Inc, Cambridge, MA, USA). Protein concentration was determined using the bicinchoninic acid (BCA) assay (Beyotime Institute of Biotechnology, Shanghai, China). This preparation of AβOs has been extensively characterized in our laboratory. To ensure the consistency of quality, we evaluated a random sample from each batch by Western blot analysis using the anti-Aβ1–42 antibody.
Aβ1-42 levels in the hippocampus were quantified using mouse ELISA kits (Invitrogen Corp, Camarillo, CA, USA) as previously described28. Briefly, the hippocampus was first homogenized in radioimmunoprecititation assay (RIPA) buffer (Beyotime Institute of Biotechnology), then the mixture was centrifuged at 27 000×g at 4 °C for 30 min, and the supernatant was collected and stored at -80 °C until use for ELISA quantifications. Aβ1–42 levels were normalized by tissue weight and/or protein amount and determined using an enhanced BCA protein assay kit (Beyotime Institute of Biotechnology).
Animals and drug treatment
Young, male Sprague-Dawley rats (220–280 g, Grade II, certificate No SCXK 2003-0007, Experimental Animal Center of Liaoning Medical University, Jinzhou, China) were used in this study. The rats were anesthetized by an intraperitoneal injection of chloral hydrate (300 mg/kg body weight) and placed in a stereotaxic apparatus. A small hole was drilled in the skull through which a guide cannula was then inserted (-0.7 mm from bregma, 1.7 mm lateral to midline, and 4.0 mm from dura) for the intracerebroventricular (icv) injection. At 24 h post-operation, the rats were icv injected with either AβOs (5 nmol in 5 μL) or vehicle (5 μL of sterile water) by means of a Hamilton microsyringe (Hamilton, GR, Switzerland). The injection lasted for 5 min, and the needle with the syringe was left in place for another 2 min to complete the drug infusion.
Atorvastatin was dissolved in sterile water containing 10% dimethyl sulfoxide (DMSO). To investigate the effects of long-term administration of atorvastatin on water maze learning deficits and synaptic impairments, we administrated the rats with 10 mg/kg atorvastatin by oral gavage once per day during 2 consecutive weeks (first administration occurred 5 d before the AβOs or vehicle injection). The dose of atorvastatin was similar to that used in Clarke's study examining the central effects of atorvastatin29. Groups treated with vehicle (sterile water with 10% DMSO) were used as the control. The behavioral experiment was performed 24 h post injection.
Morris water maze
Spatial learning and memory (acquisition and recall), which are tasks sensitive to hippocampal dysfunction, were examined using the Morris water maze task as previously described30. On the first day, the rats underwent a habituation swim for 10 s without the platform. Then, animals received a 3-d training session, during which they were required to swim to a visible platform in a room with visual wall cues. Next, the testing trial started in which the rats had to find the hidden platform using the visuospatial cues after the wall cues and platform location were switched on and the platform was submerged. This process lasted for 5 consecutive days . In each trial, rats were placed into tank at 1 of 4 designated departure points in a random order. If the rat failed to find the hidden platform within 120 s, they were guided to the platform and given a swim latency score as 120 s. The animals were allowed to stay on the platform for 20 s.
During the trials, swim latency (time to reach the platform) and the path taken by the animals to reach the platform were recorded by a video camera connected to an image analyzer. The probe trial (platform removed) was performed on d 9. All of the parameters were recorded and analyzed using a computer-operated vedio tracking software (Any-maze, Stoelting, NJ, USA). All of the experiments started at the same time every day. After behavioral testing, the animals (5 in each group) were euthanized by an intraperitoneal injection of chloral hydrate, the brains were removed, and both hippocampi of each brain were manually dissected and immediately placed in liquid nitrogen and kept frozen until processing. The rats (5 in each group) used for Nissl staining and immunohistochemical staining were anesthetized and perfused transcardially with 4% paraformaldehyde.
Cytokine protein quantification
The concentrations of IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) were determined in the hippocampus using commercially available rat ELISA assays following the manufacturer's instructions (R&D Systems; Minneapolis, MN, USA). Briefly, frozen hippocampal tissue (0.2 g) was homogenized with a glass homogenizer in 1 mL of PBS buffer (pH 7.2) containing 1 mmol/L phenylmethylsulfonyl fluoride, 1 mg/L pepstatin A, 1 mg/L aprotinin, and 1 mg/L leupeptin, and centrifuged at 12 000×g for 20 min at 4 °C. The supernatant was collected, and total protein was determined using a BCA protein assay reagent kit. Standards, controls, and samples (50 μL) were pipetted into a 96-well plate pre-coated with polyclonal antibodies specific for IL-1β, IL-6, or TNF-α, incubated at room temperature for 2 h on an orbital plate shaker, and then washed with PBS before the addition of the conjugate. After several washes, substrate solution (1:1 mixture of H2O2 and tetramethylbenzidine) was added, and the plates were incubated at room temperature in the dark for 1 h. The color reaction was stopped by an equal volume of stop solution. Absorbance was read at 450 nm. The absorbance values were corrected for protein and expressed as pg of IL-1β, IL-6, or TNF-α/mg protein.
Primary hippocampal neuron cultures
Primary cultures were obtained from the hippocampi of 0- to 24-h-old Sprague-Dawley rats as previously described31. The cultures were plated on poly-L-lysine-coated 16-mm-diameter coverslips (∼150 cells/mm2) for immunocytochemistry assays or 6-well culture plates (1×106) for Western blot analysis. Neurons were grown at 37 °C in a humidified atmosphere of 5% CO2/95% O2. Forty-eight hours after the plating, the media were removed and replaced with Dulbecco's modified eagle medium containing 3 mg/mL glutamine, 2% B-27 (Life Technologies, Gaithersburg, MD, USA), and 5 μmol/L cytosine arabinofuranoside (Sigma), which inhibit the proliferation of non-neuronal cells. One week later, the culture matured and formed functional synaptic connections. We did NeuN and glial fibrillary acidic protein (GFAP, a marker of astrocytes) immunostaining after the arabinofuranoside treatment for 3 d to confirm the neurons, and evaluated AβO-induced neuronal damage after culturing the neurons for 1 week. AβOs were directly added to the medium and the neurons were incubated for 48 h. To test the ability of atorvastatin to modify the effects of the AβOs, we added the drug 1 h before the addition of the AβOs.
Immunocytochemical evaluation of synaptotoxicity
After fixation with 4% paraformaldehyde for 30 min, neurons were permeabilized in PBS with 0.2% Triton X-100 for 5 min and incubated with 3% BSA in PBS for 30 min at room temperature for the immunocytochemical analysis of synaptophysin (a protein located in synaptic vesicles) and microtubule-associated protein-2 (MAP-2, a dendritic marker)32. The cells were incubated with a mouse monoclonal anti-MAP-2 antibody (1:500, Abcam Inc), mouse monoclonal anti-synaptophysin (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), or rabbit polyclonal anti-PSD-95 (1:200, Santa Cruz Biotechnology) overnight at 4 °C. After extensive washes with PBS, the cells were incubated with an anti-mouse or anti-rabbit secondary antibody conjugated with fluorescein (1:200, The Jackson Labs, West Grove, PA, USA). The cells were then visualized by confocal microscopy (Leica SP5, Leica Microsystems Ltd, Germany).
Western blot analysis
Western blot analysis were performed for the detection of synaptophysin, PSD-95, and p38MAPK, as previously described31. Fresh hippocampal tissue or cultured hippocampal neurons were lysed in RIPA buffer. After detergent-insoluble materials were removed by centrifugation at 12 000×g for 10 min, the protein concentration in the soluble fraction was measured using an enhanced BCA protein assay kit. Equal amounts of protein were then separated by SDS-PAGE, transferred onto nitrocellulose membranes, and probed with primary antibodies against the following proteins: mouse monoclonal anti-synaptophysin (1:500), rabbit polyclonal anti-PSD-95 (1:500), rabbit anti-phospho-p38MAPK (Thr180/tyr182, 1:1000, Cell Signaling Technology, Beverly, MA, USA), or rabbit anti-p38MAPK (1:1000, Cell Signaling Technology). After being washed with PBS, membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (1:2000, Cell Signaling Technology). Then, membranes were washed and revealed using ECL kit (Pierce, Rockford, IL, USA). The membranes were then reprobed for β-actin immunoreactivity using a mouse anti-β-actin antibody (1:2000, Cell Signaling Technology). To determine the phosphorylation ratio of p38MAPK, the membranes were reprobed with rabbit anti-p38MAPK total (1:1000, Cell SignalingTechnology). Staining intensity was quantified from 4 blots derived from four rats or 4 independent experimental trials. The density of each band was quantified using Image J software and normalized to total kinase or β-actin expression. The protein levels reported in the figures were expressed as a ratio of the band intensity for the protein of interest to that for total kinases or β-actin, which was used as loading controls.
Statistical analysis
The data are expressed as the mean±SEM and were analysed by one-way ANOVA followed by an LSD post hoc multiple-comparison test or by Student's t-test for two-group comparisons. P<0.05 was considered statistically significant.
Results
Identification of Aβ peptide solutions and the accumulation of Aβ in the hippocampus after icv administration
The Coomassie brilliant blue-stained SDS–PAGE gels and Western blot analysis of Aβ peptide solutions used in this study showed that the solutions primarily consisted of dimers (approximately 8 kDa) and monomers (Figure 1A). There was an accumulation of AβOs in the hippocampus 24 h after the icv administration of 5 nmol AβOs (104.9±16.53 pg/mg protein; n=5, P<0.01). The hippocampal AβO levels decreased (46.1±7.7 pg/mg protein; n=5) over the course of 9 d (Figure 1B).

Identification of Aβ peptide solutions and accumulation of Aβ in the hippocampus after icv administration. (A) Coomassie brilliant blue-stained SDS-PAGE gel image and anti-antibody-based Western blot analysis of Aβ1–42 showed that the solutions primarily consisted of dimers and monomers. The positions of Aβ dimmers (D) and monomers (M) are indicated by arrows on the right side of the blot. (B) Aβ1–42 accumulated in the hippocampus 24 h and 9 d after icv administration, as measured by ELISA. Data are expressed as the mean±SEM. n=5. cP<0.01 compared with the control group.
Atorvastatin mitigated AβO-induced cognitive decline in the water maze task
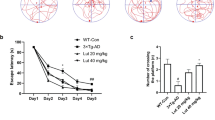
We found that rats treated with AβOs showed severe behavioral impairments in the Morris water maze task. Overall, the rats in the AβO-treated group had no difficulty learning to escape to the visible platform (d 1–3; Figure 2). Importantly, time latency to find the hidden platform was longer in the AβO-treated group compared with the vehicle-treated group, indicating an AβO-induced learning impairment (d 4–8; Figure 2). Similarly, in the spatial memory component of the test, the time spent in the target quadrant or the number of crossings over the platform location was significantly reduced in the AβO-treated group compared with the vehicle-treated group (probe trial, d 9; Figure 2), despite no alteration in swim speed (data not shown). Atorvastatin prevented AβO-induced learning and memory deficits; thus, the time latencies to find the hidden platform and the time spent in the target quadrant were similar between the AβO-treated group that received atorvastatin and the vehicle-treated group (Figure 2).

Atorvastatin prevented AβO-induced learning and memory deficits in rats. AβO-treated rats displayed longer latencies to reach the hidden platform (d 5–8) (A), as well as decreased time (B) and number of platform crossings (C) in the target quadrant during the probe trial compared with the control rats. Data are expressed as the mean±SEM of 12 rats. bP<0.05, cP<0.01 compared with the control group. eP<0.05, fP<0.01 compared with the AβO-treated group.
Atorvastatin prevented AβO-induced synaptic protein loss
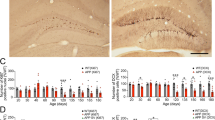
To determine whether atorvastatin can prevent AβO-induced synapse loss, we conducted an analysis of synaptic proteins. As presynaptic markers, the level of the synaptic vesicle protein synaptophysin was evaluated. As postsynaptic markers, the level of PSD-95 was evaluated. As shown in Figure 3, significant reductions in the levels of synaptophysin (Figure 3A) and PSD-95 (Figure 3B) were found in the AβO-treated rats 9 d after icv injection, suggesting a decrease in synaptic density. The oral administration of atorvastatin significantly prevented the AβO-induced decrease in the levels of synaptophysin (Figure 3A) and PSD-95 (Figure 3B) 9 d after AβO treatment; thus, the the levels of these 2 proteins were similar between the hippocampal tissues prepared from the control-treated rats and those from the AβO-treated rats that received atorvastatin, respectively. We next investigated whether atorvastatin inhibited AβO-induced dendritic and synaptic damage in cultured hippocampal neurons. As shown in Figure 4, the dendrites of AβO-treated neurons were thinner and shorter, with a frequently fragmented or “beaded” appearance. Treatment with AβOs also substantially reduced the number of synaptophysin-immunoreactive spots after the neurons exposure to AβOs (50 nmol/L) for 48 h. To quantify this AβO-induced synaptotoxicity, we used Western blot analysis, which showed a dose-dependent decrease in the density of synaptophysin and PSD-95 (Figure 5A). Similar to the in vivo hippocampal preparations, this AβO-induced decrease in the density of synaptophysin in neuronal cultures was partially prevented by atorvastatin treatment (Figure 5B). In parallel, atorvastatin was also able to partially prevent the AβO-induced decrease in the PSD-95 level in neuronal cultures (Figure 5B). These results indicate that atorvastatin is able to prevent the synaptic protein loss induced by AβOs.

Western blot analysis showing the effects of atorvastatin on the AβO-induced decrease in synaptophysin and PSD-95 protein expression in rat hippocampus. Rats were treated with AβOs (5 nmol, icv) or sterile water (control). Atorvastatin (10 mg/kg) was administered daily (starting from 5 d before AβO treatment). (A) Synaptophysin protein levels. (B) PSD-95 protein levels. The bar chart shows the semiquantitative analysis of synaptophysin and PSD-95. Data are expressed as the mean±SEM of 4 independent preparations. cP<0.01 compared with the control group. fP<0.01 compared with the AβO-treated group.

Representative images of cultured hippocampal neurons obtained via a phase-contrast microscope (scale bar, 25 μm) and immunolabeled against MAP-2 or synaptophysin (scale bar, 50 μm). Insets on the right side represent digital enlargements (scale bar, 200 μm) of the dendrite segments indicated by white boxes. Cultured hippocampal neurons at d 7 were pretreated with vehicle solution (control, 0.1% DMSO) or atorvastatin (5 μmol/L) for 1 h and then exposed to AβOs (50 nmol/L) for 48 h in the presence of vehicle or atorvastatin. Following the treatment period, phase-contrast digital images of the neurons were taken using a phase-contrast microscope. Hippocampal neurons were immunostained for MAP-2 or synaptophysin after 48 h incubation with AβOs (50 nmol/L) and analyzed by confocal microscopy.

Atorvastatin prevented the decrease in AβO-induced synaptophysin and PSD-95 in cultured hippocampul neurons. (A) Representative Western blot of synaptophysin and PSD-95. Group data showing the normalization of synaptophysin and PSD-95 proteins to β-actin protein was determined in each group from 4 experiments. (B) Representative Western blot showing that atorvastatin (1 and 5 μmol/L) prevented AβO-induced decreases in synaptophysin and PSD-95 in a concentration-dependent manner. Group data showing the normalization of synaptophysin and PSD-95 proteins to β-actin protein was determined in each group from 4 experiments. Data are expressed as the mean±SEM. bP<0.05, cP<0.01 compared with the control group. fP<0.01 compared with the AβO-treated group.
Signaling pathways involved in the neuroprotection afforded by atorvastatin against AβO-induced synaptotoxicity
Based on the fact that the activation of p38MAPK plays an important role in the intracellular mechanisms of neurodegeneration, in particular Aβ1–42-induced neurotoxicity33,34, we investigated whether p38MAPK is involved in the neuroprotective effects afforded by atorvastatin. The results showed that an icv injection of AβOs led to a significant increase in phospho-p38MAPK protein expression without a concurrent increase in the total level of this kinase. The AβO-induced increase in phospho-p38MAPK was prevented by atorvastatin treatment (Figure 6A). In addition, atorvastatin (10 mg/kg per day for 2 weeks) treatment alone did not result in a significant decrease in the basal expression of phospho-p38MAPK in rat hippocampus. To further test the involvement of p38MAPK in the atorvastatin-mediated protection against AβO-induced synaptotoxicity, we investigated the time course of AβO-induced activation of p38MAPK in cultured hippocampal neurons. The results showed that after the incubation of neurons with AβOs (50 nmol/L) for 12 h, there was a significant increase in phospho-p38MAPK protein expression. At this time point, atorvastatin treatment partially abolished the AβO-induced increase in phospho-p38MAPK protein expression in a concentration-dependent manner (Figure 6B). To test the key role of p38MAPK in the AβO-induced synaptotoxicity, we used the p38MAPK inhibitor SB203580 to block the actions of p38MAPK kinase. The results showed that SB203580 (5 μmol/L) completely prevented the AβO-induced decrease in synaptophysin and PSD-95 levels (Figure 6C). Together, these results suggest that the atorvastatin-mediated neuroprotection against AβO-induced synaptotoxicity may be through the p38MAPK pathway.

The neuroprotection afforded by atorvastatin against AβO-induced synaptotoxicity involved the p38MAPK signaling pathway. (A) Representative Western blot comparing phospho-p38MAPK in rat hippocampus. Group data showing the normalization of phospho-p38MAPK to total p38MAPK was determined in each group from 4 experiments. (B) Representative Western blot comparing phospho-p38MAPK in cultured hippocampal neurons. Cultured hippocampal neurons at d 7 were pre-incubated with vehicle solution (control, 0.1% DMSO) or atorvastatin (1, 2.5, or 5 μmol/L) for 1 h and then exposed to AβOs (50 nmol/L) for 12 h in the presence of vehicle or atorvastatin. Group data showing the normalization of phospho-p38MAPK to total p38MAPK were determined in each group from 4 experiments. (C) Representative Western blot showing that the p38MAPK inhibitor SB203580 prevented the decrease of synaptophysin and PSD-95 proteins induced by AβOs. Hippocampal neurons were pre-incubated with vehicle solution (control, 0.1% DMSO) or SB203580 (5 μmol/L) for 30 min and then exposed to AβOs (50 nmol/L) for 48 h in the presence of vehicle or SB203580. Group data showing the normalization of synaptophysin and PSD-95 proteins to β-actin protein were determined in each group from 4 experiments. Data are expressed as the mean±SEM. bP<0.05, cP<0.01 compared with the control group. eP<0.05, fP<0.01 compared with the AβO-treated group.
Atorvastatin inhibited AβO-induced overproduction of proinflammatory cytokines
To determine whether atorvastatin treatment can inhibit AβO-induced proinflammatory cytokine production, we examined protein levels of IL-1β, TNF-α, and IL-6 in the hippocampus. The results revealed that the concentration of IL-1β (Figure 7A), TNF-α (Figure 7B), and IL-6 (Figure 7C) in the hippocampus was significantly increased in the AβO-treated rats compared with the control rats 9 d after icv injection. Atorvastatin treatment significantly prevented AβO-induced increase in protein levels of IL-1β, TNF-α, and IL-6 in the hippocampus.

Atorvastatin suppressed the proinflammatory cytokine upregulation induced by AβOs. Treatment with atorvastatin resulted in a significant suppression of the AβO-induced increase in the hippocampal levels of the proinflammatory cytokines IL-1β (A), TNF-α (B), and IL-6 (C). Rats were treated with AβOs (5 nmol, icv) or sterile water (control). Atorvastatin (10 mg/kg) was administered daily starting 5 d before AβO treatment. Rats were euthanized on d 9, and hippocampal extracts were analyzed by ELISA. Data are expressed as the mean±SEM of 5 rats per group. cP<0.01 compared with the control group. fP<0.01 compared with the AβO-treated group.
Discussion
The present results provide the first demonstration that atorvastatin treatment abolishes the loss of synaptic markers (synaptophysin and PSD-95) induced by a single icv infusion of AβOs, which induce synaptotoxicity and memory dysfunction, two cardinal features of the early phase of AD. These results are relevant for the following two reasons. First, they provide additional evidence that atorvastatin has a potential neuroprotective action against the neuronal toxicity induced by AβOs. Second, they provide a clear demonstration that the neuroprotection afforded by atorvastatin is associated with the inhibition of proinflammatory cytokines, and these effects may be mediated by the p38MAPK signal pathway.
Converging lines of evidence suggest that AβOs play a role in the cognitive impairment characteristics of AD. A recent study indicated that Aβ dimers present in the water-soluble phase are strongly associated with AD-type dementia35 because this dimer was not detected in non-dementia patients. In our study, the rat Aβ peptide solutions used consisted primarily of dimers and monomers. It has been found that AβOs are highly neurotoxic and kill hippocampal neurons at nanomolar concentrations27. AβOs accumulate at synaptic sites36, where they bind to postsynaptic density complexes with a high affinity14,37 and disrupt synaptic plasticity38,39. This provides strong evidence for direct AβO toxicity to post-synaptic components, a possible physical basis of synaptic dysfunction in AD. The administration of AβOs into the lateral ventricle of rats or mice has been widely used to model neuroinflammation and to induce AD-related impairments40,41; however, this administration is unable to induce all pathological AD hallmarks, such as amyloid plaques and phospho-tau positive cells42.
In the present study, we demonstrated that a single icv injection of AβOs induces the loss of synaptic markers (synaptophysin and PSD-95) that are linked to a decline in learning and memory functions in the Morris water maze paradigm. Based on our findings, it is clear that rat Aβ1–42 oligomers also have significant neurotoxicity. This supports the hypothesis that synaptic dysfunction is a precocious core modification of AD43. Synapse loss is the most robust correlate of AD-associated cognitive deficits44. It was found that AβO attachment to synapses induces spine loss14. The repeated treatment with atorvastatin protects hippocampus against synaptotoxicity induced by a single icv infusion of AβOs. In cultured hippocampal neurons, we also found that AβOs caused reduced levels of critical dendritic and synaptic proteins. This AβO-induced decrease in the density of synaptophysin and PSD-95 in neuronal cultures was also prevented by atorvastatin treatment. Thus, it is tempting to propose that the promising beneficial effects of atorvastatin used to prevent the burden of AD may be related to the synaptoprotective effects. This proposal does not exclude the possibility that other mechanisms may also contribute to the neuroprotection against Aβ-induced neurotoxicity and memory impairment. It should be stressed that we only obtained evidence that AβOs caused synapse loss; however, the extent to which this synapse loss relates to the known AβO-induced functional impairment of hippocampal synapses remains to be determined.
In recent years, there has been increasing interest in the potential of statins for the treatment of AD, with the observation that the incidence of AD is markedly reduced in patients receiving statin therapy for hyperlipidemia20,45. The proposed mechanisms by which statins may act include a reduction in brain Aβ production through alterations in metabolism of amyloid precursor protein46 and a reduction in inflammation attributable to microglia activation45. In vitro experiments have shown that statins attenuate inflammatory responses mediated by Aβ peptides47. In vivo statin use has also resulted in robust anti-inflammatory effects48. Atorvastatin is used clinically worldwide and can cross the blood–brain barrier. In the present study, we demonstrated that the pretreatment with atorvastatin is also able to attenuate the production of inflammation cytokines IL-1β, TNF-α, and IL-6 observed in the hippocampus of AβO-injected rats, showing that atorvastatin has anti-inflammatory properties. This is consistent with many other studies that demonstrated that glial activation and neuroinflammation can be modulated by atorvastatin treatment.
TNF-α is a multifunctional cytokine that triggers a wide range of cellular responses. In the CNS, TNF-α, most likely through TNFR1 activation, regulates synapse damage and disrupts learning and memory. TNF-α has also been shown to participate in the Aβ-induced inhibition of LTP, which is most likely dependent on p38MAPK49. The inhibition of TNF-α signaling has been shown to attenuate AD-like pathology and cognitive impairments in transgenic mouse models as well as in AD patients50,51, whereas the upregulation of TNF-α has been shown to exacerbate AD pathology. Indeed, TNF-α levels have been found to be elevated within the cerebrospinal fluid (CSF) of AD patients by as much as 25-fold52. Studies in subjects with mild cognitive impairment who later progress to develop AD suggest that increased CSF TNF-α level is an early event, and its rise correlates with disease progression53. IL-1β, another proinflammatory cytokine, also appears to play an important role in AD pathogenesis and progression. First, the analysis of AD brain tissue demonstrates IL-1 overproduction, which closely corresponds to the extent of neuropathology found in a given brain region. Second, cell-based studies show that IL-1 can elicit the production of a number of detrimental molecules from microglia, astrocytes, and neurons54,55. More importantly, IL-1-mediated proinflammatory sequelae could damage neuronal connectivity via mechanisms beyond the neurotoxic effects of Aβ production. IL-1 may potentially contribute to the reorganization of the cytoskeleton, interrupt normal microtubule assembly and axon stabilization, and eventually result in the loss of synaptic proteins and synapses.
Finally, given that the inhibition of p38 activation is sufficient to prevent Aβ-induced neurotoxicity, as also observed by others33,56, atorvastatin treatment controls AβO-induced neurotoxicity through the regulation of p38MAPK phosphorylation. Activated p38MAPK is observed in human AD brain tissue57 and in AD-relevant animal models58,59, and cell culture studies strongly implicate p38MAPK in the increased production of proinflammatory cytokines in the glia activated by Aβ60. The activation of p38MAPK in neuronal cells has been associated with IL-1 and hyperphosphorylated tau in AD61. Similar to several other members of the MAPK family, p38MAPK is activated by dual phosphorylation; cytokines, including IL-1 and TNF-α, can affect this activation. In our present study, the phosphorylation of p38MAPK was significantly increased in cultured hippocampal neurons treated with AβOs. The finding that the inhibition of p38MAPK with the p38MAPK inhibitor SB203580 significantly suppressed AβO-decreased levels of synaptophysin and PSD-95 adds to the evidence of the role for this kinase in AβO-induced synaptotoxicity. This observation further confirms the previous finding that the inhibition of neuronal p38MAPK prevented decrease in synaptophysin level correlated with neuronal tau phosphorylation62.
In summary, the present observations that atorvastatin prevents synaptotoxicity in both in vitro and in vivo models pertinent to AD reinforces and extends the notion of the potential neuroprotective role of atorvastatin against the neuronal toxicity induced by Aβ peptides. These studies provide insights into the mechanisms by which statins may reduce AD pathogenesis.
Author contribution
Ying JIN designed the research; Ling-ling ZHANG, Hai-juan SUI, Bing LIANG, Han-ming WANG, Wen-hui QU, and Sheng-xue YU performed the research; Hai-juan SUI analyzed the data; and Ying JIN wrote the paper.
References
Haass C, Selkoe DJ . Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8: 101–12.
Cerpa W, Dinamarca MC, Inestrosa NC . Structure–function implications in Alzheimer's disease: effect of Abeta oligomers at central synapses. Curr Alzheimer Res 2008; 5: 233–43.
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 2003; 100: 10417–22.
Chang L, Bakhos L, Wang Z, Venton DL, Klein WL . Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer's disease drug candidate screening. J Mol Neurosci 2003; 20: 305–13.
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8: 79–84.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416: 535–9.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci 2004; 24: 10191–200.
Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R . Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 2010; 13: 190–6.
Liu J, Chang L, Roselli F, Almeida OF, Gao X, Wang X, et al. Amyloid-beta induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors. J Alzheimers Dis 2010; 22: 541–56.
El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS . PSD-95 involvement in maturation of excitatory synapses. Science 2000; 290: 1364–8.
Rao A, Kim E, Sheng M, Craig AM . Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 1998; 18: 1217–29.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 2005; 8: 1051–8.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL . Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27: 2866–75.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 2007; 27: 796–807.
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ . Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 2011; 31: 6627–38.
Liao JK, Laufs U . Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005; 45: 89–118.
Wang CY, Liu PY, Liao JK . Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med 2008; 14: 37–44.
Hosaka A, Araki W, Oda A, Tomidokoro Y, Tamaoka A . Statins reduce amyloid beta-peptide production by modulating amyloid precursor protein maturation and phosphorylation through a cholesterol-independent mechanism in cultured neurons. Neurochem Res 2013; 38: 589–600.
Kurata T, Miyazaki K, Kozuki M, Morimoto N, Ohta Y, Ikeda Y, et al. Progressive neurovascular disturbances in the cerebral cortex of Alzheimer's disease-model mice: protection by atorvastatin and pitavastatin. Neuroscience 2011; 197: 358–68.
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA . Statins and the risk of dementia. Lancet 2000; 356: 1627–31.
Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G . Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57: 1439–43.
Simons M, Schwarzler F, Lutjohann D, von Bergmann K, Beyreuther K, Dichgans J, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 2002; 52: 346–50.
Sparks DL, Sabbagh M, Connor D, Soares H, Lopez J, Stankovic G, et al. Statin therapy in Alzheimer's disease. Acta Neurol Scand Suppl 2006; 185: 78–86.
Li L, Cao D, Kim H, Lester R, Fukuchi K . Simvastatin enhances learning and memory independent of amyloid load in mice. Ann Neurol 2006; 60: 729–39.
Waters DD . Safety of high-dose atorvastatin therapy. Am J Cardiol 2005; 96: 69F–75F.
Clarke RM, O'Connell F, Lyons A, Lynch MA . The HMG-CoA reductase inhibitor, atorvastatin, attenuates the effects of acute administration of amyloid-beta1–42 in the rat hippocampus in vivo. Neuropharmacology 2007; 52: 136–45.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998; 95: 6448–53.
Cao C, Cirrito JR, Lin X, Wang L, Verges DK, Dickson A, et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer's disease transgenic mice. J Alzheimers Dis 2009; 17: 681–97.
Clarke RM, Lyons A, O'Connell F, Deighan BF, Barry CE, Anyakoha NG, et al. A pivotal role for interleukin-4 in atorvastatin-associated neuroprotection in rat brain. J Biol Chem 2008; 283: 1808–17.
Briones TL, Woods J . Chemotherapy-induced cognitive impairment is associated with decreases in cell proliferation and histone modifications. BMC Neurosci 2011; 12: 124.
Jin Y, Sui HJ, Dong Y, Ding Q, Qu WH, Yu SX, et al. Atorvastatin enhances neurite outgrowth in cortical neurons in vitro via up-regulating the Akt/mTOR and Akt/GSK-3beta signaling pathways. Acta Pharmacol Sin 2012; 33: 861–72.
Silva CG, Porciuncula LO, Canas PM, Oliveira CR, Cunha RA . Blockade of adenosine A(2A) receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis 2007; 27: 182–9.
Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation 2007; 4: 21.
Zhu X, Mei M, Lee HG, Wang Y, Han J, Perry G, et al. P38 activation mediates amyloid-beta cytotoxicity. Neurochem Res 2005; 30: 791–6.
Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010; 133: 1328–41.
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J . A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci 2009; 29: 4004–15.
Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, et al. Congo red and thioflavin-T analogs detect Abeta oligomers. J Neurochem 2008; 104: 457–68.
Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 2002; 924, 133–40.
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ . Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 2006; 572: 477–92.
Canas PM, Porciuncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci 2009; 29: 14741–51.
Yamada M, Chiba T, Sasabe J, Nawa M, Tajima H, Niikura T, et al. Implanted cannula-mediated repetitive administration of Abeta25–35 into the mouse cerebral ventricle effectively impairs spatial working memory. Behav Brain Res 2005; 164: 139–46.
Takeda S, Sato N, Niisato K, Takeuchi D, Kurinami H, Shinohara M, et al. Validation of Abeta1–40 administration into mouse cerebroventricles as an animal model for Alzheimer disease. Brain Res 2009; 1280: 137–47.
Walsh DM, Selkoe DJ . Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004; 44: 181–93.
Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem 2007; 100: 23–35.
Wolozin B . Cholesterol, statins and dementia. Curr Opin Lipidol 2004; 15: 667–72.
Petanceska SS, DeRosa S, Olm V, Diaz N, Sharma A, Thomas-Bryant T, et al. Statin therapy for Alzheimer's disease: will it work? J Mol Neurosci 2002; 19: 155–61.
Cordle A, Landreth G . 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci 2005; 25: 299–307.
Lee ST, Chu K, Park JE, Hong NH, Im WS, Kang L, et al. Atorvastatin attenuates mitochondrial toxin-induced striatal degeneration, with decreasing iNOS/c-Jun levels and activating ERK/Akt pathways. J Neurochem 2008; 104: 1190–200.
Wang Q, Wu J, Rowan MJ, Anwyl R . Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci 2005; 22: 2827–32.
Giuliani F, Vernay A, Leuba G, Schenk F . Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res Bull 2009; 80: 302–8.
McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis 2009; 34: 163–77.
Tarkowski E, Blennow K, Wallin A, Tarkowski A . Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 1999; 19: 223–30.
Tarkowski E, Andreasen N, Tarkowski A, Blennow K . Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry 2003; 74: 1200–5.
Woiciechowsky C, Schoning B, Stoltenburg-Didinger G, Stockhammer F, Volk HD . Brain-IL-1 beta triggers astrogliosis through induction of IL-6: inhibition by propranolol and IL-10. Med Sci Monit 2004; 10: BR325–30.
Akama KT, Van Eldik LJ . Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem 2000; 275: 7918–24.
Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci 2008; 28: 3521–30.
Sun A, Liu M, Nguyen XV, Bing G . P38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp Neurol 2003; 183: 394–405.
Jin Y, Yan EZ, Fan Y, Zong ZH, Qi ZM, Li Z . Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus. Acta Pharmacol Sin 2005; 26: 943–51.
Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, et al. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis 2002; 11: 257–74.
Lee YB, Schrader JW, Kim SU . p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000; 12: 874–80.
Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int 2001; 39: 341–8.
Li Y, Liu L, Barger SW, Griffin WS . Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 2003; 23: 1605–11.
Acknowledgements
This work was supported by grants from the Education Commission of Liaoning Province (LT2010064) and Liaoning Medical University (2012005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, Ll., Sui, Hj., Liang, B. et al. Atorvastatin prevents amyloid-β peptide oligomer-induced synaptotoxicity and memory dysfunction in rats through a p38 MAPK-dependent pathway. Acta Pharmacol Sin 35, 716–726 (2014). https://doi.org/10.1038/aps.2013.203
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.203
Keywords
This article is cited by
-
Neuroprotective effect of nose-to-brain delivery of Asiatic acid in solid lipid nanoparticles and its mechanisms against memory dysfunction induced by Amyloid Beta1-42 in mice
BMC Complementary Medicine and Therapies (2023)
-
Regulatory role of melatonin in Notch1 signaling pathway in cerebral cortex of Aβ1−42-induced Alzheimer’s disease rat model
Molecular Biology Reports (2023)
-
Therapeutic Effects of High-Intensity Interval Training Exercise Alone and Its Combination with Ecdysterone Against Amyloid Beta-Induced Rat Model of Alzheimer’s Disease: A Behavioral, Biochemical, and Histological Study
Neurochemical Research (2022)
-
Atorvastatin Attenuates Cognitive Deficits and Neuroinflammation Induced by Aβ1–42 Involving Modulation of TLR4/TRAF6/NF-κB Pathway
Journal of Molecular Neuroscience (2018)
-
Protective effect of melatonin on soluble Aβ1–42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus
Alzheimer's Research & Therapy (2016)
